DOI:
10.1039/C0SC00220H
(Edge Article)
Chem. Sci., 2010,
1, 369-373
Received
8th March 2010
, Accepted 25th April 2010
First published on
15th June 2010
Abstract
Dinuclear Co–salen complexes (R,R)-(S,S)-1–4 were synthesized and compared with their mononuclear counterpart rac-5 to suggest that a bimetallic mechanism is in charge for the alternating copolymerization of propylene oxide with CO2 in the absence of onium salt while a monometallic mechanism is more probable in its presence.
Introduction
In homogeneous catalysis, a bimetallic reaction mechanism is involved in a variety of metal-catalyzed reactions where two metal centers work cooperatively.1 Aiming at the improvement of catalytic activity and selectivity by proximity, well-defined dinuclear or multinuclear metal complexes have been designed and applied to catalytic reactions. For example, Jacobsen et al. synthesized bimetallic Cr–salen (basic skeleton of salenH2 = N,N′-disalicylidene-1,2-ethylenediamine) complexes2 and Co–salen complexes3 to reveal that the bimetallic mechanism is operating both in the asymmetric ring opening reactions of epoxides with TMSN3 catalyzed by Cr and with H2O catalyzed by Co. Repetitive nucleophilic ring-opening of epoxide leads to its polymerization. Mechanistic studies have also been devoted to the ring-opening polymerization of epoxide, and a bimetallic mechanism is postulated for Al-4 and Co-catalyzed5 polymerization of propylene oxide (PO).
Another class of important ring-opening reaction of epoxide is the alternating copolymerization of epoxides with CO2, which is recognized as a promising candidate for utilization of CO2 as a carbon resource.6–11 The copolymerization proceeds by the alternating two reactions: (i) CO2 insertion into a metal–alkoxide and (ii) nucleophilic ring-opening of an epoxide by the resulting carbonate. For the reaction (ii), a bimetallic mechanism was naturally anticipated because of its similarity to the above mentioned nucleophilic ring-opening reactions. Indeed, a bimetallic mechanism was indicated for the cyclohexene oxide (CHO)/CO2 copolymerization with Zn/β-diiminate.12 Following the report, a variety of dinuclear zinc13 or magnesium14 complexes were developed for high catalytic activity. On the other hand, a monometallic mechanism was suggested for the PO/CO2 and CHO/CO2 copolymerizations by using Cr–salen,10a,10c,15 and –tetramethyltetraazaannulene complexes,16 Co–salen complexes equipped with 1,5,7-triabicyclo[4,4,0]dec-5-ene pendant,11k or Al–tetraphenylporphyrin.17 Coates et al. drew both mono- and bimetallic mechanisms depending on the reaction conditions, namely, monometallic in the presence of onium salt and bimetallic in its absence.11c,18 Thus, at present, the mono- vs. bimetallic mechanism is still under argument for the epoxide/CO2 copolymerizations.
Here we indicate that the bimetallic propagation mechanism operating in the PO/CO2 copolymerization by using dinuclear cobalt–salen complexes in the absence of onium salts. Dinuclear Co complexes varying in the linker length between the coordination sites 1–4 were synthesized and compared with the mononuclear Co complex 5 (Fig. 1). Amongst the complexes examined in this work, (R,R)-(S,S)-3 with a four-methylene linker showed the highest TOF and the high activity was maintained even under the highly diluted condition of [PO]/[Co center] = 10,000. The effect of the amount of onium salts on catalytic performance was also investigated, indicating a monometallic propagation mechanism both in mononuclear and dinuclear systems in the presence of onium salts.
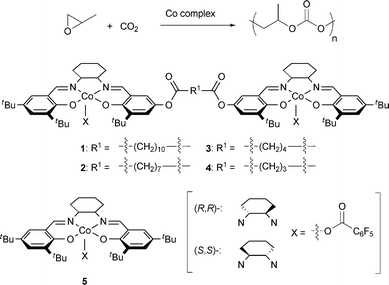 |
| | Fig. 1 Dinuclear and mononuclear Co complexes 1–5. | |
Results and discussion
Synthesis of dinuclear Co complexes 1–4
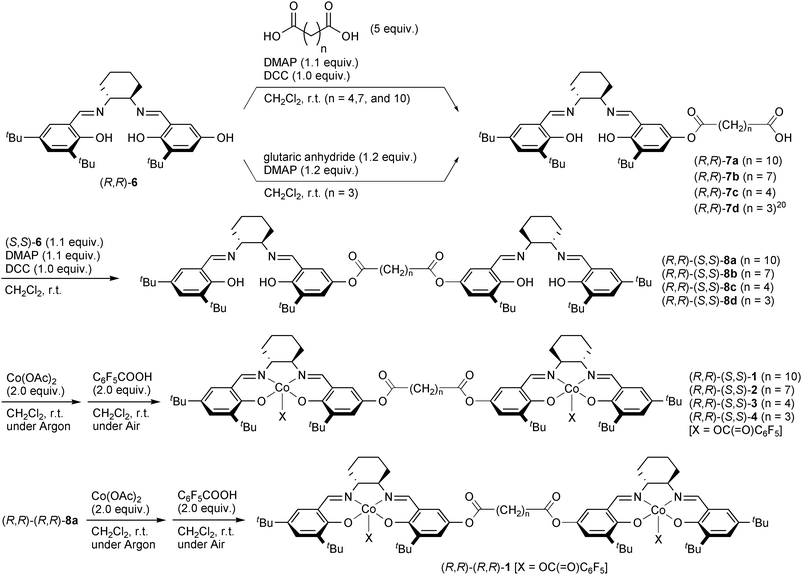
Bissalen ligand precursors were synthesized from hydroxysalen 619 and dicarboxylic acids. The synthetic route to (R,R)-(S,S)-1 is described in Scheme 1. One salen unit (R,R)-6 is first attached to dodecanedicarboxylic acid to form (R,R)-7a and another salen unit (S,S)-6 was next connected to give the bissalen ligand (R,R)-(S,S)-8a. The related compounds varying in the linker length, 8b–d were synthesized in the same procedure [(R,R)-7d was synthesized from (R,R)-6 and glutaric anhydride].20 A diastereomer of (R,R)-(S,S)-8a, (R,R)-(R,R)-8a was synthesized following the literature method.2 The bissalen ligands 8a–d thus prepared was exposed to Co(OAc)2 to form the dinuclear Co(II) complexes, which were oxidized by air to the corresponding Co(III) complexes 1–4. The complexes were applied to the PO/CO2 alternating copolymerization.
The effect of relative configuration of the two salen units
We first investigated the PO/CO2 copolymerization by using dinuclear cobalt complexes (R,R)-(R,R)-1 and (R,R)-(S,S)-1. The results are shown in Table 1 in comparison to the data taken with mononuclear racemic complex rac-5. With (R,R)-(R,R)-1, the structure of the obtained copolymer was moderately-alternating with 84% carbonate linkage and 16% ether linkage. No production of cyclic propylene carbonate was observed. The notable characteristic of the dinuclear cobalt complex is that the catalytic activity was maintained under lower concentration ([PO]/[Co] = 3,000, run 2). This tendency is contrastive to a mononuclear complex rac-5: the catalytic activity was significantly reduced under lower concentration, that is TOF = 100 at [PO]/Co = 1000 and TOF = 20 at [PO]/[Co] = 3000 (runs 14 and 15). These results suggest that the proximity of two cobalt centers allows complex (R,R)-(R,R)-1 to catalyze the copolymerization effectively even under low concentration. In other words, the copolymerization was indicated to proceed via a bimetallic mechanism. Relative stereochemistry of salen ligands significantly impact on the catalytic activity of dinuclear cobalt–salen complexes.11c Complex (R,R)-(S,S)-1, consisting of two opposite enantiomers of a cobalt–salen unit, showed much higher activity than (R,R)-(R,R)-1a (compare runs 3 and 4 with 1 and 2, respectively). The catalyst showed reasonable TOF even at PO/[Co] = 5,000 (run 5). In runs 3–5, a slight decrease in TOF was detected at the lower concentration although the drop range is much smaller than that for rac-5 (runs 14 and 15). This may imply that an intermolecular reaction of (R,R)-(S,S)-1 also takes part in product formation under higher concentration.2 In addition to higher catalytic activity, higher carbonate-linkage content was achieved by using (R,R)-(S,S)-1a. Thus, the following studies were carried out by the (R,R)-(S,S)-isomers.
| Run |
Complex |
[PO]/[Co] |
T/°C |
TOF (×102 h−1)b,c |
M
n
/g mol−1 |
M
w/Mnd |
Carbonate linkage (%)c |
|
Copolymerization conditions: propylene oxide (PO, 2.0 mL), CO2 (5.3 MPa), cobalt complex for 2 h.
Turnover frequency (TOF) = (mol of PO consumed)·(mol of Co center)−1·h−1.
Calculated by 1H NMR spectroscopy by using phenanthrene as an internal standard. In all cases, cyclic propylene carbonate was not observed.
Estimated by gel-permeation-chromatography analysis using a polystyrene standard. The molecular weight is strongly affected by an amount of concomitant water since it works as a chain-transfer reagent.11g,21 Accordingly, the Mn values were not consistent with those estimated from the yields.
The reaction was stopped in 0.5 h.
|
| 1 |
(R,R)-(R,R)-1 |
1,000 |
22 |
0.3 |
9,800 |
1.07 |
84 |
| 2 |
|
3,000 |
22 |
0.4 |
14,400 |
1.08 |
84 |
| 3 |
(R,R)-(S,S)-1 |
1,000 |
22 |
1.5 |
18,100 |
1.51 |
97 |
| 4 |
|
3,000 |
22 |
1.2 |
21,600 |
1.19 |
96 |
| 5 |
|
5,000 |
22 |
0.8 |
19,500 |
1.21 |
97 |
| 6 |
(R,R)-(S,S)-2 |
1,000 |
22 |
1.3 |
14,600 |
1.50 |
94 |
| 7 |
(R,R)-(S,S)-3 |
1,000 |
22 |
1.8 |
26,200 |
1.36 |
88 |
| 8 |
|
3,000 |
22 |
1.8 |
36,700 |
1.07 |
84 |
| 9 |
|
3,000 |
40 |
2.4 |
29,500 |
1.66 |
90 |
| 10e |
|
3,000 |
40 |
4.3 |
13,600 |
1.14 |
87 |
| 11 |
|
3,000 |
60 |
0.7 |
13,900 |
1.35 |
82 |
| 12 |
|
10,000 |
22 |
1.2 |
29,500 |
1.22 |
88 |
| 13 |
(R,R)-(S,S)-4 |
1,000 |
22 |
1.4 |
15,800 |
1.37 |
97 |
| 14 |
rac-5 |
1,000 |
22 |
1.0 |
15,100 |
1.55 |
96 |
| 15 |
|
3,000 |
22 |
0.2 |
15,000 |
1.28 |
93 |
Optimization of the linker between the two salen units
Next, we compared the complexes (R,R)-(S,S)-1–4 in order to find the best distance between the two salen units. The length of alkanedioate linker significantly influenced the catalytic activity of PO/CO2 alternating copolymerization, as was observed in the nucleophilic ring-opening of epoxides by using dinuclear Cr–salen complexes.2 Among the dinuclear Co–salen complexes, (R,R)-(S,S)-3 with hexanedioate linker demonstrated the highest activity (compare runs 3, 6, 7, and 13 in Table 1). Complex (R,R)-(S,S)-3 can keep its catalytic activity under a highly-diluted condition (run 12, [PO]/[Co] = 10,000, TOF = 120). Increase in polymerization temperature to 40 °C improved the catalytic activity up to 430 (run 10). However, a further increase in polymerization temperature (60 °C) diminished the catalytic activity. This may be due to the reduction of cobalt(III) species to cobalt(II) one considering that the color of the polymerization mixture turned to red at 60 °C.11k,22 Nevertheless, complex (R,R)-(S,S)-3 possesses a higher temperature-tolerance than the mononuclear cobalt–salen complexes (R,R)-5 (X = OAc) which showed reduced activity even at 40 °C.11a,23 In contrast, selectivity for the carbonate linkage over the ether linkage was the lowest with (R,R)-(S,S)-3 among complexes (R,R)-(S,S)-1–4 (compare runs 3, 6, 7, and 13). Thus, the proper distance between the two Co centers accelerates not only the alternating PO/CO2 copolymerization but also homopolymerization of PO.5 Such strong dependence of catalytic performance on the linker distance also supports the bimetallic mechanism in the present system.2
Structural regularity of the copolymers
Regioselectivity of PO ring-opening determines the head-to-head, head-to-tail, and tail-to-tail microstructure of the copolymer. The ratio could be calculated by 13C NMR spectra of the copolymers. In the head-to-tail structure, there exist meso and racemo microstructures depending on the asymmetric center in the main chain. Although complete peak separation for each of the triad structure mm, mr, rm, and rr was unsuccessful, we could measure the ratio of [rr]/[mm] ratio which essentially represents the r/m ratio. The head-to-tail selectivity and the [rr]/[mm] ratio for some of the representative runs in Table 1 are summarized in Table 2. The head-to-tail selectivity was all around 80% with the dinuclear complexes (R,R)-(S,S)-1–3, the value being slightly lower than those with mononuclear rac-5. It is notable that the [rr]/[mm] ratio was affected by the length of the linker between the two Co units. With (R,R)-(S,S)-1, which has a linker with ten methylene units, the [rr]/[mm] was 3.4 (run 3), slightly lower than the value 4.2 with the mononuclear counterpart rac-5 (run 14). On the other hand, complex (R,R)-(S,S)-3 showed a lower [rr]/[mm] ratio of 2.0 (run 7) and it was even lower under low concentration (runs 8 and 12). Again, this strong dependence on the linker distance supports the bimetallic propagation mechanism.
| Run |
Complex |
[PO]/[Co] |
Head-to-taila |
[rr]/[mm]a |
|
Calculated by 13C NMR spectroscopy.
|
| 3 |
(R,R)-(S,S)-1 |
1,000 |
78 |
3.4 |
| 6 |
(R,R)-(S,S)-2 |
1,000 |
79 |
3.5 |
| 7 |
(R,R)-(S,S)-3 |
1,000 |
80 |
2.0 |
| 8 |
|
3,000 |
80 |
1.7 |
| 12 |
|
10,000 |
81 |
1.1 |
| 14 |
rac-5 |
1,000 |
84 |
4.2 |
| 15 |
|
3,000 |
85 |
3.6 |
Mechanistic consideration on the bimetallic mechanism
The higher TOF and the lower [rr]/[mm] ratio with (R,R)-(S,S)-3 especially under a low concentration may be explained by the following mechanistic consideration. A proposed chain-propagation mechanism for the copolymerization is shown in Scheme 2. When rac-5 was used as a catalyst, it was proposed that (i) (R)-PO are selectively activated on cobalt complexes with a (S,S)-salen ligand (vice versa), (ii) the propagating carbonate species derived from (R)-PO preferentially attacks (S)-PO (vice versa) to give a racemo diad, and (iii) the propagating species can dissociate from the cobalt center.11c Accordingly, when complexes (R,R)-(S,S)-1–4 were employed, complex 10 should be the most favorable combination of each enantiomer of epoxide and each enantiomeric Co–salen unit. Intramolecular nucleophilic ring-opening of (R)-PO gives zwitter ionic complex 11. One possible pathway from complex 11 is migration of the propagating species to the cationic cobalt center, its subsequent CO2 insertion, and (R)-PO coordination (path a), resulting in the formation of complex 12. From 12, a meso diad will be given by the ring-opening of (R)-PO. Considering that the ratio of the meso diad is lower than that of the racemo diad (Table 2), this chain-migration seems to be a minor process. On the other hand, complex 13 may be derived from complex 11 by CO2 insertion on the (S,S)-anionic center and (S)-PO binding to the (R,R)-cationic center (path b). This causes the formation of a racemo diad. Another possible pathway (path c) is similar to path a, leading to complex 14. The difference is in the migration species: the propagating species P2O (P2 = polymer chain) migrates to the cationic cobalt center instead of the last created chain with R-configuration. From 14, again a racemo diad may be created.24
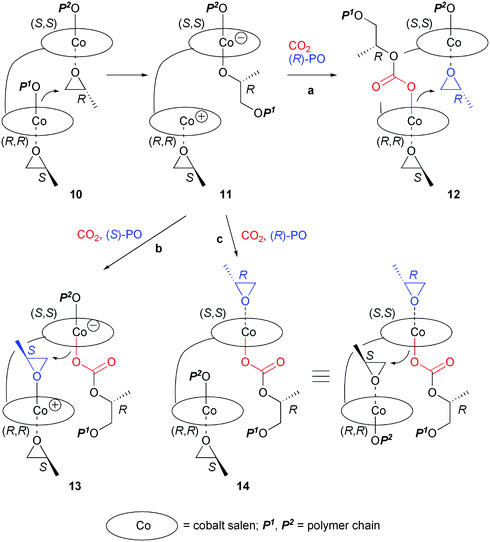 |
| | Scheme 2 Plausible propagation mechanism. | |
The highest catalytic activity of complex (R,R)-(S,S)-3 indicates that the linker length is optimally arranging the two cobalt centers into suitable positions for the bimetallic mechanism. The proximity in such an arrangement of the two cobalt centers is more effective for path a to the meso diad than path b or path c to the racemo diad, resulting in the lower [rr]/[mm] ratio. The lower carbonate-linkage content with (R,R)-(S,S)-3 may also be explained by this arrangement: a rapid PO ring-opening from 11 takes place without CO2 insertion due to the close distance between the two Co centers. On the other hand, complexes (R,R)-(S,S)-1 and 2 are more flexible because of their longer linker length. Accordingly, they produced the copolymer with a higher [rr]/[mm] ratio and a higher carbonate-linkage content in a more similar manner to those with mononuclear cobalt complex rac-5. The present results strongly support the bimetallic propagation mechanism proposed by Coates et al.11c
As described above, catalytic activity was improved by using dinuclear Co–salen complexes. The addition of onium salts to mononuclear Co–salen complexes (1.0 equiv. to Co) was also reported to enhance the catalytic activity, resulting in TOF over 500 h−1 even at ambient temperature.11b,11c Higher catalytic activity is expected under high polymerization temperatures in such a binary catalyst system.11f In fact, however, the copolymerization at 80 °C with rac-5 and bis(triphenylphosphoranylidene)iminium chloride ([PPN]Cl) resulted in the propylene carbonate (PC) formation (Table 3, run 1). In order to suppress the PC formation at high temperature, we focused on the amount of [PPN]Cl. In the copolymerization with Cr–salen complexes, the use of 0.5 equivalent of DMAP as an additive demonstrated the higher copolymer selectivity at high temperature (75 °C) than that of 1.0 equivalent.10b This tendency was found to be true of the Co–salen complex system. As shown in Table 3, the copolymer selectivity was improved with 0.5 equivalent of [PPN]Cl, resulting in high TOF over 1,000 h−1 (run 2). In this catalyst system, catalytic activity did not decrease under lower catalyst concentration (run 3). These results support that, in the presence of onium salts, the copolymerization proceeds via a monometallic propagation mechanism rather than via a bimetallic one as drawn by Coates et al.11c,18 Another important fruit of these results is that catalytic activity and selectivity of a mononuclear Co–salen complex can be easily improved by controlling the amount of onium salts.
| Run |
Complex |
[PO]/[Co] |
[PPN]Cl (equiv. to Co) |
TOF (×102 h−1)b,c |
Copolymer/PCc |
M
n
/g mol−1 |
M
w/Mnd |
Carbonate linkage (%)c |
|
Copolymerization conditions: propylene oxide (PO, 5.0 mL for runs 1, 3, and 5; 3.0 mL for runs 2 and 4), CO2 (5.3 MPa), cobalt complex, [PPN]Cl for 0.5 h.
Turnover frequency (TOF) = (mol of PO consumed)·(mol of Co center)−1·h−1.
Calculated by 1H NMR spectroscopy by using phenanthrene as an internal standard.
Estimated by gel-permeation-chromatography analysis using a polystyrene standards. The molecular weight is strongly affected by an amount of concomitant water since it works as a chain-transfer reagent.11g,21 Accordingly, the Mn values were not consistent with those estimated from the yields.
|
| 1 |
rac-5 |
10,000 |
1.0 |
3.7 |
37/63 |
12,300 |
1.34 |
99 |
| 2 |
|
3,000 |
0.5 |
11.8 |
92/8 |
20,200 |
1.59 |
>99 |
| 3 |
|
10,000 |
0.5 |
17.7 |
92/8 |
34,500 |
1.56 |
>99 |
| 4 |
(R,R)-(S,S)-3 |
3,000 |
0.5 |
12.8 |
96/4 |
30,500 |
1.46 |
>99 |
| 5 |
|
10,000 |
0.5 |
17.4 |
94/6 |
29,900 |
1.07 |
>99 |
The monometallic propagation mechanism was also indicated even when using a dinuclear Co–salen complex. A mixture of (R,R)-(S,S)-3 and [PPN]Cl (0.5 equivalent to a total amount of Co) gave high TOF over 1,000 h−1 (runs 4 and 5). The catalytic performances observed were almost identical to those with a mixture of rac-5 and [PPN]Cl (Table 3, runs 2 and 3). Accordingly, addition of onium salts probably induced the dinuclear complex to propagate the copolymer via a monometallic propagation mechanism.
Conclusions
In conclusion, we investigated the copolymerization of PO with CO2 by using dinuclear Co–salen complexes (R,R)-(S,S)-1–4 in comparison with mononuclear rac-5. Among the examined complexes, (R,R)-(S,S)-3 with four methylene linkers showed the highest TOF. The activity difference became more significant under high dilution conditions indicating that the copolymerization proceeds via a bimetallic propagation mechanism. It should be noted that catalytic activity achieved is the value in the absence of nucleophilic additives.11a In the presence of onium salts, the propagation was indicated to proceed via a monometallic mechanism11c,18 even when using a dinuclear Co complex. These data nicely agree with the previously proposed mechanism by Coates et al.11a,11c,18 The particularly interesting point in our study is that the use of 0.5 equivalent of onium salts with a mono- or dinuclear Co complex improved the high copolymer selectivity at high polymerization temperatures, thus resulting in much higher catalytic activity.
Acknowledgements
This work was supported by Grant-in-Aid for Scientific Research No. 21245023 from JSPS.
Notes and references
-
(a) E. N. Jacobsen, Acc. Chem. Res., 2000, 33, 421–431 CrossRef CAS;
(b)
M. Shibasaki and Y. Yamamoto, Multimetallic Catalysts in Organic Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2004 Search PubMed.
- R. G. Konsler, J. Karl and E. N. Jacobsen, J. Am. Chem. Soc., 1998, 120, 10780–10781 CrossRef CAS.
- J. M. Ready and E. N. Jacobsen, Angew. Chem., Int. Ed., 2002, 41, 1374–1377 CrossRef CAS.
-
(a) H. Sugimoto, C. Kawamura, M. Kuroki, T. Aida and S. Inoue, Macromolecules, 1994, 27, 2013–2018 CrossRef CAS;
(b) T. Aida and S. Inoue, Acc. Chem. Res., 1996, 29, 39–48 CrossRef CAS;
(c) W. Braune and J. Okuda, Angew. Chem., Int. Ed., 2003, 42, 64–68 CrossRef CAS;
(d) C. Billouard, S. Carlotti, P. Desbois and A. Deffieux, Macromolecules, 2004, 37, 4038–4043 CrossRef CAS.
- W. Hirahata, R. M. Thomas, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2008, 130, 17658–17659 CrossRef CAS.
- For reviews, see:
(a) K. Nakano, N. Kosaka, T. Hiyama and K. Nozaki, Dalton Trans., 2003, 4039–4050 RSC;
(b) H. Sugimoto and S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5561–5573 CrossRef CAS;
(c) G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618–6639 CrossRef CAS;
(d) D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS;
(e) G. A. Luinstra, Polym. Rev., 2008, 48, 192–219 Search PubMed.
- S. Inoue, H. Koinuma and T. Tsuruta, J. Polym. Sci., Part B: Polym. Lett., 1969, 7, 287–292 CrossRef CAS.
- T. Aida, M. Ishikawa and S. Inoue, Macromolecules, 1986, 19, 8–13 CrossRef CAS.
- For Zn catalysts, see:
(a) M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 1998, 120, 11018–11019 CrossRef CAS;
(b) D. J. Darensbourg, M. W. Holtcamp, G. E. Struck, M. S. Zimmer, S. A. Niezgoda, P. Rainey, J. B. Robertson, J. D. Draper and J. H. Reibenspies, J. Am. Chem. Soc., 1999, 121, 107–116 CrossRef CAS;
(c) D. J. Darensbourg, J. R. Wildeson, J. C. Yarbrough and J. H. Reibenspies, J. Am. Chem. Soc., 2000, 122, 12487–12496 CrossRef CAS;
(d) M. Cheng, D. R. Moore, J. J. Reczek, B. M. Chamberlain, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 8738–8749 CrossRef CAS.
- For Cr catalysts, see:
(a) D. J. Darensbourg and J. C. Yarbrough, J. Am. Chem. Soc., 2002, 124, 6335–6342 CrossRef CAS;
(b) R. Eberhardt, M. Allmendinger and B. Rieger, Macromol. Rapid Commun., 2003, 24, 194–196 CrossRef CAS;
(c) D. J. Darensbourg and R. M. Mackiewicz, J. Am. Chem. Soc., 2005, 127, 14026–14038 CrossRef CAS;
(d) D. J. Darensbourg and S. B. Fitch, Inorg. Chem., 2007, 46, 5474–5476 CrossRef CAS.
- For Co catalysts, see:
(a) Z. Q. Qin, C. M. Thomas, S. Lee and G. W. Coates, Angew. Chem., Int. Ed., 2003, 42, 5484–5487 CrossRef CAS;
(b) X. B. Lu and Y. Wang, Angew. Chem.,
Int. Ed., 2004, 43, 3574–3577 CrossRef CAS;
(c) C. T. Cohen, T. Chu and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 10869–10878 CrossRef CAS;
(d) R. L. Paddock and S. T. Nguyen, Macromolecules, 2005, 38, 6251–6253 CrossRef CAS;
(e) C. T. Cohen, C. M. Thomas, K. L. Peretti, E. B. Lobkovsky and G. W. Coates, Dalton Trans., 2006, 237–249 RSC;
(f) X. B. Lu, L. Shi, Y. M. Wang, R. Zhang, Y. J. Zhang, X. J. Peng, Z. C. Zhang and B. Li, J. Am. Chem. Soc., 2006, 128, 1664–1674 CrossRef CAS;
(g) K. Nakano, T. Kamada and K. Nozaki, Angew. Chem., Int. Ed., 2006, 45, 7274–7277 CrossRef CAS;
(h) E. K. Noh, S. J. Na, S. Sujith, S. W. Kim and B. Y. Lee, J. Am. Chem. Soc., 2007, 129, 8082–8083 CrossRef CAS;
(i) S. Sujith, J. K. Min, J. E. Seong, S. J. Na and B. Y. Lee, Angew. Chem., Int. Ed., 2008, 47, 7306–7309 CrossRef;
(j) S. J. Na, S. Sujith, A. Cyriac, B. E. Kim, J. Yoo, Y. K. Kang, S. J. Han, C. Lee and B. Y. Lee, Inorg. Chem., 2009, 48, 10455–10465 CrossRef CAS;
(k) W. M. Ren, Z. W. Liu, Y. Q. Wen, R. Zhang and X. B. Lu, J. Am. Chem. Soc., 2009, 131, 11509–11518 CrossRef CAS.
- D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2003, 125, 11911–11924 CrossRef CAS.
-
(a) B. Y. Lee, H. Y. Kwon, S. Y. Lee, S. J. Na, S. I. Han, H. S. Yun, H. Lee and Y. W. Park, J. Am. Chem. Soc., 2005, 127, 3031–3037 CrossRef CAS;
(b) Y. L. Xiao, Z. Wang and K. L. Ding, Chem.–Eur. J., 2005, 11, 3668–3678 CrossRef CAS;
(c) D. F. J. Piesik, S. Range and S. Harder, Organometallics, 2008, 27, 6178–6187 CrossRef CAS;
(d) M. R. Kember, A. J. P. White and C. K. Williams, Inorg. Chem., 2009, 48, 9535–9542 CrossRef CAS.
- Y. L. Xiao, Z. Wang and K. L. Ding, Macromolecules, 2006, 39, 128–137 CrossRef CAS.
- A monometallic mechanism was suggested for the propagation process, while a bimetallic one was proposed for the initiation process, see: D. J. Darensbourg, R. M. Mackiewicz, J. L. Rodgers, C. C. Fang, D. R. Billodeaux and J. H. Reibenspies, Inorg. Chem., 2004, 43, 6024–6034 Search PubMed.
- D. J. Darensbourg and S. B. Fitch, Inorg. Chem., 2009, 48, 8668–8677 CrossRef CAS.
- M. H. Chisholm and Z. P. Zhou, J. Am. Chem. Soc., 2004, 126, 11030–11039 CrossRef CAS.
- C. T. Cohen and G. W. Coates, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5182–5191 CrossRef CAS.
-
(a) D. A. Annis and E. N. Jacobsen, J. Am. Chem. Soc., 1999, 121, 4147–4154 CrossRef CAS;
(b) N. Madhavan, T. Takatani, C. D. Sherrill and M. Weck, Chem. Eur. J., 2009, 15, 1186–1194 CAS.
- T. S. Reger and K. D. Janda, J. Am. Chem. Soc., 2000, 122, 6929–6934 CrossRef CAS.
- H. Sugimoto, H. Ohtsuka and S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4172–4186 CrossRef CAS.
- D. J. Darensbourg and A. L. Phelps, Inorg. Chem., 2005, 44, 4622–4629 CrossRef CAS.
- The high selectivity for the copolymer was almost maintained at 45 °C. See ref. 11f.
- Another possible mechanism is that one metal center helps the formation of zwitter ionic species such as 11 and the propagation proceeds on one anionic Co center in a similar manner with a mixture of Co–salen complex and onium salt.
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.