Synthesis enables a structural revision of the Mycobacterium tuberculosis-produced diterpene, edaxadiene†
Jillian E.
Spangler
,
Cheryl A.
Carson
and
Erik J.
Sorensen
*
Department of Chemistry, Princeton University, Frick Chemical Laboratory, Princeton, NJ 08544, USA. E-mail: ejs@princeton.edu
First published on 11th June 2010
Abstract
A stereodivergent synthesis of the [3.3.1] bicyclic core of edaxadiene was completed utilizing a key intramolecular oxidative ketone allylation. Significant discrepancies between the spectroscopic data obtained for the synthetic construct and the natural isolate raised questions about the structural assignment of edaxadiene. A subsequent structural reassignment was validated by completion of a total synthesis of the correct structure of the natural product.
Tuberculosis is a pulmonary disease caused by the pathogen Mycobacterium tuberculosis. Roughly two billion people, or one-third of the world's population, are believed to be infected by the bacterium, resulting in over 1.5 million deaths each year.1 Despite these grave statistics, details about the infectivity and virulence of M. tuberculosis are only partially understood.2 Peters and co-workers recently disclosed their isolation and structural elucidation of a halimane-type diterpenoid, edaxadiene (1), a compound produced by M. tuberculosis.3 The genetic operon responsible for the production of edaxadiene is present only in pathogenic strains of mycobacteria.4 In addition, edaxadiene was demonstrated to be an in vitro inhibitor of macrophage maturation.3 These experimental observations suggest that production of edaxadiene is crucial for the infectivity and virulence of M. tuberculosis.
Our group has a growing interest in molecules related to the biology and treatment of tuberculosis.5 The biological importance as well as the scarcity of edaxadiene (1) led us to actively pursue a total synthesis of this intriguing natural product. Our goal was to produce sufficient quantities of this molecule to confirm its structural assignment and enable a search for its biomolecular target.
We recognized that at the heart of this challenge is the synthesis of a [3.3.1] bicyclic core bearing five contiguous stereogenic centers, including two all-carbon quaternary stereogenic centers. In addition, the relative configuration of the C13 stereogenic center was not assigned during structural elucidation. Although edaxadiene (1) has conservation of the halimane skeleton (Fig. 1), formation of a unique C7–C13 bond locks the conformation of the B-ring and produces a highly unfavorable 1,3-diaxial interaction between the C17 methyl group and C1 methylene that is absent in related diterpenes.6
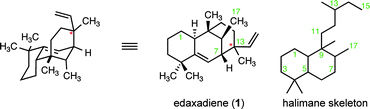 | ||
| Fig. 1 Proposed structure of edaxadiene. Stereochemistry is undefined at the starred (*) carbon. | ||
We envisioned that edaxadiene could arise from enone 3 by annulation of the leftmost ring onto the convex face of the [3.3.1] bicycle with subsequent deoxygenation of the C6 ketone (4) to form the C5–C6 olefin (Scheme 1). We reasoned that this enone (3) could be fashioned through an intramolecular ketone allylation of an enone of type 2 to forge the C7–C13 bond. Importantly, we anticipated that variation of olefin geometry and reaction conditions in this cyclization would provide access to both epimers at the unassigned C13 stereogenic center.
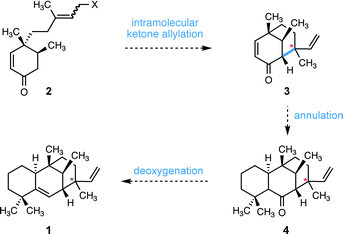 | ||
| Scheme 1 Proposed synthesis of 1via an intramolecular ketone allylation. Stereochemistry is undefined at the starred (*) carbon. | ||
Our efforts thus began with the development of a synthesis of enone 2. The thermal Diels–Alder cycloaddition of tiglaldehyde and a modified Rawal diene7 (5) provided silyl enol ether 6 (Scheme 2). Subsequent methylenation of the aldehyde provided alkene 7 in good yield on a large scale (>15 g). Regioselective hydroboration of the terminal olefin with 9-BBN furnished a B-alkyl borane that was subjected to a Suzuki–Miyaura cross-coupling reaction8 with (Z)-2-bromo-2-butene (10a) or (E)-2-bromo-2-butene (10b). The geometrical isomers 9a and 9b were obtained after deprotection of the enol ether and elimination of oxazolidinone with tetra-n-butylammonium fluoride.
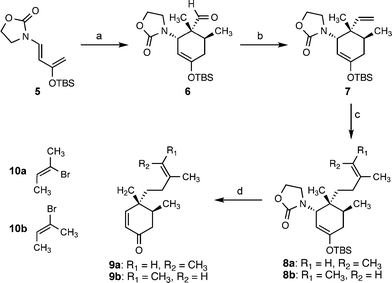 | ||
| Scheme 2 Diels–Alder-mediated synthesis of enones 9a,b. Conditions: (a) tiglaldehyde, toluene, 120 °C, 80%; (b) Ph3PCH3Br, n-BuLi, THF, 0 °C, 67%; (c) 8a: 9-BBN, THF, reflux; then 10 mol% PdCl2(dppf), aq. K3PO4, 10a, DMF, 50 °C; 8b: 9-BBN, THF, reflux; then 10 mol% PdCl2(dppf), aq. K3PO4, 10b, DMF, 50 °C; (d) 9a: TBAF, THF, 86% (2 steps); 9b: TBAF, THF, 100% (2 steps). 9-BBN = 9-borabicyclo(3.3.1)nonane; dppf = 1,1′-bis(diphenylphosphino)ferrocene; TBAF = tetra-n-butylammonium fluoride. | ||
After exploring multiple methods for constructing the C7–C13 bond to provide a [3.3.1] bicycle we found that oxidative cyclization of 9a with a bimetallic system of manganese(III) acetate and copper(II) acetate in a mixture of acetic acid and benzene (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) provided the volatile [3.3.1] bicycle 12a in moderate yield as a single stereoisomer (Scheme 3).9 This cyclization most likely proceeds through the intermediacy of an A(1,3) minimized intermediate (11a) to provide 12a. Utilizing the same reaction conditions enone 9b underwent cyclization to provide 12b, the opposite C13 epimer, albeit with a lower degree of diastereoselectivity. We were pleased to find that we could gain access to both C13 epimers by controlling the geometry of the starting trisubstituted olefin.
:1) provided the volatile [3.3.1] bicycle 12a in moderate yield as a single stereoisomer (Scheme 3).9 This cyclization most likely proceeds through the intermediacy of an A(1,3) minimized intermediate (11a) to provide 12a. Utilizing the same reaction conditions enone 9b underwent cyclization to provide 12b, the opposite C13 epimer, albeit with a lower degree of diastereoselectivity. We were pleased to find that we could gain access to both C13 epimers by controlling the geometry of the starting trisubstituted olefin.
![Synthesis of the [3.3.1] bicyclic core of 1via a stereodivergent intramolecular oxidative ketone allylation. Conditions: (a) Mn(OAc)3·2H2O, Cu(OAc)2·H2O, 80–100 °C, AcOH–benzene (1 : 3), 12a: 57%, 12b: 51% (1.4 : 1 dr); (b) NaBH4, MeOH; (c) LiAlH4, Et2O, 0 °C, 13a: 87% (2 steps), 13b: 73% (2 steps); (d) Martin sulfurane, benzene, 80 °C, 14a: 23%, 14b: 34%.](/image/article/2010/SC/c0sc00284d/c0sc00284d-s3.gif) | ||
| Scheme 3 Synthesis of the [3.3.1] bicyclic core of 1via a stereodivergent intramolecular oxidative ketone allylation. Conditions: (a) Mn(OAc)3·2H2O, Cu(OAc)2·H2O, 80–100 °C, AcOH–benzene (1:3), 12a: 57%, 12b: 51% (1.4:1 dr); (b) NaBH4, MeOH; (c) LiAlH4, Et2O, 0 °C, 13a: 87% (2 steps), 13b: 73% (2 steps); (d) Martin sulfurane, benzene, 80 °C, 14a: 23%, 14b: 34%. | ||
It was at this point in our efforts that we began to note discrepancies between the reported 13C shifts of the [3.3.1] core of edaxadiene and our synthetic constructs. To address our scepticism of the proposed structure, we converted the diastereomeric bicycles 12a,b into dienes 14a,b, bearing the full [3.3.1] bicyclic core of structure 1. Thus, stepwise reduction of each enone to the saturated alcohol (13a and 13b) was followed by dehydration to the corresponding alkene (14a and 14b) with Martin sulfurane in refluxing benzene.10 The unusually high temperature requirement for the dehydration is attributed to the equatorial disposition of the substrate alcohols. Although the high volatility and non-polarity of these compounds complicated their isolation, we were able to obtain sufficient quantities of each C13 epimer for characterization via semi-preparative gas chromatography. The spectroscopic data for each C13 epimer differed dramatically from the reported spectra of edaxadiene; most notably, the reported 13C-NMR chemical shift at C13 was substantially different (Δδ of >30 ppm) from the 13C chemical shifts observed for bicycles 14a,b (Fig. 2).
 | ||
| Fig. 2 Discrepancies in 13C-NMR shifts that indicate the structure of edaxadiene has been incorrectly assigned. | ||
These significant differences between the 13C-NMR data of the isolated 1 and our synthetic bicycles (14a and 14b) led to our re-evaluation of the reported spectroscopic data for 1. Our consideration of this data led us to propose compound 15 (Fig. 3) as the actual structure of edaxadiene. This proposal is consistent with the 13C-NMR data, most notably the deshielded nature of C13. Observation of the parent [M − H2O]+ ion by EI mass spectrometry is presumably due to the facile fragmentation of the tertiary allylic alcohol at C13 of 15. We further suspect that edaxadiene, as reported, is likely not a pure compound, but rather a diastereoisomeric mixture at the remote C13 stereogenic center, as indicated by fine doubling of the olefinic protons in the 1H spectrum of 1. Our confidence in this reassignment was bolstered by a comparison of this structure to known terpenes in the literature, which indicated that this was a previously identified halimane diterpene, nosyberkol (15), isolated in 2004 from extracts of the Red Sea sponge Raspailia sp.11 Furthermore nosyberkol (15, also referred to as isotuberculosinol) was previously speculated to be the product of the Rv3378c enzyme produced by M. tuberculosis.12
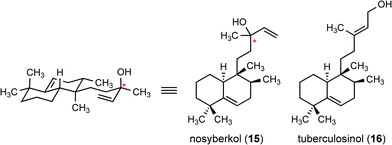 | ||
| Fig. 3 Structures of nosyberkol and tuberculosinol. Stereochemistry is unassigned at the starred (*) carbon. | ||
This proposed structural reassignment led us to develop a synthesis of nosyberkol (15) for structural verification.13 Our efforts began with an exo-selective Diels–Alder cycloaddition14 of diene 17 (available in two steps from 2,2-dimethylcyclohexanone)15 and ethyl tiglate to give the desired cycloadduct as an inseparable mixture of diastereoisomers (2:1) (Scheme 4). Subsequent reduction of the esters and separation of the primary alcohols provided the desired C10 epimer.16 Aldehyde 18 was obtained in good yield using the conditions of Parikh and Doering.17 Our attempts to perform a homologation of 18 through reaction with a phosphonate or phosphonium ylide led to no product formation; however, an aldol condensation with the sodium enolate of acetone and conjugate reduction of the resultant enone with Wilkinson's catalyst18 allowed the desired homologation to ketone 20. Our synthesis of nosyberkol was completed by the addition of vinylmagnesium bromide to the ketone to provide the desired compound as a 1.5:1 diastereomeric mixture at the tertiary allylic alcohol-bearing carbon. The spectroscopic data obtained for synthetic nosyberkol (15) were identical with those reported for both natural nosyberkol and edaxadiene (Fig. 2, see ESI†).19
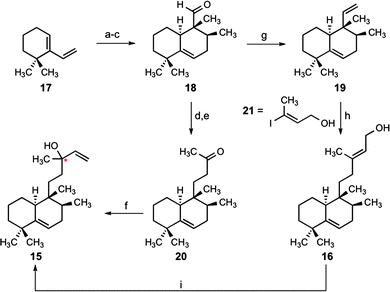 | ||
| Scheme 4 Syntheses of nosyberkol and tuberculosinol via an exo-selective Diels–Alder reaction. Conditions: (a) ethyl tiglate, neat, 160 °C, 71% (2:1 exo:endo); (b) LiAlH4, THF, 40 °C, 56% (+24% endo isomer); (c) SO3·pyridine, NEt3, CH2Cl2–DMSO, 0 °C, 86%; (d) acetone, NaHMDS, THF, −78 → 23 °C, 87%; (e) 10 mol% Rh(PPh3)3Cl, HSiEt3, CH2Cl2, 40 °C, 83%; (f) vinylmagnesium bromide, THF, 0 °C, 93% (1.5:1 dr at the starred (*) carbon); (g) Ph3PCH3Br, KHMDS, 0 °C, THF, 91%; (h) 9-BBN, THF, 80 °C; then 10 mol% PdCl2(dppf), Ph3As, Cs2CO3, 21, DMF, 73%; (i) 20 mol% CuCl2, acetone, 20% (dr = 1:1 at the starred (*) carbon). 9-BBN = 9-borabicyclo(3.3.1)nonane; dppf = 1,1′-bis(diphenylphosphino)ferrocene. | ||
We were also intrigued by the reported biomimetic conversion of tuberculosinol20 (16) (Fig. 3) into edaxadiene (15) by treatment with a mixture of copper(II) chloride and N,N′-dicyclohexylcarbodiimide (DCC).3 To examine this conversion we pursued a short synthesis of tuberculosinol from aldehyde 18. Thus methylenation of the aldehyde provided diene 19 in good yield. Subsequent regioselective hydroboration with 9-BBN and a palladium-mediated cross-coupling with (E)-3-iodobut-2-en-1-ol21 (21) (Scheme 4) provided tuberculosinol (16). On exposure to catalytic copper(II) chloride tuberculosinol (16) was converted into nosyberkol (15) (Scheme 4). We found that addition of DCC is unnecessary for this transformation and propose that this is a Lewis acid-mediated allylic transposition.22 In addition, we observed elimination of the allylic alcohol to provide a mixture of dehydrated products (34% as an E/Z mixture at the C12–C13 olefin); however, formation of the proposed structure 1 was never observed.
In conclusion, a rapid and stereodivergent synthesis of the core of the originally proposed structure of edaxadiene (1) raised questions regarding its assignment. Re-evaluation of the spectral data led to the proposal that edaxadiene is actually nosyberkol (15), a known diterpene previously isolated from Raspailia sp. Furthermore, an independent synthesis of nosyberkol has unambiguously established this structural revision.23 This synthesis of nosyberkol should provide sufficient quantities of this compound to elucidate its role in the infectivity and virulence of M. tuberculosis.
Acknowledgements
We gratefully acknowledge Princeton University, the National Science Foundation (Graduate Research Fellowship for J. E. S.), the Natural Sciences and Engineering Research Council of Canada (Postdoctoral Fellowship for C. A. C.), and the Merck Research Laboratory for supporting this work. For invaluable technical assistance during the course of this project we gratefully acknowledge Lotus Separations (preparative SFC) and John Eng (high resolution EI mass spectrometry).Notes and references
- World Health Organization, Fact Sheet 104, March 2010.
- D. G. Russell, Nat. Rev. Microbiol., 2007, 5, 39–47 Search PubMed
.
- F. M. Mann, M. Xu, X. Chen, D. B. Ulton, D. G. Russell and R. J. Peters, J. Am. Chem. Soc., 2009, 131, 17526–17527 CrossRef CAS
- K. Pethe, D. L. Swenson, S. Alonso, J. Anderson, C. Wang and D. G. Russell, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 13642–13647 CrossRef CAS
- S. D. Tilley, K. P. Reber and E. J. Sorensen, Org. Lett., 2009, 11, 701–703 CrossRef CAS
-
E. Breitmaier, Terpenes, J. Wiley-VCH, Germany, 2006 Search PubMed
- Y. Huang, T. Iwama and V. H. Rawal, J. Am. Chem. Soc., 2000, 122, 7843–7844 CrossRef CAS
- N. Miyaura, T. Ishiyama, H. Sasaki, M. Ishikawa, M. Satoh and A. Suzuki, J. Am. Chem. Soc., 1989, 111, 314–321 CrossRef
- S. A. Kates, M. A. Dombroski and B. B. Snider, J. Org. Chem., 1990, 55, 2427–2436 CrossRef CAS
- J. C. Martin and R. J. Arhart, J. Am. Chem. Soc., 1971, 93, 4327–4329 CrossRef CAS
- A. Rudi, M. Aknin, E. Gaydou and Y. Kashman, J. Nat. Prod., 2004, 67, 1932–1935 CrossRef CAS
- C. Nakano, T. Sato and T. Hoshino, Koen Yoshishu – Koryo, Terupen oyobi Seiyu Kagaku ni kansuru Toronkai, 2005, 49, 247–249 Search PubMed
- Unfortunately the published spectrum of nosyberkol (15) was provided in CDCl3, whereas the published spectrum of the proposed edaxadiene (1) was provided in C6D6, preventing a direct spectral comparison.
- D. S. de Miranda, G. J. A. de Conceição, J. Zukerman-Schpector, M. C. Guerrero, U. Schuchardt, A. C. Pinto, C. M. Rezende and J. Marsaioli, J. Braz. Chem. Soc., 2001, 12, 391–402 CAS
- S. P. Tanis and Y. M. Abdallah, Synth. Commun., 1986, 16, 251–559 CrossRef CAS
- We could further separate the two enantiomers of the exo primary alcohols to provide enantioenriched material (>99% ee) through purification with supercritical fluid chromatography (see ESI†).
- J. R. Parikh and W. E. Doering, J. Am. Chem. Soc., 1967, 89, 5505–5507 CrossRef CAS
- I. Ojima and T. Kogure, Tetrahedron Lett., 1972, 13, 5035–5038 CrossRef
- The stereochemistry of C13 was unassigned in the original isolation of nosyberkol (ref. 11). It is also unclear whether the original isolate was a single diastereoisomer at this stereogenic center.
- C. Nakano, T. Okamura, T. Sato, T. Dairi and T. Hoshino, Chem. Commun., 2005, 1016–1018 RSC
- S. Chen, R. F. Horvath, J. Joglar, M. J. Fisher and S. J. Danishefsky, J. Org. Chem., 1991, 56, 5834–5845 CrossRef CAS
- K. Arata and C. Matsuura, Chem. Lett., 1989, 1797 CAS
- Snider and co-workers independently discovered that the original structural assignment for edaxadiene is actually nosyberkol. Their studies, which also include a synthesis of nosyberkol, were published in Organic Letters (see: N. Maugel, F. M. Mann, M. L. Hillwig, R. J. Peters and B. B. Snider, Org. Lett., 2010, 12, 2626–2629 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic data; interview with Erik Sorensen. See DOI: 10.1039/c0sc00284d |
| This journal is © The Royal Society of Chemistry 2010 |