Oxygen-assisted cross-coupling of methanol with alkyl alcohols on metallic gold†‡
Bingjun
Xu
a,
Jan
Haubrich
a,
Cassandra G.
Freyschlag
b,
Robert J.
Madix
b and
Cynthia M.
Friend
*ab
aDepartment of Chemistry and Chemical Biology, Harvard University, Cambridge, MA 02138, USA. E-mail: cfriend@seas.harvard.edu; Tel: +1-617-495-4198
bSchool of Engineering and Applied Sciences, Harvard University, Cambridge, MA 02138, USA
First published on 24th June 2010
Abstract
We demonstrate for the first time that selective cross-coupling of methanol with either ethanol or n-butanol occurs below room temperature on metallic gold with no metal oxide support in a reaction sequence that occurs entirely on the surface. The esterification proceeds via activation of the alcohols by adsorbed oxygen and a sequence of reactions that involve both surface-bound alkoxys and hemiacetals as intermediates. The reaction selectivity is dictated by competing β-hydride elimination from the alkoxys. Due to the higher activation energy for β-hydride elimination from methoxy, no formate esters are formed. A molecular-scale mechanism constructed using our results is in excellent agreement with studies of heterogeneous catalysts, providing insight into selectivity control under a broad range of conditions.
Introduction
The importance of enhancing the energy efficiency of catalytic oxidation processes for chemical synthesis has spurred a great interest in the chemistry of Au-based catalysts—both heterogeneous,1–19 and homogeneous.1–3 “Green” chemical processes may be possible with Au because of the fact that many reactions occur at remarkably low temperatures and with high selectivity. For example, the oxidative coupling of methanol to the ester, methylformate, is catalyzed by unsupported nanoporous Au with nearly 100% selectivity at atomospheric pressure using O2 as an oxidant.20 Indeed, the direct correspondence between the mechanism established for the methanol-to-methylformate reaction on Au(111) containing adsorbed O and the catalytic reaction on nanoporous Au illustrate the value of fundamental studies of the surface chemistry of Au in catalysis. Selective oxidative coupling of alcohols to organic esters is of significance because of its application to the synthesis of precursors for fabrics and for the food and fragrance industries.The facility for oxidative coupling of methanol and ethanol suggests that these processes can be generalized, providing greater synthetic capability. Oxidative coupling reactions between methanol and other alcohols on supported gold catalysts have been reported in solution phase.5–7 The oxidative coupling reactions between higher molecular weight alcohols and methanol were mediated by supported Au catalysts in methanol solution with the presence of molecular oxygen. Very high selectivity (>90% in most cases) towards cross-coupling products, i.e., methyl esters, was observed. Our recent studies of methanol coupling with various aldehydes on O/Au(111) at low pressure21 as well as the coupling of methanol to acetaldehyde to form methylacetate in a catalytic process at atmospheric pressure20 were strong indicators that coupling of two different alcohols is also possible on metallic Au containing adsorbed O.
Herein, we demonstrate that cross-coupling of alcohols is efficiently mediated by atomic O bound to Au nanoparticles on the metallic Au(111) surface in the absence of a metal oxide support. The Au nanoparticles spontaneously form upon oxidation of the Au(111) surface with ozone at 200 K. The O is predominantly bound to 3-fold coordinate sites on a disordered surface in these experiments8 and it is necessary to activate the reactant alcohols. The ability to selectively convert methanol to methylformate near room temperature using a metallic nanoporous Au catalyst and O2 was also recently demonstrated.4 This fact in conjuction with the results described herein suggest that metallic Au-based materials will also be effective catalysts for selective oxidative cross-coupling of different alcohols.
Previously, we established a general mechanistic framework for oxidative coupling promoted by atomic oxygen bound to metallic Au that provides the basis for understanding the alcohol cross-coupling reactions. A predictive mechanism was constructed based on detailed studies of the oxidative self-coupling of methanol and ethanol to form esters.18,19 The recent report on selective oxidation of 2-butanol to 2-butanone on O/Au(111) is also in line with the proposed mechanism.22 We subsequently used this reaction sequence to predict and experimentally demonstrate that selective cross-coupling of alcohols with aldehydes and the acylation of amines using formaldehyde23 occur entirely on a Au surface containing atomic oxygen without the necessity of reactions in the ambient phase. Marsden et al. also reported cross-coupling between methanol and benzaldehyde over supported gold catalyst in liquid solution at temperatures as low as −78 °C, which is completely consistent with our results.17
The key features of the self-coupling reactions are (1) O–H bond dissociation in the alcohols via proton transfer to adsorbed O, yielding the corresponding alkoxys on the surface; and, (2) facile β-hydride elimination from the adsorbed alkoxy to form an adsorbed aldehyde, and (3) nucleophilic attack of the electron-deficient carbon center of the aldehyde, by the alkoxy (Scheme 1). Thus, the selective activation of the O–H bond to form the alkoxy primes the surface with the reactants necessary to produce coupling; namely, the alkoxy itself and its dehydrogenation product, the aldehyde. Once the aldehydye forms by β-H elimination, it reacts rapidly with remaining alkoxy to form a surface-bound hemiacetal, which then eliminates H to form the coupling product – the ester.
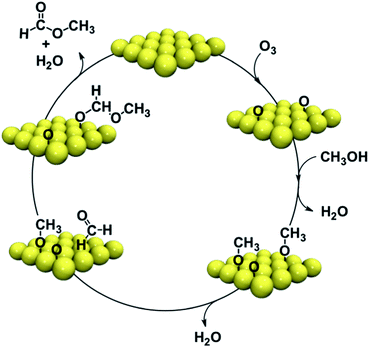 | ||
| Scheme 1 Catalytic cycle for the oxidative self-coupling reactions of methanol on gold single crystal surfaces. | ||
Though all alcohols are expected to be activated by adsorbed oxygen to form their corresponding alkoxys, alcohol cross-coupling depends upon the simultaneous existence of an alkoxy formed from one alcohol and the aldehyde from the other. The relative ease of β-H elimination of the alkoxys is therefore an important factor in determining the selectivity for cross-coupling vs. self-coupling reactions. The selectivity for cross-coupling may, thus, be quite sensitive to the respective alcohols employed. In this paper, we report for the first time the highly selective cross-coupling of methanol with two primary alcohols—ethanol and butanol—mediated by a sequence of reactions occurring entirely on the metallic gold surface. These results clearly establish that metallic Au containing surface-bound atomic O has potential as a catalyst for selectively promoting a wide range of complex cross-coupling reactions at or near room temperature.
Experimental
All experiments were performed in an ultrahigh vacuum (UHV) chamber with a base pressure below 2 × 10−10 Torr. The preparation of the clean Au(111) surface has been described elsewhere.24 The surface was first populated with 0.1 ML O (O/Au(111)) by introducing an appropriate amount of ozone at 200 K. Based on previous studies on self-coupling of alcohols,18,19 this specific surface oxygen coverage was chosen in order to minimize combustion relative to the coupling reactions. The oxygen atom coverage was calibrated by comparing the amount of O2 evolved in temperature programmed desorption to that formed for a saturation coverage of oxygen atoms.25 Oxidation of the surface in this manner leads to release of Au atoms to form nanostructures containing Au and O, most of which are smaller than 2 nm in diameter.24 This oxygen-covered Au surface is referred to here as O/Au(111).Two alcohols were premixed with a specified ratio and dosed onto the O/Au(111) surface at 150 K via a dosing tube. The volume ratio of the methanol–ethanol and methanol–butanol liquid mixtures was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 and 1:3, respectively. The compositions of the alcohol mixtures were chosen in order to yield substantial amounts of the coupling products. Sequential dosing of organic molecules was employed in some experiments, where instead of premixing, they were introduced to the surface sequentially at 150 K. Exposures, corrected for dosing enhancement, are given here in terms of Langmuir (L) (1 L = 10−6 torr-seconds); the dosing pressure was determined by uncorrected ion gauge pressures.
:2 and 1:3, respectively. The compositions of the alcohol mixtures were chosen in order to yield substantial amounts of the coupling products. Sequential dosing of organic molecules was employed in some experiments, where instead of premixing, they were introduced to the surface sequentially at 150 K. Exposures, corrected for dosing enhancement, are given here in terms of Langmuir (L) (1 L = 10−6 torr-seconds); the dosing pressure was determined by uncorrected ion gauge pressures.
Temperature programmed reaction – a method for determining the product distributions and for obtaining kinetic information – was conducted according to well-established protocols, described in detail elsewhere.24 The heating rate for all reactions was nearly constant at 5 K s−1. The reaction products were identified by quantitative mass spectrometry (Hiden HAL/3F) using fragmentation patterns obtained from authentic samples, which were found to be in general agreement with NIST reference data.26
Results
Both cross-coupling of methanol with ethanol to form methylacetate and self-coupling of ethanol to form ethylacetate were observed for reaction of a mixture of d0- methanol and d0-ethanol on O/Au(111) (Fig. 1a) (1:2 volume ratio of a liquid mixture). No self-coupling of methanol was detected. The methylacetate (CH3C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)OCH3), and ethylacetate (CH3C(O)OCH2CH3), were identified from their parent ions and by quantitative analysis of their fragmentation patterns (ESI†, Tables 1, 2). The other possible coupling products, methylformate (HC(O)OCH3) and ethylformate (HC(O)OCH2CH3), were not detectable to within the noise level of the measurement (<0.001 monolayers). Notably, except for the combustion products CO2 (∼20%) and water, no other products, such as acids or aldehydes derived from either reactant, were observed based on monitoring of key mass fragments (ESI†, Table 1); nor was residual carbon detectable. In the absence of the preadsorbed O, only reversible molecular desorption of methanol and ethanol (at 170 K and 200 K, respectively) was observed (data not shown).
O)OCH3), and ethylacetate (CH3C(O)OCH2CH3), were identified from their parent ions and by quantitative analysis of their fragmentation patterns (ESI†, Tables 1, 2). The other possible coupling products, methylformate (HC(O)OCH3) and ethylformate (HC(O)OCH2CH3), were not detectable to within the noise level of the measurement (<0.001 monolayers). Notably, except for the combustion products CO2 (∼20%) and water, no other products, such as acids or aldehydes derived from either reactant, were observed based on monitoring of key mass fragments (ESI†, Table 1); nor was residual carbon detectable. In the absence of the preadsorbed O, only reversible molecular desorption of methanol and ethanol (at 170 K and 200 K, respectively) was observed (data not shown).
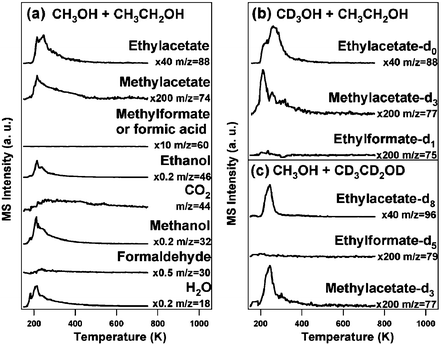 | ||
| Fig. 1 Cross-coupling between methanol and ethanol induced by temperature programmed reaction of methanol and ethanol mixtures on O/Au(111) (ΘO = 0.1 ML). (a) CH3OH and CH3CH2OH, (b) CD3OH and CH3OH, (c) CH3OH and CD3CD2OD. Surface oxygen was introduced by ozone exposure at 200 K prior to the introduction of the alcohol mixtures (6 L), which were dosed at 150 K. The contributions from cracking fragments have been subtracted for clarity. The heating rate was 5 K s−1. | ||
The identity of the products was further confirmed by the mass shifts observed for products formed from isotopically-labelled mixtures of the reactant alcohols: namely, mixtures of CD3OH/CH3CH2OH and CH3OH/CD3CD2OD (Fig. 1b, c). For reaction between CD3OH and C2H5OH the mass of the parent ion for the cross-coupling product, methylacetate, shifted from m/z 74 (CH3C(O)OCH3) to 77 (CH3C(O)OCD3) (Fig. 1b), whereas there was no shift in the peak ascribed to the ethanol self-coupling product, ethylacetate. Furthermore, there was no detectable formation of d5-ethylformate (CD3CD2OC(O)H) nor of acetaldehyde in reaction of the CH3OH/CD3CD2OD mixture (Fig.1c). Finally, neither the product distribution nor the peak temperature for peak ester formation (rate) is altered by potential kinetic isotope effects for C–H(D) bond breaking to form the acetaldehyde from the ethoxy. The isotopic distributions observed in these experiments clearly show that the C–H(D) bonds in the methyl group of the methoxy formed from methanol are retained in the products, while β-C–H(D) elimination occurs from the ethoxy.
As noted above, neither self-coupling of methanol to form methylformate nor cross-coupling to yield ethylformate was detected. Previous studies show that formaldehyde reacts readily with adsorbed methoxy to form methylformate well below 200 K,21 suggesting that in the more complex reaction conditions studied here, β-H elimination from methoxy to form formaldehyde does not compete effectively with the analogous process to yield acetaldehyde from ethoxy. However, in order to conclude that ethylformate would be expected from reaction of ethoxy and formaldehyde, this reaction was examined in detail. Indeed, following the formation of adsorbed ethoxy by reaction of ethanol with O/Au,19 coadsorption of formaldehyde produces significant amounts of ethylformate (Fig. 2). Self-coupling of ethoxy to form ethylacetate also competes with this reaction.19
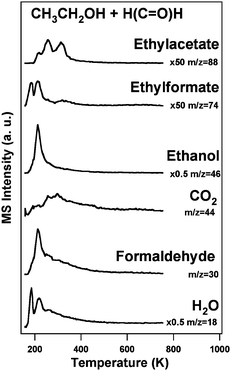 | ||
| Fig. 2 Coupling reactions spectra of ethanol and formaldehyde on O/Au(111) (ΘO = 0.1 ML). Surface oxygen was introduced by ozone exposure at 200 K prior to the introduction of the organic molecules (6 L), which were dosed at 150 K. The contributions from cracking fragments were subtracted for clarity. The heating rate was 5 K s−1. | ||
In the coupling reactions of methanol and ethanol (Fig. 1) the fact that neither methylformate nor ethylformate is formed indicates that the methoxy reacts facilely and preferentially with the acetaldehyde formed from the ethoxy, prior to any β-H elimination from adsorbed CH3O to give formaldehyde. If formaldehyde were formed, it should react with either ethoxy or methoxy adsorbed on the surface. We therefore conclude that the product distribution is determined by the more facile β-H elimination of ethoxy relative to that of methoxy which arises from the difference in bond strengths between primary and secondary C–H bonds.
Vibrational (high resolution electron energy loss (HREEL)) spectra of the adsorbed species verify that coadsorbed methoxy and ethoxy result from the exposure of O/Au to methanol and ethanol (Fig. 3a,b). The alkoxys are identified by their characteristic vibrations and the absence of the strong mode near 650 cm−1 from the OH bend in the adsorbed alcohols.19 That the O–H bonds of ethanol and methanol are activated via reaction with O/Au is indicated by the absence of this mode. While these experiments clearly show that the O–H bonds are dissociated at low temperature, the relatively low energy resolution of this technique and the intrinsic low signal-to-noise ratio due to surface roughness, does not allow us to readily distinguish methoxy and ethoxy based on small differences in vibrational frequencies for adsorbed CH3O and C2H5O.
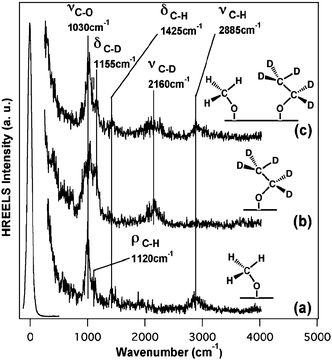 | ||
| Fig. 3 Vibrational (high resolution electron energy loss) spectra of (a) CH3O(a), (b) CD2CD3O(a) and (c) CD2CD3O(a) and CH3O(a) on O/Au(111) (ΘO = 0.1 ML) at 170 K. Surface oxygen was introduced by ozone exposure at 200 K prior to the introduction of alcohols (6 L), which were dosed at 170 K, in the cases of (a) and (b). d0-Methanol (6 L) and d6-ethanol (0.1 L) were introduced sequentially to O/Au(111) (ΘO = 0.1 ML) at 170 K. The low signal-to-noise ratio results from the roughness of the surface. | ||
We clearly demonstrated that both methoxy and ethoxy form on the surface by obtaining the vibrational spectrum after d0-methanol (CH3OH) and d6-ethanol (C2D5OD) were dosed sequentially onto O/Au(111) at 175 K. At this temperature, neither molecular methanol nor molecular ethanol remains adsorbed, and the alkoxys can be isolated. Fig. 3 shows clearly the vibrational modes expected for both the d0-methoxy and d5-ethoxy. In particular, both C–H stretch modes of methoxy and C–D stretch modes for ethoxy are observed (Fig. 3c).
We confirmed that temperature programmed heating subsequent to recording of the vibrational spectrum yields the cross-coupling and self-coupling products expected from reactions of these adsorbed alkoxys as reported above. Thus, it is clear that the coadsorbed alkoxys lead to the cross-coupling products.
In order to establish the generality of these surface-mediated cross-coupling reactions, we also examined the oxygen-assisted coupling of n-butanol and methanol. First, we showed that n-butanol self-couples to form butylbutyrate (C4H9OC(O)C3H7) (Fig. 4a). At this low initial surface oxygen coverage the selectivity for self-coupling to form butylbutyrate was 30%, the competitive product being the aldehyde, butanal (70%). Butanal and 2-butanone are not readily distinguishable from the mass fragmentation pattern as they share the only unique mass fragment (the parent ion m/z 72). m/z 72 is assigned to butanal; the formation of the ketone is unlikely since it requires a rearrangement of the carbon skeleton.
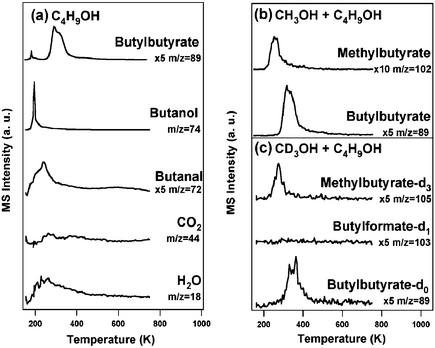 | ||
| Fig. 4 Coupling reactions on O/Au(111) (ΘO = 0.1 ML): (a) butanol (b) a 1:3 mixture of CH3OH and CH3CH2CH2CH2OH and, (c) a 1:3 mixture of CD3OH and CH3CH2CH2CH2OH. Oxygen was first deposited by introduction of ozone to the surface at 200 K prior to exposure of the alcohol mixture. Methyl n-butyrate and n-butylformate (which have the same parent ion (m/z 102)) were distinguished based on the mass shift from 102 to 105 for reaction with CH3OH vs. CD3OH, which established that only n-C3H7C(O)OCD3 is formed (not n-C4H9OC(O)D). The contributions from cracking fragments were subtracted for clarity. The heating rate is 5 K s−1. | ||
In the presence of methanol, n-butanol cross-couples to form methylbutyrate, while also forming the self-coupling product, n-butylbutyrate (Fig. 4b). Mixtures of CH3OH (or CD3OH) and n-butanol (1:3 volume ratio of a premixed liquid mixture) react on O/Au(111) to form both methylbutyrate and butylbutyrate, but no methylformate. This result generalizes the mechanistic scheme for alcohol cross-coupling to longer-chain primary alcohols (Scheme 2). Similar to the reactions of ethanol and methanol, the product distribution for n-butanol is dictated by the rapid β-H elimination to form butanal, which is then attacked by either methoxy or n-butoxy. The absence of products associated with β-C–H scission in methoxy to yield formaldehyde illustrates that this reaction does not compete favorably with that of n-butoxy. The β-H elimination from the butoxy occurs prior to that of methoxy, and the methoxy is reacted away to form the butyrates before any formaldehyde can form. In the absence of the adsorbed O, only reversible molecular desorption of methanol and butanol was observed (at 170 K and 210 K, respectively) (data not shown).
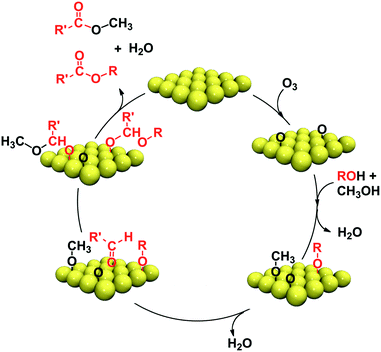 | ||
| Scheme 2 Catalytic cycle for the cross-coupling reactions of primary alcohols with methanol on gold single crystal surfaces. R′ is the alkyl fragment that remains bound to the formyl after β-H elimination from the RO group. | ||
This relative order of ease of β-hydride elimination from the alkoxys inferred from these experiments parallels that previously documented on both Cu(110) and Ag(110).27,28 The temperature at which the β-H elimination occurs decreases with increasing molecular weight of the alkoxy. For example, on Cu(110) β-H scission occurs at 355 K, 348 and 340 K for adsorbed methoxy, ethoxy and n-propoxy, respectively. Given this trend, the temperature for β-H elimination from butoxy would be expected to be significantly lower than that for methoxy on the Au(111) surface, thus dictating the product distribution observed here.
Our fundamental studies provide the basis for a catalytic cycle for the cross-coupling reactions of alcohols with methanol (Scheme 2). Based on our mechanism, primary alcohols will generally form alkoxys at low temperature by reaction with adsorbed oxygen on the Au. Once a mixture of alkoxys is formed, the products will be determined by the facility for β-H elimination from the alkoxy. Since this elimination step is slower for methanol, the methoxy will preferentially attack the other primary alkoxy, rather than self couple. We are currently investigating how the selectivity of these processes can be controlled through the manipulation of the concentration of surface species.
Our results suggest that metallic Au should be an efficient and highly selective catalyst for the vapor-phase cross-coupling of primary alcohols with methanol. Our recent studies of nanoporous Au have established that mechanisms based on fundamental studies at low pressure provide the basis for predicting reaction mechanisms and selectivity at atmospheric conditions.20
The results provided herein provide a basis for understanding the coupling reactions between methanol and other higher molecular weight alcohols done in solution phase, where only the cross-coupling products were observed.17,29,30 Since the coupling reactions were conducted in methanol solution, it is expected that the self-coupling reaction of the solute (higher molecular weight) alchohols was not observed because of (1) the facile β-H elimination from the solute alcohols, (2) the relatively high concentration of methanol in solution and (3) the relatively slow rate of conversion of methanol to formaldehyde. We further anticipate that analogous cross-coupling reactions will be possible over nanoporous Au catalysts at atmospheric pressures—a topic planned for future study.
The excellent correspondence with solution phase reactions and our low pressure studies can be attributed to several key features of the gold surface: (1) it is inert unless atomic oxygen is present; (2) the dissociation probability of molecular oxygen (or other oxygen sources) is very low on gold, which makes the steady-state surface coverage of atomic oxygen low; (3) OH is not stable with respect to disproportionation and water binds to gold surface very weakly. Therefore, water is easily displaced from the surface by the reactant alcohols. The mechanistic model for the cross-coupling of alcohols derived herein thus affords a more complete understanding of these esterification reactions in the liquid-phase.
Because both metallic copper and silver form alkoxys from alcohols in the presence of adsorbed oxygen, it is possible that they may also promote these coupling reactions. Indeed self-coupling of methanol to methylformate on Ag(110) was reported by Madix and Wachs.28 However, this matter has not been studied further to the best of our knowledge. We are currently investigating such reactions.
Conclusions
Both ethanol and n-butanol cross-couple with methanol in a process that occurs entirely on a Au surface and is mediated by adsorbed atomic oxygen. The cross-coupling esterification competes effectively with the self-coupling reactions. The selectivity for cross-coupling is attributed to the slow rate of β-C–H dissociation in methoxy relative to ethoxy or butoxy, which favors the formation of methyl esters. We conclude that this reaction pattern holds for reactions of other aliphatic alcohols with methanol over metallic gold and that it establishes a general mechanistic framework for catalytic cross-coupling using metallic Au and O2.Notes and references
- A. Abad, A. Corma and H. Garcia, Chem.–Eur. J., 2008, 14, 212 CrossRef CAS
.
- A. Abad, P. Concepcion, A. Corma and H. Garcia, Angew. Chem., Int. Ed., 2005, 44, 4066 CrossRef CAS
- S. Biella and M. Rossi, Chem. Commun., 2003, 378 RSC
- S. Carrettin, P. McMorn, P. Johnston, K. Griffin, C. J. Kiely and G. J. Hutchings, Phys. Chem. Chem. Phys., 2003, 5, 1329 RSC
- D. I. Enache, D. W. Knight and G. J. Hutchings, Catal. Lett., 2005, 103, 43 CrossRef CAS
- J. L. Gong and C. B. Mullins, J. Am. Chem. Soc., 2008, 130, 16458 CrossRef CAS
- T. Hayashi, T. Inagaki, N. Itayama and H. Baba, Catal. Today, 2006, 117, 210 CrossRef CAS
- W. Hou, N. A. Dehm and R. W. J. Scott, J. Catal., 2008, 253, 22 CrossRef CAS
- P. Fristrup, L. B. Johansen and C. H. Christensen, Catal. Lett., 2008, 120, 184 CrossRef CAS
- P. Fristrup, L. B. Johansen and C. H. Christensen, Chem. Commun., 2008, 2750 RSC
- B. Jorgensen, S. E. Christiansen, M. L. D. Thomsen and C. H. Christensen, J. Catal., 2007, 251, 332 CrossRef CAS
- F. Z. Su, J. Ni, H. Sun, Y. Cao, H. Y. He and K. N. Fan, Chem.–Eur. J., 2008, 14, 7131 CrossRef CAS
- L. C. Wang, L. He, Q. Liu, Y. M. Liu, M. Chen, Y. Cao, H. Y. He and K. N. Fan, Appl. Catal., A, 2008, 344, 150 CrossRef CAS
- X. G. Wang, H. Kawanami, N. M. Islam, M. Chattergee, T. Yokoyama and Y. Ikushima, Chem. Commun., 2008, 4442 RSC
- A. Abad, C. Almela, A. Corma and H. Garcia, Tetrahedron, 2006, 62, 6666 CrossRef CAS
- I. Nielsen, E. Taarning, K. Egeblad, R. Madsen and C. Christensen, Catal. Lett., 2007, 116, 35 CrossRef CAS
- C. Marsden, E. Taarning, D. Hansen, L. Johansen, S. K. Klitgaard, K. Egeblad and C. H. Christensen, Green Chem., 2008, 10, 168 RSC
- B. Xu, X. Liu, J. Haubrich, R. J. Madix and C. M. Friend, Angew. Chem., Int. Ed., 2009, 48, 4206 CrossRef CAS
- X. Y. Liu, B. J. Xu, J. Haubrich, R. J. Madix and C. M. Friend, J. Am. Chem. Soc., 2009, 131, 5757 CrossRef CAS
- A. Wittstock, V. Zielasek, J. Biener, C. M. Friend and M. Baumer, Science, 2010, 327, 319 CrossRef CAS
- B. Xu, X. Liu, J. Haubrich and C. M. Friend, Nat. Chem., 2010, 2, 61 Search PubMed
- T. Yan, J. Gong and C. B. Mullins, J. Am. Chem. Soc., 2009, 131, 16189 CrossRef CAS
- B. Xu, L. Zhou, R. J. Madix and C. M. Friend, Angew. Chem., Int. Ed., 2009, 49, 394
- B. K. Min, A. R. Alemozafar, D. Pinnaduwage, X. Deng and C. M. Friend, J. Phys. Chem. B, 2006, 110, 19833 CrossRef CAS
- N. Saliba, D. H. Parker and B. E. Koel, Surf. Sci., 1998, 410, 270 CrossRef CAS
- Mass Spectra in NIST Chemistry WebBook, NIST Standard Reference Database Number 69, ed. S. E. Stein, P. J. Linstrom and W. G. Mallard, Institute of Standards and Technology, Gaithersburg MD, p. 20899 Search PubMed.
- M. Bowker and R. J. Madix, Surf. Sci., 1982, 116, 549 CrossRef CAS
- I. E. Wachs and R. J. Madix, Surf. Sci., 1978, 76, 531 CrossRef CAS
- S. K. Klitgaard, A. T. DeLa Riva, S. Helveg, R. M. Werchmeister and C. H. Christensen, Catal. Lett., 2008, 126, 213 CrossRef CAS
- R. L. Oliveira, P. K. Kiyohara and L. M. Rossi, Green Chem., 2009, 11, 1366 RSC
Footnotes |
| † Electronic supplementary information (ESI) available: Mass spectrometry data. See DOI: 10.1039/c0sc00214c |
| ‡ Support of this work by the U.S. Department of Energy, Basic Energy Sciences, under grant No. DE-FG02-84-ER13289 is gratefully acknowledged. JH also acknowledges support of the Alexander von Humboldt Foundation through a Feodor Lynen fellowship. |
| This journal is © The Royal Society of Chemistry 2010 |