Stereoselectivity in oxyallyl–furan (4 + 3) cycloadditions: control of intermediate conformations and dispersive stabilisation in cycloadditions involving oxazolidinone auxiliaries†
Elizabeth H.
Krenske
a,
K. N.
Houk
*a,
Andrew G.
Lohse
b,
Jennifer E.
Antoline
b and
Richard P.
Hsung
*b
aDepartment of Chemistry and Biochemistry, University of California, Los Angeles, CA 90095, USA. E-mail: houk@chem.ucla.edu
bDivision of Pharmaceutical Sciences and Department of Chemistry, University of Wisconsin, Madison, WI 53705, USA. E-mail: rphsung@pharmacy.wisc.edu
First published on 25th June 2010
Abstract
Chiral oxazolidinones were previously thought to control cycloaddition stereoselectivity by steric crowding of one face of the substrate. We have discovered that in (4 + 3) cycloaddition reactions of oxyallyls, the stereoinduction is caused instead by stabilising CH–π interactions that lead to reaction at the more crowded face of the oxyallyl. Density functional theory calculations on the (4 + 3) cycloadditions of oxazolidinone-substituted oxyallyls with furans establish unexpected transition state conformations and a new explanation of selectivity.
Introduction
Amino acid-derived oxazolidinones are used extensively as chiral auxiliaries, following their introduction by Evans.1 They have been applied to various cycloaddition reactions, including Diels–Alder2–5and 1,3-dipolar6 cycloadditions. We have found that they also provide useful stereoselectivities in the (4 + 3) cycloadditions of dienes with oxyallyls. These (4 + 3) cycloadditions are a key step in the transformation shown in Scheme 1. Oxidation of an allenamide with DMDO generates an oxyallyl, which is then trapped with a furan.7,8 This sequence is a valuable synthetic procedure for formation of 7-membered carbocycles,9,10 which had previously been achieved using various other chiral, donor-substituted oxyallyl cations.11,12 Use of 4-phenyloxazolidinone as the auxiliary (R = Ph) provided the diastereomeric endo cycloadducts I and II in a ratio of 82![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :18,13 and the ratio increased to ≥96:4 when ZnCl2 was included in the reaction mixture.7
:18,13 and the ratio increased to ≥96:4 when ZnCl2 was included in the reaction mixture.7
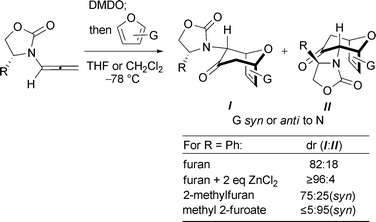 | ||
| Scheme 1 Oxazolidinone-directed asymmetric (4 + 3) cycloadditions of oxyallyls with furans. | ||
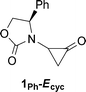
The transformation shown in Scheme 1 is believed to involve the oxyallyl 1 (Scheme 2). The Cα–N bond in 1 is expected to have substantial double bond character, leading to the possibility of E and Z isomers.
 | ||
| Scheme 2 Isomers of the oxyallyl 1Ph. | ||
The mechanism by which oxazolidinones control cycloaddition stereoselectivity was examined by Evans et al. during studies of Diels–Alder reactions (Scheme 3). These reactions were catalysed by ionisable aluminium-containing Lewis acids such as Et2AlCl.2 Evans showed that the stereoinduction arose through steric control. Chelation of the N-acyloxazolidinone 2 with AlEt2+ was suggested to produce the active dienophile, 3, which is constrained to have a geometry in which the enone and the oxazolidinone ring lie within the same plane. Cycloaddition at the less-crowded face of 3, opposite the iPr group, leads to the major product.
 | ||
| Scheme 3 Evans' oxazolidinone-directed asymmetric Diels–Alder reactions. | ||
We originally supposed that a similar mode of stereocontrol was present in our oxyallyl–furan cycloadditions. Taking into account the E and Z isomers of 1, there are four plausible transition-state geometries. These are shown in Scheme 4. The less-crowded geometries, A and C, were presumed to be preferred over the more-crowded B and D. Out of A and C, geometry C was thought to be preferred, since it would lead to the major product observed experimentally. Moreover, C has a suitable geometry for forming a chelate with ZnCl2, and the stereoselectivity was greater in the presence of ZnCl2.
 | ||
| Scheme 4 | ||
However, the cycloadditions of 1Ph with substituted furans do not agree with this model. The reaction of 1Ph with 2-methylfuran afforded the same major product as furan itself (I), but the reaction of 1Ph with methyl 2-furoate provided the opposite diastereomer (II) as the major cycloadduct.8 This reversal of stereoselectivity cannot easily be explained by the Evans-type mechanism.
We have undertaken a computational study, and report here that the factors that control stereoselectivity in oxyallyl (4 + 3) cycloadditions are different from those established for Diels–Alder reactions involving the same oxazolidinone auxiliary.
The cycloaddition of 1Ph with furan is found to involve only the E conformer—geometries A and B shown in Scheme 4. This is the case even in the presence of ZnCl2. The stereoselectivity is controlled not by steric repulsion between furan and the Ph group, but by “steric attraction”, where a stabilising CH–π interaction between furan and Ph favours cycloaddition at the more hindered π-face (geometry B). The sole involvement of 1-E, and the presence or absence of attractive CH–π interactions at the TS, correctly predict the stereoselectivities for other oxazolidinones. The reversal of stereoselectivity observed with methyl 2-furoate is also explained by a mechanism involving only the E conformer of the oxyallyl species.
Results and discussion
Electronic structure of donor-substituted oxyallyls
Density functional theory calculations were used to characterise the electronic structure of the oxazolidinone-substituted oxyallyls 1. The parent oxyallyl, 4 (Fig. 1), is a diradical, but is not an energy minimum. Highly-correlated calculations and photoelectron spectroscopy show it to have an open-shell singlet ground state, which lies 1.3 kcal mol−1 below the triplet and undergoes a barrierless ring closure to cyclopropanone.14,15 The diradical character of 4 is reflected quite well by B3LYP calculations.16–19 At the B3LYP/6-311G(2d,p) level, the singlet diradical of 4 lies 10.2 kcal mol−1 below the closed-shell singlet (which has one imaginary vibrational frequency). Although a DFT treatment is not ideal for the singlet diradical, and it predicts the singlet diradical to lie 0.8 kcal mol−1 above the triplet, this error is relatively small.![Energies of different electronic states of oxyallyls 4 and 5a–c [ΔE, kcal mol−1, B3LYP/6-311G(2d,p)].](/image/article/2010/SC/c0sc00280a/c0sc00280a-f1.gif) | ||
| Fig. 1 Energies of different electronic states of oxyallyls 4 and 5a–c [ΔE, kcal mol−1, B3LYP/6-311G(2d,p)]. | ||
In the presence of a donor substituent, the closed-shell singlet of the oxyallyl (zwitterion) is stabilised. For both 5a (X = NMe2) and 5b (X = OMe), B3LYP/6-311G(2d,p) calculations indicate a closed-shell ground state, which is stable with respect to an open-shell representation. The sulfur-substituted analogue 5c is more similar to 4 in retaining a singlet diradical ground state, but the closed-shell singlet lies only 2.9 kcal mol−1 higher in energy.
The oxazolidinone-substituted oxyallyl 1Ph has electronic properties similar to 5a and 5b. B3LYP/6-311G(2d,p) calculations indicate a negligible singlet instability (0.1 kcal mol−1). The substituted structure, 1Ph-E, is a local minimum on the B3LYP/6-31G(d) potential energy surface, but attempts to locate 1Ph-Z led instead to the corresponding cyclopropanone. The cyclopropanone 1Ph-Ecyc is 4.8 kcal mol−1 more stable than 1Ph-E (ΔH), and is the global minimum among all of the conformational and structural isomers of 1Ph. Details of these related species are given in the ESI.†
The geometry of 1Ph-E is shown in Fig. 2. The CC bond lengths in 1Ph-E are similar to those in 5a (also shown), and indicate that there is substantial electron delocalisation from nitrogen onto the allyl cation. The Cα–Cβ distance (1.46 Å) is 0.04 Å shorter than the CC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O–CCC bond in methyl vinyl ketone (6). The Cβ–Cω distance (1.40 Å) is only 0.02 Å longer than the CC bond of acetone enolate (7). The Cα–N bond (1.33 Å) is intermediate in length between a C–N and a CN bond.
O–CCC bond in methyl vinyl ketone (6). The Cβ–Cω distance (1.40 Å) is only 0.02 Å longer than the CC bond of acetone enolate (7). The Cα–N bond (1.33 Å) is intermediate in length between a C–N and a CN bond.
![Structure and molecular orbitals of 1Ph-E, and related compounds [bond lengths in Å, B3LYP/6-31G(d)].](/image/article/2010/SC/c0sc00280a/c0sc00280a-f2.gif) | ||
| Fig. 2 Structure and molecular orbitals of 1Ph-E, and related compounds [bond lengths in Å, B3LYP/6-31G(d)]. | ||
Fig. 2 also shows diagrams of the HOMO and LUMO of 1Ph-E.20 The HOMO is enolate-like, and has a larger coefficient on Cω than on Cα. The LUMO has both allyl cation and iminium π* character, with a larger coefficient on Cα than Cω. All of these features indicate that 1Ph-E is best regarded as an iminium enolate.
Transition states for (4 + 3) cycloadditions
Detailed earlier studies by Cramer and Barrows21 have revealed that the cycloadditions of oxyallyl cations with dienes can take place by several different mechanisms. For example, the highly electrophilic 2-hydroxyallyl cation was calculated to undergo (4 + 3) cycloaddition with furan by a two-step process. In comparison, the combination of enolate and iminium character in 1Ph results in behaviour that is less markedly electrophilic. Transition states for the endo (4 + 3) cycloaddition of 1Ph with furan are depicted in Fig. 3. The reaction is a concerted process, and bond development at the TS is more advanced at the nucleophilic carbon, Cω.![Transition structures and activation energies for the cycloaddition of 1Ph with furan [298.15 K, B3LYP/6-31G(d)].](/image/article/2010/SC/c0sc00280a/c0sc00280a-f3.gif) | ||
| Fig. 3 Transition structures and activation energies for the cycloaddition of 1Ph with furan [298.15 K, B3LYP/6-31G(d)]. | ||
Although only the E isomer was located as an energy minimum for 1Ph, cycloaddition transition states can be located for either the E or Z geometry. The latter are substantially higher in energy.22 For the E geometry, two concerted transition states were found—TSA and TSB—which lead to cycloadducts II and I, respectively. For the Z geometry, a concerted TS was located when furan adds to the more crowded π-face (TSD), but addition to the less crowded face is a stepwise process (TSC). Both TSC and TSD lie at high energies, due to electrostatic repulsion between the oxyallyl and carbonyl oxygens.
The Z transition states (C and D) are ≥14 kcal mol−1 higher in energy than the E transition states (A and B); hence, cycloaddition takes place exclusively via1Ph-E. Unexpectedly, however, it is 0.2 kcal mol−1 easier (ΔΔH‡) for furan to add to the more crowded face of 1Ph-E (TSB) than to the less crowded face (TSA). The corresponding free energy difference is similar, ΔΔG‡ = 0.3 kcal mol−1. The small energy difference between TSA and TSB is in good qualitative agreement with the experimental stereoselectivity of 82:18 in favour of I. The selectivity is affected little by the choice of basis set. Single-point calculations with the 6-311G(2d,p) and 6-311+G(2df,p) basis sets yield predictions of 0.4 kcal mol−1 and 0.5 kcal mol−1, respectively.
Surprisingly, similar observations are made for the ZnCl2-mediated cycloadditions of 1Ph.23 Unlike Evans' dienophile–AlEt2+ complex, the ZnCl2 complex of 1Ph is 6.2 kcal mol−1 more stable as the monodentate form, 1Ph-E·ZnCl2 (Scheme 5), than as the chelate 1Ph-Z·ZnCl2.24 The cycloaddition transition structures for 1Ph-E·ZnCl2 and 1Ph-Z·ZnCl2 are analogous to those for uncomplexed 1Ph, except more asynchronous (see ESI†). Coordination to ZnCl2 lowers the activation energies by as much as 18.8 kcal mol−1, but the exclusive participation of the E geometry is maintained. TSs for 1Ph-Z·ZnCl2 lie ≥5.2 kcal mol−1 above those for 1Ph-E·ZnCl2. Cycloaddition at the more crowded face is again preferred for 1Ph-E·ZnCl2, the selectivity being ΔΔH‡ = 1.1 kcal mol−1. This value is 0.9 kcal mol−1 greater than in the thermal reaction, consistent with the higher experimental stereoselectivity (≥96:4).
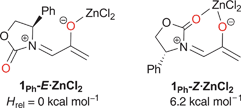 | ||
| Scheme 5 | ||
Despite their quite different surface areas and dipole moments, the structures and energies of TSA and TSB are affected little by solvation, as modeled by CPCM calculations.25 Free energies of solvation were calculated for TSA and TSB with B3LYP and several basis sets, and with additional solution-phase structure optimisations. The stereoselectivities for the thermal reactions in solution ranged from 0 to −0.4 kcal mol−1 (ΔΔG‡), compared with −0.3 kcal mol−1 in the gas phase. Details are provided in the ESI.†
The low energy of TSB appears to arise from an attractive interaction between furan and Ph. These two groups are suitably arranged for an edge-to-face CH–π interaction, with H-3 on the furan lying approximately 2.85 Å from the centre of the Ph ring. The interaction includes an electrostatic component, as indicated by a slightly (0.02e) more positive Mulliken charge on H-3 in TSB than in TSA, and is expected to include a dispersion component as in other aryl–aryl interactions.
Because the B3LYP functional does not generally model dispersion energies accurately,26 we employed several other methods to quantify the role of dispersion in TSA/B. Grimme's simple empirical formula (B3LYP-D, 2006), which sums the interatomic interactions in a pairwise fashion,27 predicts that the total dispersion energy, Edisp, is 1.5 kcal mol−1 larger (more stabilizing) in TSB than in TSA. The furan–Ph interaction is 2.4 kcal mol−1 larger in TSB than in TSA.
The B3LYP-D dispersion correction strongly favours TSB, and indeed overestimates the stereoselectivity.28 Grimme and Antony's B97-D functional,27,29 which is explicitly parameterised to provide a more sophisticated treatment of short- and long-range interactions, with the aug-cc-pVTZ basis set indicates a 2.2 kcal mol−1 preference for TSB. A similar result is obtained with Truhlar and Zhao's M06-2X functional,30 which includes attractive terms mimicking dispersion. The π-facial discrimination between TSA and TSB at the M06-2X/6-31G(d) level is ΔΔE‡ = 1.4 kcal mol−1, and this value increases to 2.2 kcal mol−1 upon full geometry optimisation. Although these newer functionals have not been as extensively benchmarked for cycloaddition transition states as B3LYP, and indeed overestimate the stereoselectivity, they are consistent in their support for a stabilising CH–π interaction in TSB.
Stereoselectivities for other oxazolidinones
A combination of steric repulsion and attractive CH–π interactions determines the stereoselectivity induced by other oxazolidinones. We calculated transition states analogous to TSA and TSB for 4-Bn- and 4-iPr-5,5-Ph2-substituted oxazolidinones, and determined their experimental dr values.31 The predicted and observed selectivities are listed in Table 1.


When the 4-phenyl substituent on the oxazolidinone is replaced by a benzyl group, the possibility of a stabilising CH–π interaction is eliminated and replaced by a repulsive furan–alkyl interaction. This is illustrated in Fig. 4. The opposite stereoisomer is now favoured by 0.1 kcal mol−1. Experimentally, II is indeed obtained as the major product, with a dr of 23:77 (I:II). An even higher selectivity in favour of II is obtained with Seebach et al.'s5 4-iPr-5,5-Ph2-oxazolidinone. In this case, the favoured TS benefits from both the minimisation of repulsive interactions between furan and the iPr group, and a stabilising CH–π interaction between furan and the C-5 Ph group trans to iPr. The calculated selectivity of 2.3 kcal mol−1 in favour of II agrees with the higher experimental selectivity for II (dr 6:94). In the absence of the two phenyl rings at C-5, the diastereoselectivity is only 45:55.7 These correlations accentuate the powerful impact that a non-bonding force such as a CH–π interaction can exert on the stereochemical course of cycloadditions.
 | ||
| Fig. 4 Control of stereoselectivity by a combination of stabilising furan–Ph interactions and destabilising furan–alkyl interactions for 1Ph, 1Bn, and 1i-Pr/Ph. Note: for 1i-Pr/Ph, C-4 of the oxazolidinone has the S configuration, whereas 1Ph and 1Bn are R. | ||
Reversal of stereoselectivity for methyl 2-furoate
The E-only mechanism predicts that the cycloadditions of 1Ph with furan and methyl 2-furoate should give opposite stereoselectivities. The four stereo- and regioisomeric TSs for cycloaddition of 1Ph-E with methyl 2-furoate are shown in Fig. 5. The TS leading to the experimental major product, syn-II (TSE), lies ≥1.9 kcal mol−1 below the other three TSs. The syn regioselectivity is expected, as it is favoured by the reactants' frontier orbital coefficients. The reversal of stereoselectivity can be ascribed to repulsive interactions between the Ph group and the 2-COOMe group. In TSG, H-3 on the furan lies only 2.81 Å from the midpoint of the Ph ring than in TSB, but the electrostatic repulsion between the Ph π cloud and the carbonyl group at C-2 of the furan outweighs the stabilising effect of CH–π attraction. Cycloaddition therefore takes place preferentially through the less crowded syn TS.![Transition structures and activation energies for the cycloaddition of 1Ph with methyl 2-furoate [298.15 K, B3LYP/6-31G(d)].](/image/article/2010/SC/c0sc00280a/c0sc00280a-f5.gif) | ||
| Fig. 5 Transition structures and activation energies for the cycloaddition of 1Ph with methyl 2-furoate [298.15 K, B3LYP/6-31G(d)]. | ||
Conclusions
Density functional theory calculations make it clear that only the E isomer of the oxyallyl 1 undergoes cycloaddition with furans. The Z isomer is subject to severe electrostatic repulsion between the oxyallyl and carbonyl oxygen atoms, and this destabilisation is not overcome even upon coordination to ZnCl2. Santos et al. have recently reported related calculations on Evans' Diels–Alder reactions (Scheme 3), and suggest that rather than taking place via the chelate, these reactions instead involve a complex of 2 in which the carbonyl groups are antiparallel (i.e. E), and each is bound to an AlEt2Cl.32 They proposed that the stereoselectivity can then be traced to repulsive interactions between the Et groups on the Lewis acid and the iPr group on the oxazolidinone. Antiparallel conformations of N-acyloxazolidinones have also been invoked in oxazolidinone-directed nitrile ylide cycloadditions6 and dynamic kinetic resolutions of α-halo carbonyl compounds.33 Santos' calculations confirmed, however, that Evans' original sterically-controlled mechanism of stereoinduction would still be valid within a stoichiometric regime where the chelate would exist.32The origin of stereoinduction in the (4 + 3) cycloadditions of 1Ph is quite distinct from these processes, and could be relevant to various other asymmetric reactions. CH–π interactions are well known to influence conformational equilibria, crystal packing, the structures of biological molecules, and molecular recognition.34,35 Previous computational studies have shown that CH–π interactions are responsible for the endo selectivity of the Diels–Alder reaction of butadiene with cyclopropene36 and the hetero-Diels–Alder reactions of ortho-xylylenes with benzaldehyde.37 The calculated selectivities in these cases amounted to 1–2 kcal mol−1.
CH–π interactions appear to be general design elements in asymmetric reactions. We recently discovered a similar example of this form of stereocontrol, in (4 + 3) cycloadditions of α-methylbenzyloxy-substituted siloxyallyl cations.38 Like 1Ph, these oxyallyl cations are relatively rigid species, and the aromatic group is held roughly perpendicular to the molecular plane. Furan was found to add to the aryl-containing face with a selectivity of approximately 1 kcal mol−1. It is likely that further examples of stereocontrol via CH–π interactions will be reported in the future.
Experimental
Density functional theory calculations were performed using the Gaussian 0339 and Gaussian 0940 packages. The nature of each stationary point was checked by means of frequency calculations, and transition states were further verified by IRC calculations.41 Zero-point energy and thermal corrections were derived from the B3LYP or M06-2X frequencies, but not scaled. The effects of solvation were simulated by means of CPCM calculations25 using UAKS radii (Gaussian 03).Acknowledgements
We thank the NIH, NSF, and Australian–American Fulbright Commission for generous financial support (GM-36700 to KNH, GM66055 to RPH) and the NCSA, UCLA ATS, and UCLA IDRE for computer resources. We also thank Dr Victor G. Young, Jr. of The University of Minnesota for solving X-ray single-crystal structures.Notes and references
- D. A. Evans, Aldrichimica Acta, 1982, 15, 23 CAS; G. Zappia, G. Cancelliere, E. Gacs-Baitz, G. Delle Monache, D. Misiti, L. Nevola and B. Botta, Curr. Org. Synth., 2007, 4, 238 Search PubMed; D. J. Ager, I. Prakash and D. R. Schaad, Aldrichimica Acta, 1997, 30, 3 CAS; D. A. Evans, G. Helmchen and M. Rüping, in Asymmetric Synthesis – The Essentials, ed. M. Christmann and S. Bräse, Wiley-VCH, Weinheim, 2nd edn, 2008, pp. 3–9 Search PubMed.
- D. A. Evans, K. T. Chapman and J. Bisaha, J. Am. Chem. Soc., 1984, 106, 4261 CrossRef CAS; D. A. Evans, K. T. Chapman and J. Bisaha, Tetrahedron Lett., 1984, 25, 4071 CrossRef CAS; D. A. Evans, K. T. Chapman, D. T. Hung and A. T. Kawaguchi, Angew. Chem., Int. Ed. Engl., 1987, 26, 1184 CrossRef; D. A. Evans, K. T. Chapman and J. Bisaha, J. Am. Chem. Soc., 1988, 110, 1238 CrossRef CAS.
- M. R. Banks, A. J. Blake, J. I. G. Cadogan, A. A. Doyle, I. Gosney, P. K. G. Hodgson and P. Thorburn, Tetrahedron, 1996, 52, 4079 CrossRef CAS.
- D. A. Evans, D. H. Brown Ripin, J. S. Johnson and E. A. Shaughnessy, Angew. Chem., Int. Ed. Engl., 1997, 36, 2119 CrossRef CAS.
- T. Hintermann and D. Seebach, Helv. Chim. Acta, 1998, 81, 2093 CrossRef CAS; C. Gaul, B. W. Schweizer, P. Seiler and D. Seebach, Helv. Chim. Acta, 2002, 85, 1546 CrossRef CAS.
- M. P. Sibi, T. Soeta and C. P. Jasperse, Org. Lett., 2009, 11, 5366 CrossRef CAS.
- H. Xiong, R. P. Hsung, C. R. Berry and C. Rameshkumar, J. Am. Chem. Soc., 2001, 123, 7174 CrossRef CAS.
- J. E. Antoline and R. P. Hsung, Synlett, 2008, 739 CAS.
- I. V. Hartung and H. M. R. Hoffmann, Angew. Chem., Int. Ed., 2004, 43, 1934 CrossRef CAS.
- M. Harmata, Acc. Chem. Res., 2001, 34, 595 CrossRef CAS.
- C. B. W. Stark, S. Pierau, R. Wartchow and H. M. R. Hoffmann, Chem.–Eur. J., 2000, 6, 684 CrossRef CAS.
- D. I. MaGee, E. Godineau, P. D. Thornton, M. A. Walters and D. J. Sponholtz, Eur. J. Org. Chem., 2006, 3667 CrossRef CAS.
- We use the term “endo” to denote the relationship between the diene and the oxyallyl oxygen in I/II and the TSs leading to them. Hoffmann has used the term “compact” to describe this type of (4 + 3) cycloaddition geometry. See: H. M. R. Hoffmann, Angew. Chem., Int. Ed., 1973, 12, 819 Search PubMed.
- M. B. Coolidge, K. Yamashita, K. Morokuma and W. T. Borden, J. Am. Chem. Soc., 1990, 112, 1751 CrossRef CAS.
- T. Ichino, S. M. Villano, A. J. Gianola, D. J. Goebbert, L. Velarde, A. Sanov, S. J. Blanksby, X. Zhou, D. A. Hrovat, W. T. Borden and W. C. Lineberger, Angew. Chem., Int. Ed., 2009, 48, 8509 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785 CrossRef CAS.
- The singlet diradical was approximated by use of the guess = mix keyword in Gaussian 03 (ref. 39).
- Orbitals were calculated at the HF/6-31G//B3LYP/6-31G(d) level.
- C. J. Cramer and S. E. Barrows, J. Org. Chem., 1998, 63, 5523 CrossRef CAS; C. J. Cramer and S. E. Barrows, J. Phys. Org. Chem., 2000, 13, 176 CrossRef CAS.
- Activation energies were calculated with respect to the most stable isomer 1Ph-Ecyc.
- The LANL2DZ basis set and ECP were used for Zn: W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284 Search PubMed.
- At the B3LYP/6-31G(d) level, the AlMe2+ chelate of the Me analogue of 2 (where iPr is replaced by Me) is 24.6 kcal mol−1 more stable than a monodentate complex where the carbonyls are anti and Al is bound to the acryloyl oxygen.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995 CrossRef CAS; V. Barone, M. Cossi and J. Tomasi, J. Comput. Chem., 1998, 19, 404 CrossRef CAS.
- M. J. Allen and D. J. Tozer, J. Chem. Phys., 2002, 117, 11113 CrossRef CAS; U. Zimmerli, M. Parrinello and P. Koumoutsakos, J. Chem. Phys., 2004, 120, 2693 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2004, 25, 1463 CrossRef CAS; S. Grimme, J. Comput. Chem., 2006, 27, 1787 CrossRef.
- Grimme27 recommends the use of polarised triple-zeta AO basis sets as a minimum for DFT-D calculations. The dispersion-corrected value of ΔΔE‡(TSA − TSB) obtained with the 6-31G(d) basis set (1.8 kcal mol−1) is very similar to the value obtained when the electronic energies are calculated with the 6-311G(2d,p) basis set at the same geometries (1.9 kcal mol−1).
- J. Antony and S. Grimme, Phys. Chem. Chem. Phys., 2006, 8, 5287 RSC.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157 CrossRef CAS; Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- Note: In the original report (ref. 7), the absolute stereochemistry of the major and minor products obtained using the 4-Bn- and 4-iPr-5,5-Ph2-oxazolidinone auxiliaries were assumed to be the same as those obtained with 4-Ph-oxazolidinone (the latter having been determined by X-ray crystallography). Our present DFT predictions, however, prompted us to determine the absolute stereochemistries for these two auxiliaries. Following the (4 + 3) cycloadditions, reduction of the cycloheptenone carbonyl group and acylation of the resulting alcohols afforded nitrobenzoates, which where characterised by X-ray crystallography. The absolute stereoselectivities in both cases were found to be opposite from those originally supposed. Details are provided in the ESI†.
- S. M. Bakalova, F. J. S. Duarte, M. K. Georgieva, E. J. Cabrita and A. G. Santos, Chem.–Eur. J., 2009, 15, 7665 CrossRef CAS.
- A. G. Santos, S. X. Candeias, C. A. M. Afonso, K. Jenkins, S. Caddick, N. R. Treweeke and D. Pardoe, Tetrahedron, 2001, 57, 6607 CrossRef; A. G. Santos, J. Pereira, C. A. M. Afonso and G. Frenking, Chem.–Eur. J., 2005, 11, 330 CrossRef.
- M. Nishio, M. Hirota and Y. Umezawa, The CH/π Interaction: Evidence, Nature, and Consequences, Wiley-VCH, New York, 1998 Search PubMed; M. Nishio, Y. Umezawa, M. Hirota and Y. Takeuchi, Tetrahedron, 1995, 51, 8665 Search PubMed; M. Nishio, Tetrahedron, 2005, 61, 6923 CrossRef CAS.
- R. E. Gillard, F. M. Raymo and J. F. Stoddart, Chem.–Eur. J., 1997, 3, 1933 CrossRef CAS.
- M. Sodupe, R. Rios, V. Branchadell, T. Nicholas, A. Oliva and J. J. Dannenberg, J. Am. Chem. Soc., 1997, 119, 4232 CrossRef CAS.
- G. Ujaque, P. S. Lee, K. N. Houk, M. F. Hentemann and S. J. Danishefsky, Chem.–Eur. J., 2002, 8, 3423 CrossRef CAS.
- E. H. Krenske, K. N. Houk and M. Harmata, Org. Lett., 2010, 12, 444 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 03, Revision C.02, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154 CrossRef CAS; C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the structural isomers of 1Ph and higher-energy cycloaddition pathways; studies of solvation effects; calculated geometries and energies; complete citations for ref. 39 and 40; and experimental and crystallographic details for the determination of absolute dr values. CCDC reference numbers 775189 and 775190. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00280a |
| This journal is © The Royal Society of Chemistry 2010 |