Controlling the specific enrichment of multi-phosphorylated peptides on oxide materials: aluminium foil as a target plate for laser desorption ionization mass spectrometry†
Liang
Qiao
a,
Hongyan
Bi
a,
Jean-Marc
Busnel
a,
Mohamad
Hojeij
a,
Manuel
Mendez
a,
Baohong
Liu
b and
Hubert H.
Girault
*a
aLaboratoire d'Electrochimie Physique et Analytique, Ecole Polytechnique Fédérale de Lausanne, Station 6, CH-1015, Lausanne, Switzerland. E-mail: hubert.girault@epfl.ch
bDepartment of Chemistry, Institute of Biomedical Sciences, Fudan University, Shanghai, 200433, P.R. China
First published on 25th June 2010
Abstract
A specific multi-phosphopeptide enrichment method is presented to complement current phosphopeptide isolation strategies that normally bias mass spectrometry analysis towards the detection of mono-phosphopeptides. An adsorption model is developed that shows how the physical parameters of adsorption such as specific surface area, adsorption time, etc. control the extraction of mono- and multi-phosphorylated peptides on oxide materials such as alumina. Commercial alumina particles, alumina supported on aluminium particles and alumina on aluminium foils were used to enrich multi-phosphopeptides. The synthesized phosphopeptides and tryptic digests of casein and milk were employed as samples to validate the adsorption simulation. Both experimental and theoretical results show how the selective enrichment of multi-phosphopeptides can be achieved when using extractors with a high specific affinity for phosphate groups but with a relatively small loading capacity or specific surface area. We also show that a commercial aluminium foil represents an ideal substrate to enrich multi-phosphorylated peptides for laser desorption ionisation mass spectrometry.
Introduction
Phosphorylation is one of the most important post-translational modifications of proteins. This reversible procedure regulates a wide range of biological processes, such as transmitting signals, labeling proteins for degradation, and controlling cellular growth.1–5 Structural characterization of phosphoproteins is of great interest for understanding these biological pathways.1 Among various strategies, mass spectrometry (MS) has emerged as the premier tool for examining protein phosphorylation.5–8 However, the low abundance of phosphorylated proteins and the difficulty of ionizing phosphorylated samples during MS-analysis make the study of phosphorylation still an analytical challenge.9–11During the past years, many techniques for isolating phosphorylated proteins or peptides from cell lysates and protein digests have been developed to simplify the analysis of protein phosphorylation. Among various chemical and affinity-based strategies,12–17 solid extraction methods have drawn wide interest due to their effectiveness as well as simplicity. One widely used solid extraction approach, namely immobilized metal affinity chromatography (IMAC),18–24 was introduced in 1975 by Porath et al.,24 relying on the interactions between metal–ligand complexes and phosphate groups. Another popular solid extraction approach is based on the special affinities between phosphate groups and metal oxides, such as ZrO2,5,6 TiO2,3,7 Nb2O5,25 and Al2O3.4,26 With these two principles, many studies have been reported to enrich phosphopeptides by using on-column,27 in-solution7 or on-target28,29 strategies.
For most of the extraction methods reported, both mono- and multi-phosphorylated peptides are collected together. Multi-phosphopeptides being more difficult to ionize than mono-phosphorylated ones, MS analysis normally detects rather exclusively mono-phosphorylated species from the enriched mixtures, neglecting many multi-phosphopeptides that are, however, very important for understanding biological pathways.30,31 To date, there are only a few reports of the specific analysis of multi-phosphorylated peptides.30,32–36
Here, we present a very simple yet very selective enrichment method for multi-phosphopeptides on commercial aluminium foils. Multi-phosphopeptides hold a stronger affinity toward solid extracting phases such as oxides compared with mono-phosphorylated ones due to the larger number of phosphate groups and therefore one should expect them to be preferentially enriched. However, the final enrichment is also regulated by adsorption/desorption kinetics and the loading capacity of extracting phases. Using a classic Langmuir isotherm to model the solid phase extraction processes, the physical factors influencing the enrichment results were studied. Commercial porous alumina particles, alumina supported on aluminium particles and alumina on aluminium foils were used as solid phase extractors with different specific surface areas. The synthesized phosphopeptides with different phosphorylation states were used to estimate the extraction rate and loading capacity of each extractor. Alumina on Al particles and foils, which have a relatively small specific surface area, were further used to selectively extract multi-phosphopeptides from the tryptic digests of α- and β-casein and drinking milk (Table 1). As a result, the obtained selectivity is very good and comparable with the best known methods using more complex functional materials.35
| Peptide sequence | Modifications | (M + H)+ |
|---|---|---|
| a PHOS stands for phosphorylation, and MSO stands for methionine sulfoxide. b The peptide sequences were confirmed by both mass measurements and literature reports except the MSO modified ones which were confirmed by mass measurements only. | ||
| TVDMESTEVFTKKTK, α-S2-casein (153–167) | 1PHOS, 1MSO | 1839.8 |
| DIGSESTEDQAMEDIK, α-S1-casein (58–73) | 2PHOS | 1927.7 |
| DIGSESTEDQAMEDIK, α-S1-casein (58–73) | 2PHOS, 1MSO | 1943.7 |
| YKVPQLEIVPNSAEER, α-S1-casein (119–134) | 1PHOS | 1951.9 |
| FQSEEQQQTEDELQDK, β-casein (48–63) | 1PHOS | 2061.8 |
| NVPGEIVESLSSSEESITR, β-casein (22–40) | 4PHOS | 2352.9 |
| QMEAESISSSEEIVPNSVEQK, α-S1-casein (74–94) | 3PHOS, 1MSO | 2577.0 |
| NTMEHVSSSEESIISQETYK, α-S2-casein (17–36) | 4PHOS | 2619.0 |
| NTMEHVSSSEESIISQETYK, α-S2-casein (17–36) | 4PHOS, 1MSO | 2634.9 |
| QMEAESISSSEEIVPNSVEQK, α-S1-casein (74–94) | 4PHOS | 2640.9 |
| VNELSKDIGSESTEDQAMEDIK, α-S1-casein (52–73) | 3PHOS | 2678.0 |
| VVRNANEEEYSIGSSSEESAEV, α-S2-casein (58–79) | 4 PHOS | 2704.9 |
| QMEAESISSSEEIVPNSVEQK, α-S1-casein (74–94) | 5PHOS | 2720.9 |
| QMEAESISSSEEIVPNSVEQK, α-S1-casein (74–94) | 5PHOS, 1MSO | 2736.9 |
| KNTMEHVSSSEESIISQETYK, α-S2-casein (16–36) | 4PHOS | 2747.1 |
| EKVNELSKDIGSESTEDQAMEDIK, α-S1-casein (50–73) | 3PHOS | 2935.2 |
| EKVNELSKDIGSESTEDQAMEDIK, α-S1-casein (50–73) | 3PHOS, 1MSO | 2951.2 |
| ELEELNVPGEIVESLSSSEESITR, β-casein (17–40) | 4PHOS | 2966.2 |
| NANEEEYSIGSSSEESAEVATEEVK, α-S2-casein (61–85) | 4PHOS | 3008.0 |
| NANEEEYSIGSSSEESAEVATEEVK, α-S2-casein (61–85) | 5PHOS | 3088.0 |
| RELEELNVPGEIVESLSSSEESITR, β-casein (16–40) | 4PHOS | 3122.3 |
| KNTMEHVSSSEESIISQETYKQEK, α-S2-casein (16–39) | 4PHOS | 3132.2 |
| QMEAESISSSEEIVPNSVEQKHIQK, α-S1-casein (74–98) | 4PHOS | 3147.2 |
Experimental
Materials
Acetonitrile (ACN, >99.5%), 2,5-dihydroxybenzoic acid (DHB, >98%), α-casein (from bovine milk, >70%), β-casein (from bovine milk, >90%), and β-insulin oxidized from bovine insulin (90%) were obtained from Sigma-Aldrich (Steinheim, Germany). Angiotensin I (98%) was purchased from Bachem (Switzerland). α_syn (107–140)_pY125 (90%, APQEGILEDMPVDPDNEApYEMPSEEGYQDYEPEA) and α_syn (121–140)_pY125pS129 (90%, DNEApYEMPpSEEGYQDYEPEA) were synthesized by the Laboratoire de neurobiologie moléculaire et neuroprotéomique of Ecole Polytechnique Fédérale de Lausanne. Trifluoroacetic acid (TFA, >99.8%) was purchased from Acrös Organics (Geel, Belgium). Trypsin (Mw. 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 Da, bovine pancreas) was obtained from AppliChem GmbH (Darmstadt, Germany). Aluminium (99%, powder) was obtained from Acros (Chemie Brunschwig AG). Aluminium oxide (99.99%; >95% α-form) was purchased from Fluka AG. All these reagents were used as received without further purification. Fresh milk and aluminium foil were purchased from a local grocery store. Deionized water (18.2 MΩ cm) was produced by an alpha Q-Millipore System (Zug, Switzerland).
800 Da, bovine pancreas) was obtained from AppliChem GmbH (Darmstadt, Germany). Aluminium (99%, powder) was obtained from Acros (Chemie Brunschwig AG). Aluminium oxide (99.99%; >95% α-form) was purchased from Fluka AG. All these reagents were used as received without further purification. Fresh milk and aluminium foil were purchased from a local grocery store. Deionized water (18.2 MΩ cm) was produced by an alpha Q-Millipore System (Zug, Switzerland).
Microscope and nitrogen adsorption
Transmission electron microscopy (TEM) measurements of aluminium and alumina powders were performed on a Philips CM20 operated at 200 KV. Small volumes of sample suspension were deposited on copper mesh grids and covered with carbon coating films. The grids were then dried under a nitrogen atmosphere in a glove box. The shape and the size distributions were obtained by analyzing at least 100 particles. Nitrogen gas adsorption/desorption isotherms were obtained by using a Micromeritics Gemini. Samples were pretreated by heating at 200 °C under vacuum for 1 h to remove H2O. The Brunauer–Emmett–Teller (BET) method was employed to measure surface areas.Tryptic digestion of α-casein, β-casein and drinking milk
α-Casein or β-casein was dissolved in ammonium bicarbonate aqueous solution (25 mM, pH ∼ 8) with a final concentration of 200 ng μl−1, and then thermo-denatured at 100 °C for 5 min. After cooling under ambient conditions, the proteins were digested with trypsin for 12 h at 37 °C (enzyme–protein ratio of 1:30 (w/w), in 25 mM ammonium bicarbonate buffer). Digestion of milk was performed according to a reported method.28 30 μl fresh milk was diluted in 900 μl 25 mM NH4HCO3 aqueous solution. The solution was then centrifugated at 16000 rpm for 15 min. The supernatant was saved for tryptic digestion. After denaturation at 100 °C for 5 min, the supernatant was incubated with 30 μg trypsin for 12 h at 37 °C for proteolysis.
In-solution enrichment of phosphopeptides
The in-solution enrichment of phosphopeptides was performed in a TFA buffer (50% ACN, 49.9% H2O, 0.1% TFA) with various peptide-concentrations and extractor-amounts under ambient conditions with shaking. After 1 h or various incubation-times, suspensions were centrifugated, and the supernatant removed. The particles were then washed three times with TFA buffer (10 min incubation, 20 μl buffer). Finally, the particles was dispersed in 2 μl water, deposited and dried on a MALDI target plate. With an overlay of DHB matrix (10 mg ml−1 DHB in 1% H3PO4 aqueous solution), adsorbed samples were directly analyzed on a Bruker Microflex MALDI-TOF MS.On-plate enrichment of phosphopeptides with aluminium foil
Aluminium foil was employed as a disposable layer and assembled on a MALDI target plate. 1 μl protein tryptic digest in TFA buffer (0.1% TFA in H2O without ACN) was deposited on the Al foil. The deposition and incubation were all performed in a small box saturated with H2O vapour to avoid evaporation. After 30 min, samples were washed away. Afterwards, the Al foil surface was washed with TFA buffer three times, for 10 min each. Finally, the DHB matrix was dropped as an overlayer before MALDI-TOF MS analysis.Data analysis and mathematical modelling
All mass spectrometry experiments were run in positive ion mode on a Bruker Microflex equipped with a nitrogen laser (wavelength 337 nm). Although the acidic phosphopeptides should theoretically have a better ionization efficiency in the negative ion mode than the positive ion mode, it was observed in our lab as well as in other labs37,38 that the negative ion mode cannot practically enhance the ionization of phosphopeptides. The laser intensity was adjusted to obtain good resolution and signal-to-noise ratio (S/N). MS characterizations were repeated several times for each sample to avoid the randomicity of MALDI process. Mass spectrometric data analysis was performed with flexAnalysis from Bruker and the FindMod tool available on the ExPASy (Expert Protein Analysis System) proteomics server of the Swiss Institute of Bioinformatics (SIB) (http://expasy.org/). The masses and intensities of special peaks were sent to the FindMod tool for database searching, where experimentally measured peptide masses were compared with the theoretical peptides calculated from a specified Swiss-Prot/TrEMBL entry to finish the peak identification. Mathematical modeling was performed with Maple 13 (Waterloo Maple Inc.) and Wolfram Mathematica 7.Results and discussion
Simulation of phosphopeptide extraction with a Langmuir isotherm
Mathematical simulation is employed to study the adsorption/desorption kinetics and equilibrium of phosphopeptide extraction processes on solid extractors. Because the extractor phase is normally very large compared to peptides, we model the enrichment procedure by using a classic adsorption/desorption Langmuir isotherm on a flat substrate. As shown in Scheme 1, we consider here a site for peptide adsorption on the solid extractor holding many oxide anchors to interact with the different phosphate groups. We also consider that a peptide is adsorbed as soon as a single link between a phosphate group and an anchor in the empty site is established. In the case of multi-phosphopeptides, additional links will take place with time on the same site, assumed to be with the same binding rate kf. Successive breaking of links, also considered at the same rate kb, finally result in desorption of the peptide. It is assumed that the sizes of all peptides on the extractors are more-or-less the same to simplify the model.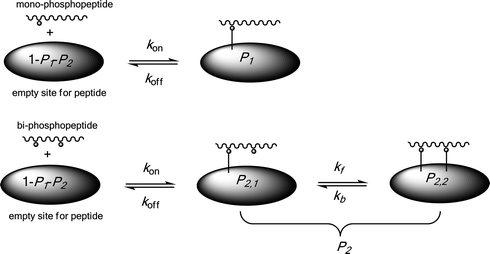 | ||
| Scheme 1 Schematic illustration of phosphopeptide adsorption/desorption processes on a solid extractor. | ||
The kinetic laws for such a model for a mixture of mono- and bi-phosphopeptides read:
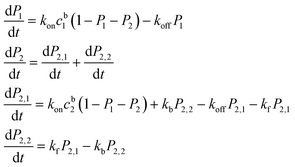 | (1) |
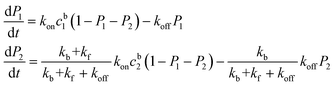 | (2) |
Because the affinity between the phosphate group and alumina is strong, it is reasonable to assume kf ≫ kb and kf ≫ koff. The equations can then be simplified as:
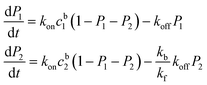 | (3) |
Similarly, the kinetic law for a mixture of mono-, bi-, tri- and tetra-phosphopeptides reads:
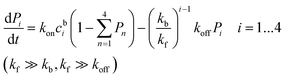 | (4) |
The relationship between cb, the peptide surface coverage ratio P and the initial peptide concentration c0 is:
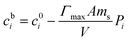 | (5) |
According to eqn (5), the kinetics eqn (4) can be written as:
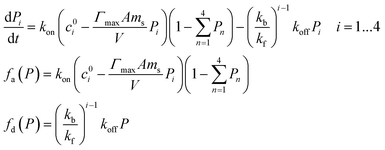 | (6) |
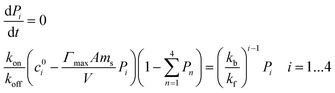 | (7) |
A simulation shown in Fig. 1a illustrates that specific extraction of multi-phosphopeptides from a given peptide-mixture can be realized by adjusting the amount of the extractor. The constant condition for Fig. 1a is Γmax = 10−5 mol m−2, A = 0.1 m2 g−1, V = 10−5 l, kon/koff = 106 M, ci0 = 10−6 M, and kb/kf = 0.01. However, the equilibrium enrichment result is also influenced by the assumed constants. Because kf should be much larger than kb, we keep the assumption that kb/kf = 0.01. The influence of AΓmax can be observed by comparing Fig. 1a and 1b. Keeping all constants the same except increasing A to 1 m2 g−1, the amount of extractor must be decreased by 10 times in order to selectively enrich only multi-phosphopeptides from the same sample. kon/koff can influence the extracted amounts of mono-phosphopeptides when the extractor is in excess, whereas the largest amount of extractors being employed for selective extraction of multi-phosphopeptides is always the same under various kon/koff ratios, as shown in ESI SI-1.† Similarly, selective extraction of only tetra-phosphopeptides or tri- and tetra-phosphopeptides should also be achieved by adjusting the amounts of extractors and influenced by Γmax, A, and kon/koff in a similar way.
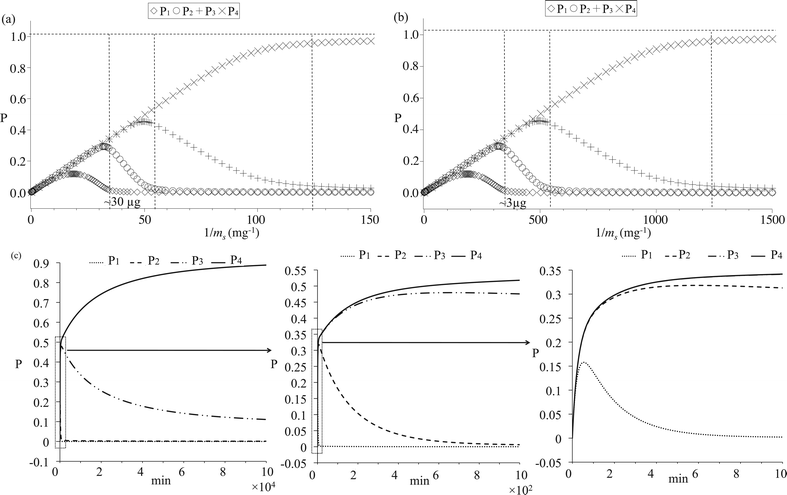 | ||
| Fig. 1 (a, b) Simulated phosphopeptide adsorption equilibrium results as functions of extractor-amounts, (a): extractors with small specific surface area, (b): extractors with large specific surface area; (c) Simulated phosphopeptide adsorption kinetics with zooming-out from left to right. In figure c left, P1 is overlapped with P2. In figure c right, P3 is overlapped with P4. | ||
Extraction kinetics are simulated in Fig. 1c to verify the feasibility of approaching the enrichment equilibrium. The constant condition is kon = 106 min−1 M−1, koff = 1 min−1, kb/kf = 0.01, Γmax = 10−5 mol m−2, A = 0.1 m2 g−1, ci0 = 10−6 M, V = 10−5 l and ms = 10−5 g. According to the equilibrium simulation in Fig. 1a, under such a constant condition, tetra-phosphopeptides should be selectively extracted. In Fig. 1c, it is found that adsorption rates are very fast for all the phosphopeptides, while desorption rates for bi-, tri- and tetra-phosphopeptides are much slower than that of mono-phosphorylated peptides due to the very small value of kb/kf. Therefore, from a kinetic viewpoint, selective enrichment of multi-phosphopeptides can be easily achieved while the separation of bi- and tri-phosphopeptides from tetra-phosphopeptides is rather impossible when there is a moderate value for koff. In the following experiments, it will be shown that bi-, tri- and tetra-phosphopeptides were always enriched together from α-casein and milk digests, validating the present kinetics simulation.
Combining the equilibrium and kinetics simulations, we can infer the following conclusions:
1. Selective adsorption of multi-phosphopeptides from a given sample can be realized by using any solid extractors having special affinities for the phosphate group via adjusting the employed amounts of extractors and incubation time.
2. Particles with large specific surface area and loading capacity should show bad selectivity while big particles with small specific surface area should be suitable for the selective enrichment of multi-phosphopeptides.
3. Separation of tetra-phosphopeptides from other multi-phosphopeptides is possible at equilibrium but not feasible in a time resolved manner, and all multi-phosphopeptides should be extracted with a similar rate during experiments.
4. Fast or moderate kinetics are preferred for mono-phosphopeptide desorption. Then, porous materials are not suitable for the selective enrichment of multi-phosphopeptides as their 3-D structures may limit the mass transport.
Adsorption kinetics and equilibrium with standard synthesized peptide mixture
Alumina and aluminium powder were used to try to selectively enrich multi-phosphopeptides by following the conclusions inferred from the simulation. Metallic aluminium is very reactive with atmospheric oxygen. A thin passivation layer of alumina (∼4 nm thickness) can form in about 100 ps on an exposed aluminium surface according to the literature.39 Therefore, for both extractors, the affinities towards phosphorylated peptides are always based on the specific interaction between alumina and the phosphate groups.26 In the following paragraphs, we shall use the core–shell nomenclature to describe thin alumina layers on aluminium, i.e. Al@Al2O3. Fig. 2 shows TEM images of alumina and Al@Al2O3 powder. It was found that the particle size of Al@Al2O3 is ∼20 μm, while the diameter of alumina is only several hundred nanometres. Nitrogen adsorption experiments show that the specific surface area of Al@Al2O3 is only 0.11 m2 g−1 while the specific surface area of Al2O3 powder is 4.5 m2 g−1, consistent with the TEM observations, Table 2. Considering that these two extractors have identical chemical properties, Γmax should be similar for both alumina and Al@Al2O3. Therefore, it can be estimated that Al@Al2O3, which holds a very small specific surface area, is more suitable for the selective extraction of multi-phosphopeptides.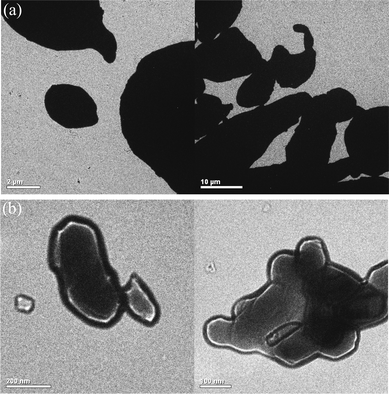 | ||
| Fig. 2 TEM images of the commercial (a) Al@Al2O3 powder and (b) alumina powder. | ||
| Extractor | BET surface area/m2 g−1 | Average particle diameter/μm |
|---|---|---|
| Al2O3@Al | 0.11 | 21 |
| Al2O3 | 4.49 | 0.33 |
A simple standard peptide-mixture was firstly used to validate this model. The mixture was composed of four peptides in equal molar amounts, including angiotensin I, oxidized β-insulin, α_syn(107–140)_pY125 (P1) and α_syn(121–140)_pY125pS129 (P2). We chose here α_syn(121–140)_pY125pS129 instead of α_syn(107–140)_ pY125pS129 as the multi-phosphopeptide, because the dephosphorylated fragment, i.e. the mono-phosphorylated α_syn(107–140), can be generated from bi-phosphorylated α_syn(107–140) during the MALDI-TOF analysis, thereby influencing the detection of α_syn(107–140)pY125. Fig. 3a shows a mass spectrum of the peptide-mixture (900 nM each peptide) without any pre-treatment, where angiotensin I dominates the spectrum and the peaks of the phosphopeptides are very weak. Fig. 3b and 3c show mass spectra of peptides enriched from the mixture (900 nM each peptide, 20 μl) by 20 μg Al@Al2O3 and 20 μg alumina powders respectively. The incubation time was 1 h to make sure that enrichment equilibrium of P1 had been approached. On the spectrum, almost only doubly phosphorylated peptide α_syn(121–140)_pY125pS129 (m/z: 2527.4) is observed in the case of using Al@Al2O3 as the extractor. In contrast, several intensive peaks of mono-phosphopeptides also appear on the spectrum in the case of using alumina powder as the extractor. The peak with m/z of 3940.4 corresponds to the singly charged α_syn(107–140)_pY125. The peaks with m/z of 2284.2 and 1594.5 correspond to by-products generated during the synthesis of α_syn(121–140)_pY125pS129, which are indeed mono-phosphopeptides of DNEAEMPpSEEGYQDYEPEA and PpSEEGYQDYEPEA respectively. The experimental results are consistent with the estimates made previously by considering the simulation and BET results, proving that it is reasonable to predict the performance of extractors in phosphopeptide enrichment by using the current model.
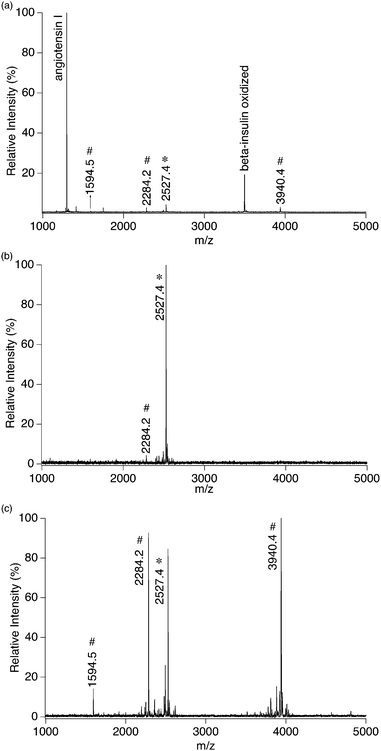 | ||
| Fig. 3 (a) Mass spectrum of 900 nM four-peptide-mixture without any pre-treatment, and mass spectra of peptides extracted from 20 μl 900 nM four-peptide-mixture by (b) ∼20 μg Al@Al2O3 powder and (c) ∼20 μg Al2O3 powder; *: multi-phosphopeptide related peaks; #: single-phosphopeptide related peaks. | ||
Phosphopeptide enrichments were also performed using various extractor-amounts to further confirm the deductions obtained from simulation. The concentrations of peptide mixture were fixed at 900 nM, the volume was fixed at 20 μl and the amounts of Al@Al2O3 were varied from 4 μg to 200 μg. The incubation time was always 1 h to make sure that the enrichment equilibrium of mono-phosphopeptides had been approached. As shown in the ESI SI-2,† nearly only multi-phosphopeptide α_syn(121–140)_pY125pS129 was observed in the mass spectra when the amount of Al@Al2O3 was less than 40 μg. A weak signal corresponding to α_syn(107–140)_pY125 was obtained when the amount of Al@Al2O3 was 100 μg. Whereas the enrichment had no specificity towards multi-phosphopeptides when the amount of Al@Al2O3 was as high as 200 μg. This result agrees with the simulation shown in Fig. 1a.
Adsorption–desorption kinetics were evaluated and it was found that the equilibrium for mono-phosphopeptides could be approached in 6 min in the case of using the 4-peptide-mixture as a sample and Al@Al2O3 as the extractor (ESI SI-4†). When the incubation time was only 1 min, a weak peak of α_syn(107–140)_pY125 could be observed. However, when the incubation time was longer than 6 min, almost only signals corresponding to α_syn(121–140)_pY125pS129 were obtained, agreeing with the simulation in Fig. 1c.
It should also be possible to selectively enrich multi-phosphopeptides by using alumina powders according to the extraction model. Considering that the specific surface area of alumina powder is ∼45 times larger than that of Al@Al2O3, very low amounts of Al2O3 particles should be used for specifically extracting multi-phosphopeptides. Indeed, as shown in Fig. SI-3,† selective isolation of multi-phosphopeptides from 20 μl of 900 nM four-peptide-mixture can be partially achieved when the quantity of alumina powder is lowered to 2 μg, agreeing with the simulation in Fig. 1b. However, such a small extractor amount is inconvenient to handle. Therefore, only Al@Al2O3 was further used to enrich multi-phosphopeptides from protein tryptic digests in the following parts.
Specific in-solution enrichment of multi-phosphopeptides using Al@Al2O3 powder
Following success in standard peptides, Al@Al2O3 powder was further used to extract multi-phosphopeptides from protein digests. Tryptic digest of β-casein was employed as the first sample. According to the literature, there are normally several mono-phosphopeptides and one tetra-phosphopeptide in the tryptic digests of β-casein. Fig. 4a shows a mass spectrum of peptides enriched from 20 μl 850 nM β-casein digest by 20 μg Al@Al2O3 powder. It is obvious that almost only one peptide with four phosphate groups (Mw 3121.3) is observed. The peak with m/z of 1561.7 can be attributed to a double charged ion of the same tetra-phosphorylated peptide,28 while the peak with m/z of 3035.7 corresponds to a dephosphorylated fragment of the peptide, generated during the MS analysis. This MS result is very different from that obtained by using previously reported phosphopeptide enrichment methods. In most cases, peaks corresponding to single phosphorylated peptides of β-casein with m/z of 2061.8 and 2556.1 would be very intensive whilst the peak with m/z of 3122.3 is normally quite weak or even unobservable.7,28,40,41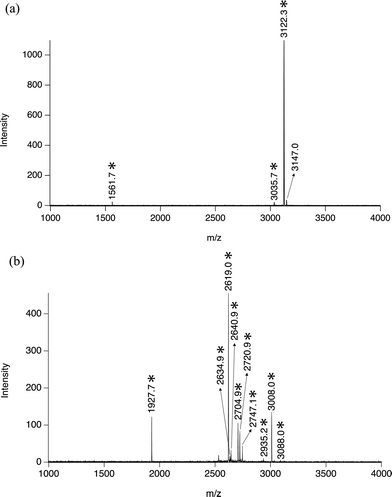 | ||
| Fig. 4 (a) Mass spectrum of peptides extracted from 20 μl 850 nM β-casein digest by ∼20 μg Al@Al2O3 powder; and (b) mass spectrum of peptides extracted from 20 μl 850 nM α-casein digest by ∼20 μg Al@Al2O3 powder; *: multi-phosphopeptide related peaks. | ||
Similar specific multi-phosphopeptide enrichment was also achieved when using α-casein digest as the sample. As shown in Fig. 4b, from 20 μl 850 nM tryptic digest, 10 multi-phosphopeptides were enriched by 20 μg Al@Al2O3 and identified by mass spectrometry, while no mono-phosphopeptide was observed, Fig. 4b, Table 1. Indeed, it is rare to find so many multi-phosphorylated peptides from α-casein digest with a single MS run and no pre-separation. Considering that the total concentration of multi-phosphopeptides here should be much larger than 900 nM, some samples were wasted in this case.
The specific multi-phosphopeptide enrichment strategy was also applied to milk protein digests (∼1 μM protein concentration, 20 μl). As shown in Fig. 5, 11 multi-phosphorylated peptides from milk digests were enriched by 20 μg Al@Al2O3 and identified, while no mono-phosphopeptides were obtained, further proving the unique specificity. This result is very different from that obtained by using a strategy developed previously for analyzing milk phosphoproteins: only 3 multi-phosphopeptides and 5 mono-phosphorylated ones were detected.28
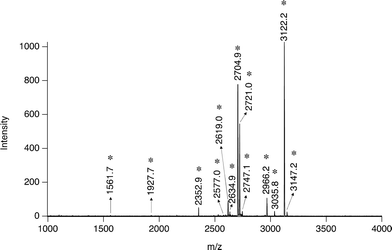 | ||
| Fig. 5 Mass spectrum of peptides extracted from 20 μl tryptic milk digest (∼1 μM protein concentration) by ∼20 μg Al@Al2O3 powder; *: multi-phosphopeptide related peaks. | ||
In this section, it has been proven that the equilibrium/kinetics controlled multi-phosphopeptide enrichment strategy is also successful in analyzing tryptic digests of standard protein and simple protein mixtures. With these results, it is logical to predict that this approach can also be used in the study of protein phosphorylation of complex samples, such as cell lysates. In order to extract as many multi-phosphopeptides as possible and avoid the co-enrichment of mono-phosphopeptides, the total amount of multi-phosphopeptides should be estimated and suitable quantities of extractors should be calculated. If the amount of multi-phosphopeptide is really low in a sample, extractors with very small specific surface area can be used.
On-plate enrichment of multi-phosphopeptides using an aluminium foil
From the analysis viewpoint, on-plate enrichment of phosphopeptides is interesting because the handling of samples is minimized. Holding the same chemical properties as that of aluminium powders and a flat surface, bare aluminium foil is an ideal candidate to act as a disposable layer on the MALDI target plate with the function of selectively extracting multi-phosphopeptides from deposited solutions. Similar to Al@Al2O3 powder, the surface of Al foil is also covered with a thin layer of oxide. Therefore, we shall continue with the core –shell terminology and use the expression Al@Al2O3 foil.Indeed, we have reported recently that commercial aluminium foil can be used as a disposable layer for the MALDI target plate with the advantages of convenience and low cost.42 Specifically, we printed the aluminium foil with TiO2 nanoparticles (20 nm) for on-plate enrichment of mono-phosphopeptides.42 Here, we use the bare Al@Al2O3 foil to on-plate enrich multi-phosphorylated peptides without any modification. As shown in Fig. 6a and b, the on-plate enrichment method is quite successful. 1 Multi-phosphopeptide from 85 fmol β-casein digest and 14 multi-phosphopeptides from 85 fmol α-casein digest were enriched and identified separately, while only 1 weak peak corresponding to single phosphorylated peptide was observed in each spectrum, illustrating the high specificity of the strategy. The detection limit influenced by both extraction efficiency and MS sensitivity is about 20 fmol, as shown in Fig. 6c and 6d, where 1 multi-phosphorylated peptide from β-casein digest and 5 multi-phosphorylated peptides from α-casein digest were identified.
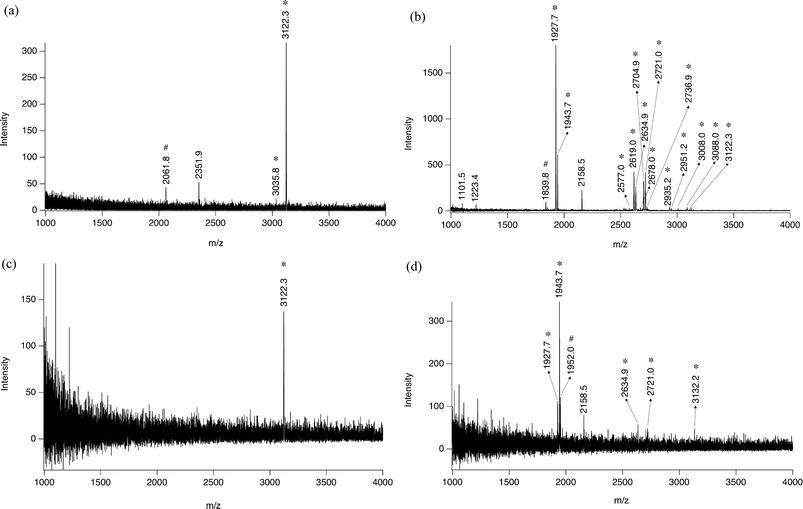 | ||
| Fig. 6 Mass spectra from (a) 1 μl 85 nM tryptic β-casein digest, (b) 1 μl 85 nM tryptic α-casein digest, (c) 1 μl 20 nM tryptic β-casein digest, and (d) 1 μl 20 nM tryptic α-casein digest after 30 min in situ phosphopeptide enrichment on Al@Al2O3 foil; *: multi-phosphopeptide related peaks; #: single-phosphopeptide related peaks. | ||
We have recently reported that the rotogravure-printed TiO2 spots on Al foil substrate are better at adsorbing multi-phosphopeptides compared to screen-printed TiO2 spots.42 Now with the present model, we can attribute this phenomenon to the difference in specific surface area. On rotograved TiO2 spots, we have a low coverage of TiO2 nanoparticles with part of the aluminium foil substrate still partially exposed, whereas the screen printed spots are a few microns thick forming a mesoporous structure with a very high binding capacity. The present model corroborates the previous observations that rotograved films resulted in a co-enrichment of multi- and mono-phosphopeptides, and that screen-printed resulted mainly in the enrichment of mono-phosphopeptides.
In spite of the advantages, such as facility and high selectivity, the present multi-phosphopeptide analysis method is still limited by the inherent drawbacks of MALDI-MS. Because of its poor reproducibility and limited compatibility with various tandem MS techniques, MALDI-MS is not suitable for complex biosample analysis or quantification analysis, and therefore we did not evaluate the adsorption percentages of multi-phosphopeptides. HPLC-ESI-MS combined with the current multi-phosphopeptide enrichment method could be more interesting with a view to proteome research. However, compared to ESI, MALDI holds the advantage in performing in situ ionization. With the Al foil or the previously reported TiO2–Al foil, it is possible to extract in situ mono- and multi-phosphopeptides from a piece of tissue, respectively, and to run MALDI-MS-imaging on these Al foils to investigate the surface phosphorylated species distribution.
Conclusion
In conclusion, specific and efficient strategies for enriching multi-phosphopeptides have been developed based on an adsorption kinetics model. This work can be extended to the fabrication of aluminium powder filled columns or microchips for multi-phosphopeptide affinity chromatography, optimizing experimental conditions for orderly enriching of mono- and multi-phosphorylated peptides, and searching for other extractors for the specific adsorption of multi-phosphopeptides.Acknowledgements
We thank the Swiss National Science Foundation for supporting the project “Analytical tools for proteome analysis and redoxomics (200020–127142)”, the NSFC (20775016, 20925517), the Laboratoire de technologie des poudres at Ecole Polytechnique Fédérale de Lausanne for the BET analysis, and the Laboratoire de neurobiologie moléculaire et neuroprotéomique of Ecole Polytechnique Fédérale de Lausanne for the standard phosphopeptide synthesis.Notes and references
- T. Hunter, Philos. Trans. R. Soc. London, Ser. B, 1998, 353, 583–605 CrossRef CAS.
- M. Hjerrild and S. Gammeltoft, FEBS Lett., 2006, 580, 4764–4770 CrossRef CAS.
- M. R. Larsen, T. E. Thingholm, O. N. Jensen, P. Roepstorff and T. J. D. Jorgensen, Mol. Cell. Proteomics, 2005, 4, 873–886 CrossRef CAS.
- F. Wolschin, S. Wienkoop and W. Weckwerth, Proteomics, 2005, 5, 4389–4397 CrossRef CAS.
- H. J. Zhou, S. Y. Xu, M. L. Ye, S. Feng, C. Pan, X. G. Jiang, X. Li, G. H. Han, Y. Fu and H. Zou, J. Proteome Res., 2006, 5, 2431–2437 CrossRef.
- C. Y. Lo, W. Y. Chen, C. T. Chen and Y. C. Chen, J. Proteome Res., 2007, 6, 887–893 CrossRef.
- C. T. Chen and Y. C. Chen, Anal. Chem., 2005, 77, 5912–5919 CrossRef CAS.
- X. Q. Xu, C. H. Deng, M. X. Gao, W. J. Yu, P. Y. Yang and X. M. Zhang, Adv. Mater., 2006, 18, 3289–3293 CrossRef CAS.
- R. Aebersold and D. R. Goodlett, Chem. Rev., 2001, 101, 269–295 CrossRef CAS.
- J. Reinders and A. Sickmann, Proteomics, 2005, 5, 4052–4061 CrossRef CAS.
- G. R. Blacken, M. Volny, T. Vaisar, M. Sadilek and F. Turecek, Anal. Chem., 2007, 79, 5449–5456 CrossRef CAS.
- Z. A. Knight, B. Schilling, R. H. Row, D. M. Kenski, B. W. Gibson and K. M. Shokat, Nat. Biotechnol., 2003, 21, 1047–1054 CrossRef CAS.
- Y. Oda, T. Nagasu and B. T. Chait, Nat. Biotechnol., 2001, 19, 379–382 CrossRef CAS.
- L. J. Zhang, Y. W. Xu, H. J. Lu and P. Y. Yang, Proteomics, 2009, 9, 4093–4097 CrossRef CAS.
- Y. W. Xu, L. J. Zhang, H. J. Lu and P. Y. Yang, Anal. Chem., 2008, 80, 8324–8328 CrossRef CAS.
- W. A. Tao, B. Wollscheid, R. O'Brien, J. K. Eng, X. J. Li, B. Bodenmiller, J. D. Watts, L. Hood and R. Aebersold, Nat. Methods, 2005, 2, 591–598 CrossRef CAS.
- H. L. Zhou, J. D. Watts and R. Aebersold, Nat. Biotechnol., 2001, 19, 375–378 CrossRef CAS.
- L. M. Nuwaysir and J. T. Stults, J. Am. Soc. Mass Spectrom., 1993, 4, 662–669 CrossRef CAS.
- M. C. Posewitz and P. Tempst, Anal. Chem., 1999, 71, 2883–2892 CrossRef CAS.
- A. Stensballe, S. Andersen and O. N. Jensen, Proteomics, 2001, 1, 207–222 CrossRef CAS.
- S. B. Ficarro, M. L. McCleland, P. T. Stukenberg, D. J. Burke, M. M. Ross, J. Shabanowitz, D. F. Hunt and F. M. White, Nat. Biotechnol., 2002, 20, 301–305 CrossRef CAS.
- C. S. Raska, C. E. Parker, Z. Dominski, W. F. Marzluff, G. L. Glish, R. M. Pope and C. H. Borchers, Anal. Chem., 2002, 74, 3429–3433 CrossRef CAS.
- T. S. Nuhse, A. Stensballe, O. N. Jensen and S. C. Peck, Mol. Cell. Proteomics, 2003, 2, 1234–1243 CrossRef.
- J. Porath, J. Carlsson, I. Olsson and G. Belfrage, Nature, 1975, 258, 598–599 CrossRef CAS.
- S. B. Ficarro, J. R. Parikh, N. C. Blank and J. A. Marto, Anal. Chem., 2008, 80, 4606–4613 CrossRef.
- C. T. Chen, W. Y. Chen, P. J. Tsai, K. Y. Chien, J. S. Yu and Y. C. Chen, J. Proteome Res., 2007, 6, 316–325 CrossRef.
- N. Sugiyama, T. Masuda, K. Shinoda, A. Nakamura, M. Tomita and Y. Ishihama, Mol. Cell. Proteomics, 2007, 6, 1103–1109 CrossRef CAS.
- L. Qiao, C. Roussel, J. J. Wan, P. Y. Yang, H. H. Girault and B. H. Liu, J. Proteome Res., 2007, 6, 4763–4769 CrossRef CAS.
- F. Tan, Y. J. Zhang, J. L. Wang, J. Y. Wei, P. B. Qin, Y. Cai and X. H. Qian, Rapid Commun. Mass Spectrom., 2007, 21, 2407–2414 CrossRef CAS.
- J. Kim, D. G. Camp and R. D. Smith, J. Mass Spectrom., 2004, 39, 208–215 CrossRef CAS.
- S. Liu, C. J. Zhang, J. L. Campbell, H. X. Zhang, K. K. C. Yeung, V. K. M. Han and G. A. Lajoie, Rapid Commun. Mass Spectrom., 2005, 19, 2747–2756 CrossRef CAS.
- S. Kim, H. Choi and Z. Park, Mol. Cells, 2007, 23, 340–348 CAS.
- H. Wang, J. C. Duan, L. H. Zhang, Z. Liang, W. B. Zhang and Y. K. Zhang, J. Sep. Sci., 2008, 31, 480–487 CrossRef CAS.
- T. E. Thingholm, O. N. Jensen, P. J. Robinson and M. R. Larsen, Mol. Cell. Proteomics, 2008, 7, 661–671.
- C. Chang, C. Wu, Y. Wang and H. Chang, Anal. Chem., 2008, 80, 3791–3797 CrossRef.
- J. J. Wan, K. Qian, L. Qiao, Y. H. Wang, J. L. Kong, P. Y. Yang, B. H. Liu and C. Z. Yu, Chem.–Eur. J., 2009, 15, 2504–2508 CrossRef CAS.
- J. L. Wolfender, F. X. Chu, H. Ball, F. Wolfender, M. Fainzilber, M. A. Baldwin and A. L. Burlingame, J. Mass Spectrom., 1999, 34, 447–454 CrossRef CAS.
- K. Janek, H. Wenschuh, M. Bienert and E. Krause, Rapid Commun. Mass Spectrom., 2001, 15, 1593–1599 CrossRef CAS.
- T. Campbell, R. K. Kalia, A. Nakano, P. Vashishta, S. Ogata and S. Rodgers, Phys. Rev. Lett., 1999, 82, 4866–4869 CrossRef CAS.
- Y. Li, Y. Liu, J. Tang, H. Lin, N. Yao, X. Shen, C. Deng, P. Yang and X. Zhang, J. Chromatogr., A, 2007, 1172, 57–71 CrossRef CAS.
- H. K. Kweon and K. Hakansson, Anal. Chem., 2006, 78, 1743–1749 CrossRef CAS.
- H. Y. Bi, L. Qiao, J. M. Busnel, V. Devaud, B. H. Liu and H. H. Girault, Anal. Chem., 2009, 81, 1177–1183 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Mathematical simulation and adsorption equilibrium and kinetics investigations. See DOI: 10.1039/c0sc00224k |
| This journal is © The Royal Society of Chemistry 2010 |