The role of functional group concentration in solvation thermodynamics†
Niklaas J.
Buurma
a,
Joanne L.
Cook
a,
Christopher A.
Hunter
*a,
Caroline M. R.
Low
b and
Jeremy G.
Vinter
c
aKrebs Institute, Department of Chemistry, University of Sheffield, Sheffield, UK S3 7HF. E-mail: C.Hunter@shef.ac.uk; Fax: +44 114 2229346; Tel: +44 114 2229456
bDrug Discovery Facility, Biochemistry, Imperial College, London, UK SW7 2AY
cCresset Biomolecular Discovery, BioPark Hertfordshire, Broadwater Road, Welwyn Garden City, UK AL7 3A
First published on 11th June 2010
Abstract
High throughput experiments using a molecular recognition probe reveal a simple relationship between solvent functional group concentration and selective solvation. The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 association constant for the H-bonding interaction between tri-n-butylphosphine oxide and 4-phenylazophenol was measured in 1088 different alkane–ether mixtures using a UV-Vis plate reader. Although the stability of the complex decreased with increasing concentration of the more polar ether cosolvent as expected, the results show that it is the functional group composition rather than the constitution of the solvent molecules or the properties of the bulk liquid that determines the solvation thermodynamics. Thus the solvent properties of a simple ether can be reproduced by an appropriate mixture of a polyether and an alkane that has the same net concentration of ether oxygen functional groups. The results suggest that solvation may be understood at the molecular level simply by considering the polarities and the concentrations of the functional groups present in the solvent, because these are the parameters that affect local solvation interactions with the solutes.
:1 association constant for the H-bonding interaction between tri-n-butylphosphine oxide and 4-phenylazophenol was measured in 1088 different alkane–ether mixtures using a UV-Vis plate reader. Although the stability of the complex decreased with increasing concentration of the more polar ether cosolvent as expected, the results show that it is the functional group composition rather than the constitution of the solvent molecules or the properties of the bulk liquid that determines the solvation thermodynamics. Thus the solvent properties of a simple ether can be reproduced by an appropriate mixture of a polyether and an alkane that has the same net concentration of ether oxygen functional groups. The results suggest that solvation may be understood at the molecular level simply by considering the polarities and the concentrations of the functional groups present in the solvent, because these are the parameters that affect local solvation interactions with the solutes.
Introduction
Solvent plays a critical role in a wide range of phenomena, from biomolecular recognition to organic synthesis, but a molecular level understanding of solvation phenomena has remained elusive.1–6 The thermodynamics of solvation governs the efficiency and selectivity of both covalent and non-covalent chemical processes, but our ability to make accurate and reliable predictions is hampered by the complexity of the liquid phase. If we are to be able to realise molecular engineering in the context of areas like medicine and nanotechnology, a quantitative understanding of the details of the relationship between non-covalent interactions and chemical structure will be essential. In particular, the ability to pick the right solvent or solvent combination to optimise a process in chemical engineering, the selectivity of a reaction, the properties of a macromolecular structure or supramolecular assembly will be key.The pragmatic approach to dealing with solvent effects is to exploit empirical parameters that characterise the bulk liquid or the interaction with a spectroscopic probe in order to quantify properties such as polarity, polarisability and hydrogen bonding ability.4–6 The advantage of these parameters is that they often require a single measurement to characterise a given solvent, offering a convenient method for ranking solvents with respect to a particular property. However, non-covalent interactions are governed by equilibrium thermodynamics, and so, although parameters based on spectroscopic probes or reaction kinetics can be used to formulate models of solution equilibria, they must be parameterised for each case.7–9 Moreover, solvent mixtures often display non-linear properties, which are difficult to relate to the properties of the constituent solvents.10–16 Preferential solvation of a solute means that the composition of the solvent shell does not always reflect the composition of the bulk liquid and is likely to be solute-dependent, which limits the application of spectroscopic probes to understanding solvation effects in solvent mixtures.
Supramolecular complexes have been exploited for understanding solvents effects on molecular recognition, but the complexity of these systems complicates dissection of the many thermodynamic contributions.17–24 We recently introduced a new molecular recognition based approach to studying solvent effects.25 The method provides quantitative thermodynamic information on the effect of the solvent on non-covalent interactions and has been applied to a range of competitive solvents as well as solvent mixtures.26 Direct measurement of the equilibria in complex mixtures of solutes and solvents should lead to new insight into phenomena such as preferential solvation and provide the basis for predictive models. The disadvantage of the approach is that it requires complete titrations to study each solvent environment, so the full potential will only be realised if high throughput automated experiments can be implemented. Here, we describe a high throughput UV-Vis absorption experiment, which uses a molecular recognition probe to characterise the thermodynamic properties of solvent mixtures.
Results and discussion
The 1:1 complex formed between tri-n-butylphosphine oxide, 1, and 4-phenylazophenol, 2, provides a convenient spectroscopic probe of H-bonding even in competitive solvents like ethers (Fig. 1). Tri-n-butylphosphine oxide is an exceptionally strong H-bond acceptor (β = 10.2) and has no significant UV-Vis absorption over 300 nm, so it can be used at very high concentrations as the guest in UV-Vis titration experiments.27 In addition, 1 has no polar H-bond donor sites, and 31P NMR dilution studies in n-octane showed no evidence of self-interaction at concentrations up to 10 mM. 4-Phenylazophenol is a good H-bond donor, and although the azo nitrogens of 2 are reasonable H-bond acceptors, UV-Vis dilution studies in n-octane showed no evidence of self-interaction at concentrations up to 100 mM. 2 has an intense UV-Vis absorption band between 300 and 400 nm, which is sensitive to H-bonding interactions with the hydroxyl group. Thus the absorption maximum of 2 changes from 339 nm in n-octane to 349 nm in di-n-hexyl ether, due to the phenol–ether H-bond (Fig. 1). However, addition of 1 leads to a further shift in the absorption maximum to 356 nm, which is characteristic of the 1·2 complex and independent of the solvent (Fig. 1). The phenolate anion obtained by deprotonation of 2 absorbs at 420 nm, and this was not observed in any of the experiments reported here.
![UV-Vis absorption spectra for titration of 1 into 2 (a) in n-octane, [2] = 50 μM and (b) in di-n-hexyl ether, [2] = 20 μM. The spectrum of unbound 2 is shown in blue and the 1·2 complex in red. Chemical structures of 1 and 2 are also shown.](/image/article/2010/SC/c0sc00209g/c0sc00209g-f1.gif) | ||
| Fig. 1 UV-Vis absorption spectra for titration of 1 into 2 (a) in n-octane, [2] = 50 μM and (b) in di-n-hexyl ether, [2] = 20 μM. The spectrum of unbound 2 is shown in blue and the 1·2 complex in red. Chemical structures of 1 and 2 are also shown. | ||
In order to investigate a wide range of different solvent compositions, we have developed an automated titration protocol for a UV-Vis plate reader. Solutions of 1 and 2 were loaded onto a 96-well quartz microplate, such that the concentration of host, 2, was constant and the concentration of guest, 1, spanned five orders of magnitude. This gives the experiment an excellent dynamic range, and the binding isotherm is generated in a straightforward manner by simply recording the absorbance of the plate. Mixed solvent titration data were generated by sequential addition of aliquots of the two solvents generating 34 × 48 point binding isotherms in 2.5 h.
Whilst the automated experiment allows rapid collection of data, there are potential drawbacks associated with the methodology described above. The microplate is open to the atmosphere, so volatile solvents can evaporate and hygroscopic solvents can absorb water. The problem of evaporation was minimised by using high boiling point solvents, and the results of conventional manual titrations are in agreement with the automated experiments, suggesting that interactions of the microplate samples with the atmosphere have a limited effect on the measured association constants (Table 1). However, when the association constant is low (log K < 2), the error in the automated experiment is significant.
| Solvent | Manual | Automated |
|---|---|---|
| n-Octane | 5.1 ± 0.2 | 5.0 ± 0.7 |
| Di-n-hexyl ether | 2.7 ± 0.2 | 2.6 ± 0.1 |
| Di-n-octyl ether | 2.5 ± 0.4 | 2.3 ± 0.3 |
| Di(ethyleneglycol)dibutyl ether | 2.0 ± 0.2 | 1.6 ± 0.2 |
| Tetra(ethyleneglycol)dimethyl ether | 1.7 ± 0.2 | 1.1 ± 0.5 |
A typical binding isotherm is shown in Fig. 2a. The increase in absorbance between guest concentrations of 10−4 and 10−3 M is due to the formation of the 1·2 complex. At very high concentrations of 1, there is an increase in absorbance with guest concentration, which we attribute to a weaker second binding event. The data were therefore fit to a 1:1 isotherm with a linear correction to account for the second phase of the titration. Fig. 2b illustrates typical data obtained from a complete automated experiment in mixtures of n-octane and di-n-hexyl ether. Data from three different runs of the experiment show that reproducibility is good, allowing accurate association constants to be determined routinely for a wide range of solvent conditions. In the automated experiment, the errors in individual association constants (±50%) are larger than those obtained in a typical manual titration (±10%), but the trends over a complete data set are clear (Fig. 2b).
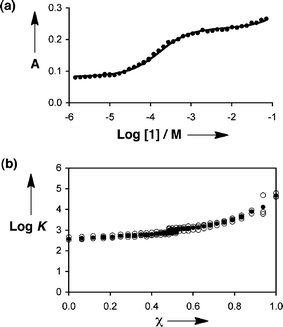 | ||
| Fig. 2 Data from automated titration of 1 into 2. (a) A binding isotherm: UV-Vis absorption at 390 nm, A, in an 88:12 v/v mixture of n-octane and di-n-hexyl ether. (b) Association constants (log K) obtained in mixtures of n-octane and di-n-hexyl ether plotted as a function of the volume fraction of n-octane (χ). The average of three different runs of the experiment is shown (filled circles) along with the results of the individual runs (open circles). | ||
Automated titrations were carried out using the 1·2 complex in mixtures of alkanes and ethers to give 1088 association constants for different solvent compositions (see ESI†). In order to obtain data in solvent mixtures very dilute in ether, the experiment was carried out using 2%, 5% and 10% by volume mixtures of ether and n-octane as one solvent, and pure n-octane as the other. The range of solvent polarity of these mixtures as measured by the dielectric constant is between 2.0 and 7.9, or between 31 and 39 as measured by the ET(30) parameter.28
The association constant is highest in pure alkanes and lowest in pure polyethers. Fig. 2b shows that the stability of the 1·2 complex decreases with increasing ether content, and this trend was observed in all of the experiments. For mixtures of different ethers, the changes in log K as a function of solvent composition were smaller, and there was almost no variation in log K in mixtures of alkanes. For a particular ether–alkane combination, the variation in log K with composition is almost linear with ether concentration, but there is a significant variation from one ether to another. For example, the value of log K observed for pure di-n-hexyl ether is the same as that observed for a 4:1 mixture of n-octane and tetraethylene glycol dimethyl ether. Tetraethylene glycol dimethyl ether contains five times as many oxygen binding sites as di-n-hexyl ether, which is striking compared with the fact that a five-fold dilution of tetraethylene glycol dimethyl ether is required to produce similar solvent properties to di-n-hexyl ether. This result implies that it is the concentration of oxygen binding sites rather than the concentration of the ether cosolvent that governs the behaviour of this system. In other words, the molecular constitution is of secondary significance, rather it is the functional group composition of the liquid mixture that is the key determinant of the solvation thermodynamics.
Fig. 3a shows the stability of the 1·2 complex plotted as a function of the molecular concentration of the ether cosolvent for all of the systems studied. The association constant decreases with ether concentration, but there is no clear cut trend, and the data become very scattered at high ether concentrations. In contrast, if we allow for the different numbers of oxygen binding sites present in the polyethers, the data converge to provide a much clearer picture of the behaviour of the system. Fig. 3b shows the stability of the 1·2 complex plotted as a function of the concentration of ether oxygen, [O], present in the solvent mixture. There is some scatter in the raw data with a number of outliers (open grey data points), which is a feature of the high-throughput approach, but the overall trend in Fig. 3b is unambiguous, and the behaviour is remarkably simple (filled black data points).
![(a) Relationship between the association constant for the 1·2 complex, log K, and the molecular concentration of ether cosolvent, [ether], for 1088 mixtures of alkanes and ethers (see ESI). (b) Relationship between the association constant for the 1·2 complex, log K, and the concentration of ether oxygen functional groups, [O], for 1088 mixtures of alkanes and ethers (see ESI). Open grey circles are the results of the individual experiments, filled black circles are the average values over a window of 0.25 units on the log K scale with error bars at the 95% confidence limit. Log K for the pure n-octane experiment is shown as a dotted black line, and the solid black line is the best fit for the data points for which log [O] > −1.7 ([O] > 20 mM). The filled grey circles (inset) show how the wavelength of the absorption maximum of 2, λmax, varies as a function of [O] (data from a titration of di-n-hexyl ether into an n-octane solution of 2).](/image/article/2010/SC/c0sc00209g/c0sc00209g-f3.gif) | ||
| Fig. 3 (a) Relationship between the association constant for the 1·2 complex, log K, and the molecular concentration of ether cosolvent, [ether], for 1088 mixtures of alkanes and ethers (see ESI†). (b) Relationship between the association constant for the 1·2 complex, log K, and the concentration of ether oxygen functional groups, [O], for 1088 mixtures of alkanes and ethers (see ESI†). Open grey circles are the results of the individual experiments, filled black circles are the average values over a window of 0.25 units on the log K scale with error bars at the 95% confidence limit. Log K for the pure n-octane experiment is shown as a dotted black line, and the solid black line is the best fit for the data points for which log [O] > −1.7 ([O] > 20 mM). The filled grey circles (inset) show how the wavelength of the absorption maximum of 2, λmax, varies as a function of [O] (data from a titration of di-n-hexyl ether into an n-octane solution of 2). | ||
At low [O], the stability of the complex is identical to that in pure alkanes (dotted line at log K ≈ 5 in Fig. 3b). Under these conditions, the concentration of ether oxygen is too low to compete with the bulk alkane solvent for interaction with 2, even though alkanes are very poor H-bond acceptors.29,30 Once [O] exceeds 20 mM, there is a uniform decrease in the stability of the 1·2 complex, as solvation of 2 by ether becomes more and more significant. In this region of the plot, the data follow a straight line with a slope of −1.2. In other words, the reaction order with respect to ether oxygen is approximately −1, and the association constant, K, is inversely proportional to the concentration of ether oxygen in the solvent mixture, [O].
This experiment provides a clear indication of the mechanism by which solvation effects modulate H-bonding interactions. There is a simple competition between solute–solvent interactions and H-bonding interactions between solutes. It appears that neither the bulk properties of the medium, such as dielectric constant, nor the details of the molecular structure of the solvent have an impact on the thermodynamic properties of H-bonding interactions. The solvent effects can be understood at the molecular level based on the polarity of the solvent functional groups, as quantified by the H-bond parameters, α and β,25,27 and the concentrations of the solvent functional groups. The system chosen for study here shows particularly simple behaviour, because alkanes and ethers both have no significant H-bond donor ability, and alkanes have no significant H-bond acceptor ability, so solvent effects are dominated by interactions with the ether oxygen H-bond acceptors.
Solvation of the H-bond donor 2 can be studied independently. Thus the association constant for the 1:1 complex formed between di-n-hexyl ether and 2 in n-octane was measured by a conventional UV-Vis titration, K = 28 ± 4 M−1. This means that in n-octane, 2 does not start to bind to ether solvents until the concentration of ether is around 4 mM, and 2 will be 50% solvated by ether when the ether concentration reaches 40 mM. This is consistent with the onset of ether solvation effects on the 1·2 association constant at a concentration of about 20 mM (intersection of the dotted and solid lines in Fig. 3b). The inset in Fig. 3b shows how the wavelength of the maximum in the UV-Vis absorption spectrum of 2 varies in the course of the titration of 2 with di-n-hexyl ether in n-octane. The fully bound value of 346 nm is reached at an ether concentration of 0.4 M and does not change as more ether is added. In contrast, increasing the concentration of ether from 0.4 to 4 M does affect the solvation thermodynamics, and the stability of the 1·2 complex falls by an order of magnitude over this range. Spectroscopic probes report on the solvation state of the solute, and so the difference between 90% and 99% solvated is hard to detect. In contrast, molecular recognition probes directly measure these differences in population and so provide a different kind of information.
Experimental
Association constants were determined using a BMG Labtech Fluorostar Optima plate reader with a Hellma 96-well quartz microplate. In a typical experiment, the microplate contained two titrations; the first in n-octane (S1) in wells 1–48 (titration 1), the second in an ether or polyether (S2) in wells 49–96 (titration 2). In addition to mixtures of pure solvents, dilute solutions (10 vol%, 5 vol% and 2 vol%) of the solvent of interest dissolved in n-octane were also used as S2. These solvent mixtures were prepared by volume. For example, a 2 vol% solution of di-n-hexyl ether in n-octane was prepared by transferring 2 mL of di-n-hexyl ether to a 100 mL volumetric flask, and the flask was then filled with n-octane. Two host stock solutions were prepared from accurately weighed samples of 2 (5 mg) dissolved in S1 and S2 in 25 mL volumetric flasks, to give a concentration of 1 mM in each. Five guest stock solutions were prepared by dissolving an accurately weighed sample of 1 (0.5 g) in S1 or S2 in a 5 mL volumetric flask to give a 0.46 M stock solution of 1 (stock solution 1). A serial dilution was carried out, whereby 570 μL of stock solution 1 was transferred to a 5 mL volumetric flask, which was then filled with S1 or S2 to give stock solution 2 (5.2 × 10−2 M). Each new stock solution was diluted in the same way to give a further three stock solutions, with concentrations of 6.0 × 10−3 M, 6.8 × 10−4 M and 7.7 × 10−5 M. Two sets of guest solutions were prepared, one in dissolved in S1, the other set dissolved in S2. These solutions were loaded onto the 96-well quartz microplate using four purpose written protocols with the UV-Vis plate reader. The first three protocols dealt with the pipetting of stock solutions of 1 and 2 onto the microplate. The first protocol pipetted the two most dilute solutions of 1 into wells 31–48, The next protocol pipetted the next two solutions of 1 into wells 11–30, then the third protocol pipetted the most concentrated solution of 1 into wells 1–10. Then 15 μL of a solution of 2 (1 mM) were pipetted into wells 1–48. This procedure was repeated for wells 49–96 (titration 2), using the stock solutions dissolved in S2. The final protocol was designed to top up each well with the relevant solvent to give a total volume of 150 μL in each well. There were repeats in some of the guest concentrations to highlight any possible errors in stock solution concentrations or pipetting. For example, wells 9 and 11 had the same concentration of 1. This was achieved by adding 15 μL of stock solution 1 to well 9 and 129 μL of stock solution 2 to well 11.The filling procedure was followed by addition of pure solvents to collect data for mixtures of S1 and S2. The absorbance of each well was measured at 6 wavelengths (260 nm, 280 nm, 340 nm, 390 nm, 420 nm and 600 nm) to obtain titration data in pure S1 and S2. After this 10 μL aliquots of pure S2 were added into each well of titration 1 (dissolved in S1), and 10 μL aliquots of pure S1 were added into each well of titration 2 (dissolved in S2). After addition the plate was agitated to ensure mixing, and then the instrument recorded the absorbance in each well. This procedure was repeated until all the wells were filled (320 μL), to give binding isotherms over the entire volume fraction range from pure S1 to pure S2. The output is an Excel spreadsheet, which was analysed using the Solver routine to optimise a binding constant for each solvent composition (K), the extinction coefficient of the free host (εfree), the extinction coefficient of the bound host (εbound) and the extinction coefficient that describes the second weak binding event (εG) to give a calculated absorbance (Acalc) that matches the experimental absorbance (Aexpt). Dilution of the solutes during the solvent titration is taken into account in the analysis, but Aexpt is not affected by this dilution, because there is a corresponding increase in the path length as the well is filled. The data are fit by minimising the sum of the residuals between Acalc and the experimental absorbance (Aexpt) for every solvent composition in every well of the plate (eqn (1) and (2)).
| Acalc = εG[G] + εfree[Hfree] + εbound[Hbound] | (1) |
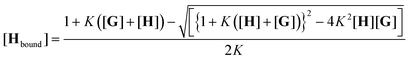 | (2) |
The script for operation of the plate reader and the Excel spreadsheet for analysis of the data are available from the authors on request.
Conclusions
This paper introduces a new high throughput approach to the study of solvation phenomena using molecular recognition probes. These systems provide quantitative thermodynamic information on the interactions of solvents with solutes, and this information is qualitatively different from that obtained using the conventional spectroscopic probes that have been used to derive standard scales of solvent polarity. Spectroscopy reports on the solvation state of the probe rather than the thermodynamic driving forces involved in, for example, selective solvation, which are strongly concentration dependent, as demonstrated here. The system studied in this edge article shows that solvation may be understood at the molecular level based on both the polarity and the concentrations of functional groups present in the solvent. The constitution of the solvent molecules is less important than the functional group composition, so that the solvent properties of a simple ether can be reproduced by an appropriate mixture of a polyether and an alkane (Fig. 4). The behaviour in ether–alkane mixtures is particularly simple, because the ether oxygen is the only functional group of note. However, the experimental approach that we have developed opens the way for the characterisation of more complex systems.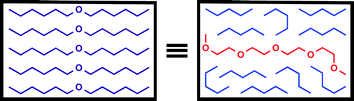 | ||
| Fig. 4 Solvent effects on H-bonding are determined by functional group composition rather than molecular constitution, so that the stability of the 1·2 complex is the same in a long chain ether as in an appropriate mixture of a polyether and an alkane. | ||
Acknowledgements
We thank the EPSRC, BBSRC and James Black Foundation for funding.Notes and references
- C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, 3rd edn, Wiley-VCH Verlag GmbH, 2004 Search PubMed.
- Y. Marcus, The Properties of Solvents, John Wiley & Sons, Chichester, 1998 Search PubMed.
- A. Zaks and A. M. Klibanov, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 3192–3196 CAS.
- M. J. Kamlet and R. W. Taft, J. Am. Chem. Soc., 1976, 98, 377–383 CrossRef CAS.
- A. R. Katritzky, K. Jug and D. C. Oniciu, Chem. Rev., 2001, 101, 1421–1449 CrossRef CAS.
- Y. Marcus, Chem. Soc. Rev., 1993, 22, 409–416 RSC.
- M. H. Abraham, R. Kumarsingh, J. E. Cometto-Muniz, W. S. Cain, M. Roses, E. Bosch and M. L. Diaz, J. Chem. Soc., Perkin Trans. 2, 1998, 2405–2411 RSC.
- M. H. Abraham, C. E. Green, W. E. Acree, C. E. Hernandez and L. E. Roy, J. Chem. Soc., Perkin Trans. 2, 1998, 2677–2681 RSC.
- M. Plass, K. Valko and M. H. Abraham, J. Chromatogr., A, 1998, 803, 51–60 CrossRef CAS.
- Z. Maksimovic, C. Reichardt and A. Spiric, Fresenius' Z. Anal. Chem., 1974, 270, 100–104 CAS.
- E. Gebicka and W. J. Lenoble, Tetrahedron Lett., 1983, 24, 2427–2428 CrossRef CAS.
- A. Bagno, F. Rastrelli and G. Saielli, Prog. Nucl. Magn. Reson. Spectrosc., 2005, 47, 41–93 CrossRef CAS.
- Y. Umebayashi, K. Matsumoto, M. Watanabe and S. Ishiguro, Phys. Chem. Chem. Phys., 2001, 3, 5475–5481 RSC.
- T. M. Krygowski, P. K. Wrona, U. Zielkowska and C. Reichardt, Tetrahedron, 1985, 41, 4519–4527 CrossRef CAS.
- Y. Marcus, J. Chem. Soc., Perkin Trans. 2, 1994, 1015–1021 RSC.
- M. H. Abraham, P. L. Grellier and R. A. McGill, J. Chem. Soc., Perkin Trans. 2, 1988, 339–345 RSC.
- S. B. Ferguson, E. M. Sanford, E. M. Seward and F. Diederich, J. Am. Chem. Soc., 1991, 113, 5410–5419 CrossRef CAS.
- E. Klein, Y. Ferrand, N. P. Barwell and A. P. Davis, Angew. Chem., Int. Ed., 2008, 47, 2693–2696 CrossRef CAS.
- H. J. Schneider, R. Kramer, S. Simova and U. Schneider, J. Am. Chem. Soc., 1988, 110, 6442–6448 CrossRef CAS.
- J. C. Adrian and C. S. Wilcox, J. Am. Chem. Soc., 1991, 113, 678–680 CrossRef.
- B. Siegel and R. Breslow, J. Am. Chem. Soc., 1975, 97, 6869–6870 CrossRef CAS.
- R. P. Bonar-Law and J. K. M. Sanders, J. Am. Chem. Soc., 1995, 117, 259–271 CrossRef CAS.
- T. Mizutani, K. Wada and S. Kitagawa, J. Org. Chem., 2000, 65, 6097–6106 CrossRef CAS.
- G. A. Breault, C. A. Hunter and P. C. Mayers, J. Am. Chem. Soc., 1998, 120, 3402–3410 CrossRef CAS.
- J. L. Cook, C. A. Hunter, C. M. R. Low, A. Perez-Velasco and J. G. Vinter, Angew. Chem., Int. Ed., 2007, 46, 3706–3709 CrossRef CAS.
- J. L. Cook, C. A. Hunter, C. M. R. Low, A. Perez-Velasco and J. G. Vinter, Angew. Chem., Int. Ed., 2008, 47, 6275–6277 CrossRef CAS.
- C. A. Hunter, Angew. Chem., Int. Ed., 2004, 43, 5310–5324 CrossRef CAS.
- C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS.
- R. Cabot, C. A. Hunter and L. M. Varley, Org. Biomol. Chem., 2010, 8, 1455–1462 RSC.
- R. Cabot and C. A. Hunter, Org. Biomol. Chem., 2010, 8, 1943–1950 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1: Solvent mixtures studied by automated titration of 1 into 2. See DOI: 10.1039/c0sc00209g |
| This journal is © The Royal Society of Chemistry 2010 |