Different electronic structure of phosphonyl radical adducts of N-heterocyclic carbenes, silylenes and germylenes: EPR spectroscopic study and DFT calculations†‡
Dennis
Sheberla
a,
Boris
Tumanskii
*a,
Adam C.
Tomasik
b,
Amitabha
Mitra
b,
Nicholas J.
Hill
b,
Robert
West
*b and
Yitzhak
Apeloig
a
aSchulich Faculty of Chemistry and the Lise Meitner-Minerva Center for Computational Quantum Chemistry, Technion-Israel Institute of Technology, Haifa, 32000, Israel. E-mail: tboris@tx.technion.ac.il
bDepartment of Chemistry, University of Wisconsin, Madison, Wisconsin 53706, USA. E-mail: west@chem.wisc.edu
First published on 26th May 2010
Abstract
Stable N-heterocyclic carbenes and germylenes were allowed to react with a phosphonyl radical, (i-PrO)2(O)P˙ (7), generated by photolysis of [(i-PrO)2(O)P]2Hg. The products were identified by EPR spectroscopy. An unsaturated carbene (1) and germylene (3) react with 7 at the divalent atom to give unstable radical products (τ½ = 0.2 s). A benzo-annulated carbene (4) and a saturated germylene (6) react with 7 to give more active radicals. An unsaturated (2) and a saturated silylene (5) undergo rapid reaction (in the dark) with [(i-PrO)2(O)P]2Hg to yield unusual silyl phosphites. In these cases only secondary radicals were observed. DFT (PBE0/TZVP//B3LYP/6-31+G(d)) calculations of the radical adducts of the different (C, Si, Ge) unsaturated N-heterocyclic divalent species with the phosphonyl radical show that the unpaired electron is delocalized over the five-membered ring; the spin density on the central atoms decreases in the order C, 39% > Si, 14% > Ge, 2%. These trends can be understood in terms of a zwitterionic structure of the radical adducts. The calculations of the radical adducts of 4, 5 and 6 with 7 indicate larger spin density on the central atom, 47%, 58% and 42% on C, Si, Ge, respectively.
Introduction
Divalent compounds of group 14 elements are among the most important reactive intermediates.1 For a long time species involving divalent carbon and its heavier congeners could only be directly observed by spectroscopic techniques, either in the gas phase2 or in low-temperature matrices,3 but could not be isolated in macroscopic amounts at room temperature. Considerable progress in the chemistry of these compounds was achieved with the synthesis and isolation of stable N-heterocyclic carbenes4a–4c and later of its heavier analogs.4d–4i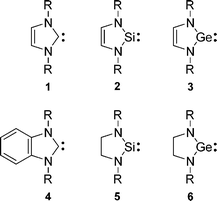
These discoveries have stimulated research on the electronic structure of these novel divalent species and on the reasons for their unusual stability.5
Carbene 1 and its heavier group 14 congeners have lone pair electrons as well as a low-lying vacant pπ orbital, which implies that they can act both as electron donors, e.g.: in carbene–, silylene–, germylene–transition metal complexes,6 and as electron acceptors, e.g.: in reduction by alkali metals.7 These species therefore may possess high reactivity towards free radicals with different donor–acceptor properties. Interaction of the divalent species with radicals to form a new bond and change their valency from two to three can involve, in the first step, a single electron transfer. An analogous case of interaction of free radicals with compounds containing lone pair electrons may be found in the radical chemistry of phosphites or phosphines, where radical addition to trivalent organophosphorus species leads to formation of four-valent, phosphorus-centered intermediates (eqn (1)).8
| X3P: + Y˙ → X3P˙–Y | (1) |
Although many reactions of stable N-heterocyclic divalent compounds have now been described,9 few reports on the chemistry of radicals of these compounds are available.10 Several radical adducts of unsaturated 1–3 with free radicals of different chemical nature, such as O-, P-, Re-, Co-, Mo-centered radicals (eqn (2)), have been studied by EPR and DFT calculations and were reported recently by us10a–c and by other groups.10d–k These adducts represent a new type of neutral radicals stabilized by various degrees of delocalization, which depend on the central divalent atom.10a–c The first isolations of diamagnetic products of free radical addition to silylenes and germylenes have been reported recently.10h,i
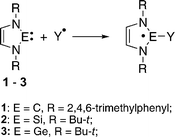 | (2) |
We present here a first systematic study of phosphonyl radical (i-PrO)2(O)P˙ (7) adducts of N-heterocyclic stable carbenes, silylenes and germylenes by EPR spectroscopy and DFT calculations. We consider the following: (i) spectral features of the radical adducts, (ii) the effect of the divalent atom E (E![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, Si, Ge) on the spin density distribution in radical adducts, (iii) the effect of a saturated or unsaturated N-heterocycle on the electronic and molecular structure of the generated adducts. Additionally, we report a new type of reaction of silylenes 2 and 5 with phosphonyl mercury which yields silyl phosphites.
C, Si, Ge) on the spin density distribution in radical adducts, (iii) the effect of a saturated or unsaturated N-heterocycle on the electronic and molecular structure of the generated adducts. Additionally, we report a new type of reaction of silylenes 2 and 5 with phosphonyl mercury which yields silyl phosphites.
Results and discussion
In the following sections we discuss the structures and EPR spectra of radical adducts of 1–6 with the phosphonyl radical 7. Selected calculated geometrical parameters are given in Table 1 and the hfc constants and spin density distribution are provided in Table 2.| Radical | E | Σθ(E) | E–P | E–Na | N–Cb | C–C′ |
|---|---|---|---|---|---|---|
| a Average of E–N and E–N′. b Average of N–C and N′–C′. | ||||||
| 1a | C | 352.3 | 1.781 | 1.421 | 1.395 | 1.349 |
| 4a | C | 340.9 | 1.836 | 1.423 | 1.399 | 1.416 |
| 2a | Si | 284.2 | 2.400 | 1.841 | 1.365 | 1.374 |
| 5a | Si | 313.0 | 2.339 | 1.754 | 1.472 | 1.533 |
| 3a | Ge | 270.0 | 2.493 | 1.961 | 1.348 | 1.390 |
| 6a | Ge | 303.6 | 2.463 | 1.856 | 1.463 | 1.529 |
| a(E) exp | a(E) calc | SD(E) | a(P) exp | a(P) calc | SD(P) | a(2N) expa | a(2N) calca | SD(2N) b | |
|---|---|---|---|---|---|---|---|---|---|
| a Average value on two atoms. b Sum of values on two atoms. c No experimental data. d Calculated data for model 4a′. e Not observed due to low signal-to-noise ratio. | |||||||||
| 1a | 25.6 | 21.9 | 39.3% | 48.7 | 34.9 | 7.5% | 4.7 | 3.1 | 34.3% |
| 2a | —c | −75.0 | 14.4% | — | 243.5 | 10.5% | — | 4.4 | 45.8% |
| 3a | 15.0 | −13.9 | 2.4% | 186.0 | 182.9 | 8.1% | 6.4 | 5.3 | 54.2% |
| 4a | n/oe | 46.8 | 46.7% | 81.0 | 122.2 | 7.1% | 3.6 | 2.5 | 27.3% |
| 5a | — | −192.3 | 57.8% | — | 264.0 | 11.6% | — | 3.7 | 16.8% |
| 6a | n/o | −78.3 | 42.0% | 199.6 | 302.1 | 14.5% | 7.6 | 4.6 | 23.7% |
Radical 7 is pyramidal and the unpaired electron occupies an orbital with nearly sp3 hybridization.11a (the EPR spectrum and a DFT calculated structure of 7 are presented in the ESI, Fig. S1†). The radical addition product of phosphonyl radicals in which the phosphorus atom is β to the radical center yield large aP values (40–140 G), which are easy to detect. Thus, the EPR spectra can provide important information about the structures of the radical adducts.10c,11b–d
a) Carbene adducts
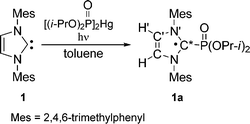 | (3) |
The following hyperfine coupling (hfc) constants were measured for radical 1a (g = 2.0027, τ½ = 7.1 s): a(31P) = 48.7 G, a(214N) = 4.7 G, a(21H′) = 1.1 G, a(13C*) = 25.6 G (Table 2). According to DFT calculations, the central carbon atom in 1a is slightly pyramidal, Σθ(C*) = 352° and the heterocycle is almost planar. The calculated Löwdin atomic spin density of 1a shows that 39% of the spin density is localized on the central carbon atom (C*) and 37% on the other ring atoms.
 | (4) |
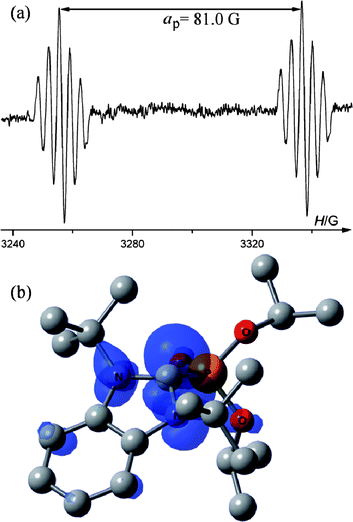 | ||
| Fig. 1 (a) EPR spectrum of 4a recorded under UV-irradiation at 298 K, (b) DFT calculated spin density of 4a′ at 0.003 au contour level. Calculated g-factor = 2.0032. | ||
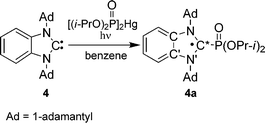
To investigate the structure and the spin density distribution in radical 4a DFT quantum mechanical calculations were carried out on a model radical (4a′), in which 1-adamantyl groups were replaced by tert-butyl groups. The heterocycle of 4a′ is nearly planar (∠C*NCC′ = −5.6° and ∠C*N′C′C = 6.7°), with a more pyramidal central carbon atom (Σθ(C*) = 341°) than that in 1a (Table 1). Calculations show that 47% of the spin density is localized on the central carbon atom, and 36% of the spin density is distributed on the other atoms of the heterocycle and annulated aromatic ring (Table 2). The difference between the calculated (aP = 122 G) and experimental (aP = 81.0 G) 31P hfc constant can be attributed to conformational and vibrational averaging and general method error.
b) Germylene adducts
 | (5) |
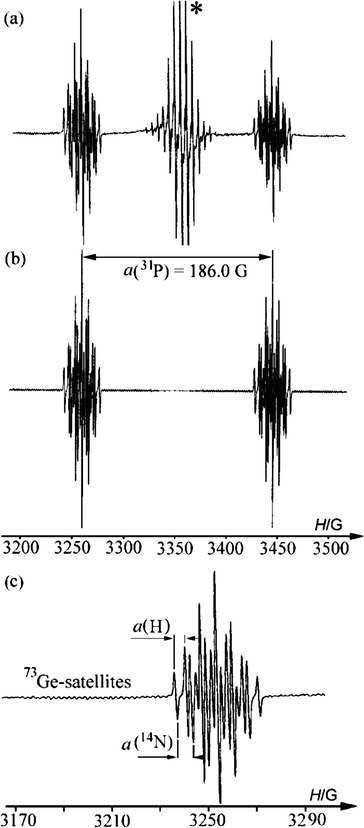 | ||
| Fig. 2 (a) EPR spectrum of a toluene solution of 3a recorded under UV-irradiation at 298 K (asterisk marks lines belonging to side products12), (b) simulated spectrum of 3a, (c) expanded low field multiplet. | ||
When the irradiating light is turned off a fast (and reversible) decrease in the intensity of the EPR signal of 3a is observed (τ½ = 0.2 s) (Fig. S3†). The rate of decay of 3a is not markedly dependent on temperature (230–290 K) and we believe that radical 3a decays via dimerization (dimer structure unknown). We estimate the dimerization rate constant as 2kdim = 5 × 103 M−1 s−1.
According to DFT calculations, the heterocycle of 3a is nearly planar (∠GeNCC′ = −2.4° and ∠GeN′C′C = 1.4°), with a significant pyramidality at the germanium atom, Σθ(Ge) = 270.0°. The calculated g-factor (2.0027), hfc constants for 79Ge, 14N, 1H, 31P and the calculated spin density (Fig. 3, Table 2) show good agreement with the experimental EPR data of 3a, indicating that only 2% of the spin density is localized on the germanium atom, and 81% of the spin density is distributed over the other atoms of the heterocycle. The calculations indicate that 12% of the spin density is localized on the phosphonyl group, accounting for the high hyperfine splitting by the 31P nucleus. Thus, the spin density is transferred from germanium into the five-member ring and into the phosphonyl group, leading to an unusual zwitterionic structure (eqn (5)). An analogous case of a nearly complete transfer of spin density away from a heteroatom occurs in the phosphorus-centered radical ˙PPh(OEt)2(OBu-t), where the spin density is delocalized onto the aromatic ring.13
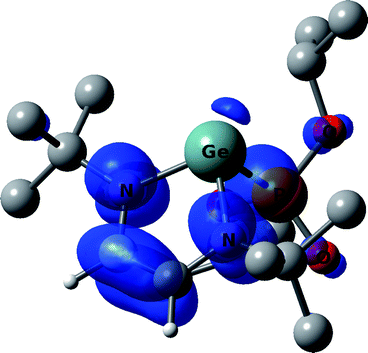 | ||
| Fig. 3 DFT calculated spin density of 3a at 0.003 au contour level. Calculated g-factor, absolute values of hfc constants and Löwdin atomic spin densities (in parentheses) at the particular nucleus of radical 3a: g = 2.0027, a(13C) = 1.0 G (14.8%), a(13C′) = 2.2 G (11.9%), a(1H) = 4.1 G (−1.0%), a(1H′) = 3.5 G (−1.0%). | ||
The hyperfine coupling of germanium in 3a, a(73Ge) = 15.0, is much smaller than that in the stable pyramidal radical [(Me3Si)2N]3Ge˙ (171.0 G),14 and in fact even smaller than reported for the planar radical (t-Bu2MeSi)3Ge˙ (20.0 G).15 The relatively low 73Ge hyperfine coupling constant in radical 3a is consistent with delocalization of the unpaired electron away from the Ge atom. Similar to 3a, a low 73Ge hyperfine coupling constant (9.6 G) was observed for the isoelectronic Cl-substituted germanium radical 3b, which was isolated as deep red crystals in the reaction of the anion radical of 1,2-bis[(2,6-diisopropylphenyl)imino]acenaphthene and GeCl2.10j We believe that in this case, full stabilization of the radical was achieved by additional delocalization of the unpaired electron over the annulated acenaphthene fragment of the molecule (a DFT calculated structure of 3b is presented in the ESI, Fig. S4†).
 | (6) |
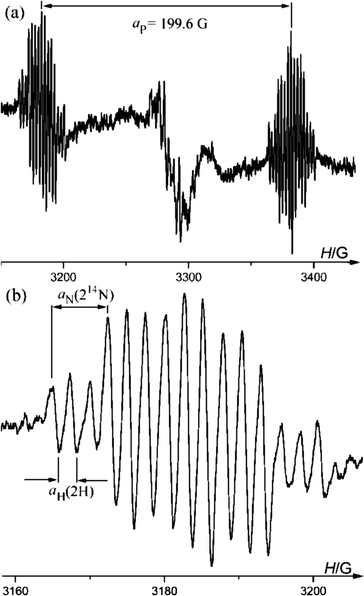 | ||
| Fig. 4 (a) EPR spectrum of 6a recorded under UV-irradiation at 298 K, (b) expanded low field multiplet. | ||
After the irradiating light is turned off the signal of 6a immediately disappears. The signal intensity of 6a is lower by an order of magnitude than the signal of 3a at a similar reactant concentration and condition of UV-irradiation. This fact indicates that the rate of 6a dimerization is markedly faster than for 3a.
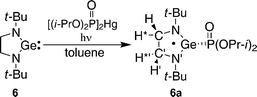
DFT calculations of hfc constants in radical 6a predict a somewhat larger aP = 302.1 G. The calculated spin density distribution shows that in radical 6a 42% of the spin density is localized on the central germanium atom, and 24% of the spin density is distributed on the 14N atoms (Fig. 5), in contrast to the spin density distribution in 3a in which only 2% of the spin density resides on the Ge. Such a spin distribution difference between 3a and 6a is reasonable, because 6a has no conjugated π-system which can delocalize the spin.
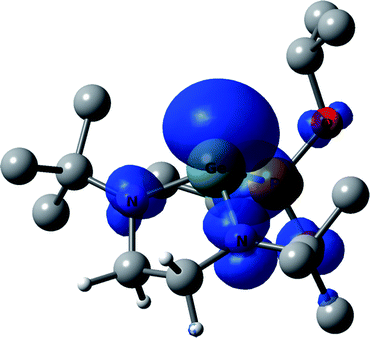 | ||
| Fig. 5 DFT calculated spin density of 6a at 0.003 au contour level. Calculated g-factor, absolute values of hfc constants and Löwdin atomic spin densities (in parentheses) at the particular nucleus of radical 6a: g = 2.0031, a(13C) = 0.5 G (0.7%), a(13C′) = 0.4 G (−0.2%), a(1H) = 0.8 G (−0.1%), a(1H*) = 2.3 G (0.2%), a(1H′) = 5.6 G (0.7%), a(1H*′) = 7.5 G (0.9%). | ||
c) Silylene adducts
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio begins immediately even at 200 K and gives an unexpected red-yellow Hg-containing product (2b), which was characterized by 1H, 13C, 31P and 29Si NMR spectroscopy (Scheme 1). 2b is not stable at r.t. and completely disappears after 12 h (in the dark) forming a new compound 2c, also observed by NMR spectroscopy. Additionally, traces of (i-PrO)2(O)PH have been observed by NMR spectroscopy, indicating that radical processes are involved. Both products 2b and 2c are very air and moisture sensitive. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) of the reaction mixture shows signals corresponding to 2c and of the Hg-elimination product of 2b (Fig. S5†).
:2 ratio begins immediately even at 200 K and gives an unexpected red-yellow Hg-containing product (2b), which was characterized by 1H, 13C, 31P and 29Si NMR spectroscopy (Scheme 1). 2b is not stable at r.t. and completely disappears after 12 h (in the dark) forming a new compound 2c, also observed by NMR spectroscopy. Additionally, traces of (i-PrO)2(O)PH have been observed by NMR spectroscopy, indicating that radical processes are involved. Both products 2b and 2c are very air and moisture sensitive. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) of the reaction mixture shows signals corresponding to 2c and of the Hg-elimination product of 2b (Fig. S5†).
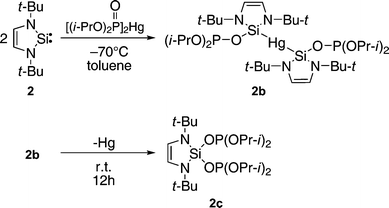 | ||
| Scheme 1 | ||
Reactions of silylene with dialkylphosphite and its derivatives are new and interesting, however evidently, we cannot use [(i-PrO)2(O)P]2Hg for formation of Si–P bonds in radical addition reactions.
The EPR monitoring of a toluene solution containing [(i-PrO)2(O)P]2Hg and 2 in the dark and under UV irradiation reveals only minor secondary products of various radical reactions (for details see ESI, Fig. S6–S7†).
Although we could not observe the primary radical adducts (2a) of the phosphonyl radical with 2, we carried out computations for radical 2a in order to compare it with the analogous adducts of the carbenes and germylenes discussed above. DFT calculations of the structure (Scheme 1) and hfc constants in 2a predict a(31P) = 243.5 G. Fig. 6 shows the calculated structure of 2a and its spin density. The heterocycle of 2a is planar (∠SiNCC′ = −0.5° and ∠SiN′C′C = 0.5°), with a pyramidal central silicon, Σθ(Si*) = 284.2°. The calculated spin densities show that in primary radical 2a 14% of the spin density is localized on the central silicon atom and 65% of the spin density is distributed on the other atoms of the heterocycle. The spin delocalization pattern is similar to that in radical 3a (E = Ge), but with slightly higher spin density on the central silicon atom.
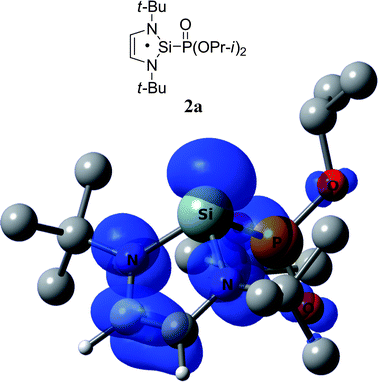 | ||
| Fig. 6 DFT calculated spin density of 2a at 0.003 au contour level. Calculated g-factor, absolute values of hfc constants and Löwdin atomic spin densities (in parentheses) at the particular nucleus of radical 2a: g = 2.0032, a(13C) = 0.9 G (13.4%), a(13C′) = 1.6 G (9.5%), a(1H) = 3.0 G (−0.86%), a(1H′) = 2.4 G (−0.61%). | ||
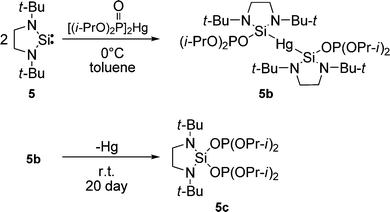 | ||
| Scheme 2 | ||
As we did for 2a, we studied computationally the radical adduct (5a) of phosphonyl radical 7 to silylene 5. The DFT calculations predict a(31P) = 264.0 G. The calculated spin density distribution shows that in radical 5a 58% of the spin density is localized on the central silicon atom, and 19% of the spin density is distributed over the other atoms of the heterocycle (Fig. 7).
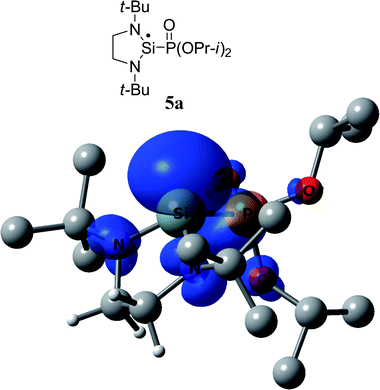 | ||
| Fig. 7 DFT calculated spin density of 5a at 0.003 au contour level. Calculated g-factor, absolute values of hfc constants and Löwdin atomic spin densities (in parentheses) at the particular nucleus of radical 5a: g = 2.0032, a(13C) = 2.3 G (0.9%), a(13C′) = 1.1 G (0.1%), a(1H) = 0.7 G (0.06%), a(1H*) = 2.9 G (0.29%), a(1H′) = 1.7 G (0.24%), a(1H*′) = 5.0 G (1.1%). | ||
Conclusions
Radical reactions of phosphonyl radicals with stable N-heterocyclic carbenes, silylenes and germylenes were studied by EPR spectroscopy and by DFT calculations. The calculations show that for unsaturated radicals 1a, 2a and 3a, the degree of pyramidality at the radical center increases in the order: C (Σθ = 352°) ≪ Si (Σθ = 284°) < Ge (Σθ = 270°), which is accompanied by a decrease in the spin density on the central atom (C (39.3%) ≫ Si (14.4%) > Ge (2%)) and an increased spin delocalization over the 5-membered ring (C ≪ Si < Ge, highest delocalization). These trends can be understood in terms of a larger contribution of zwitterionic resonance structure B (eqn (7)) along the series Ge > Si ≫ C. This trend is consistent with the strength of np-SOMO(E) → π*(N–CC–N) conjugation which also follows the order Ge > Si > C, as the SOMO(E) energy in e.g., H3E˙ increases along the series C (−10.5 eV) ≪ Si (−9.1 eV) < Ge (−8.9 eV) (UHF/6-311+G//UB3LYP/6-311+G(d)). In all these trends large differences are observed between C and Si while the differences between Si and Ge are small.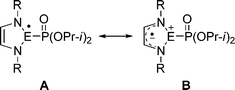 | (7) |
In saturated radicals 5a and 6a, lacking the possibility for cyclic electron delocalization, the calculated spin density at the central atom is much higher (Si, 58%; Ge, 42%) than in the corresponding unsaturated radicals 2a and 3a, as expected.
Silylenes, in contrast to carbenes and germylenes react spontaneously with [(i-PrO)2(O)P]2Hg to yield unusual mercury-containing silyl phosphites. This new reaction opens interesting synthetic perspectives (e.g. synthesis of metallosilyl phosphite compounds).
The geometry, spin density distribution and reactivity of radical adducts of group 14 imidazol-2-ylidene and imidazolin-2-ylidene demonstrate a strong dependence on the nature of the heterocycle and on the divalent central atom. Such dependence is unprecedented in radical chemistry. We are continuing to explore this interesting new class of radical reactions.
Experimental section
Standard Schlenk techniques in a vacuum (10−2 torr) were used for the preparation and manipulations of all samples.Typical EPR experiment
The phosphonyl radical 7 was generated photolytically in situ from [(i-PrO)2(O)P]2Hg. UV-irradiation of solutions containing the N-heterocyclic metallylenes and [(i-PrO)2(O)P]2Hg in a Pyrex tube within the cavity of the EPR spectrometer gave rise to the radical adducts of metallylenes whose spectra were recorded. The typical concentration of the metallylenes was 10−2 M and of Hg[P(O)(OPr-i)2]2 was 3 × 10−3 M.Materials
The synthesis of 2,4d3,4h4,4b5,4f64h and [(i-PrO)2(O)P]2Hg17 is described elsewhere. Toluene and benzene were degassed, kept on t-BuLi in a vacuum and distilled prior to use.EPR spectroscopy
EPR spectra were recorded on a Bruker EMX-10/12 X-band (ν = 9.4 GHz) digital EPR spectrometer equipped with a Bruker N2-temperature controller. Samples were irradiated with the focused and filtered (λ > 300 nm) or unfiltered (λ > 250 nm) light of a high-pressure mercury lamp (1 kW) (ARC lamp power supply model 69920) in the resonator of the EPR spectrometer.EPR spectra were recorded at microwave power 0.5–1.0 mW, 100 kHz magnetic field modulation of 0.05–1.00 G amplitude. Digital field resolution was 2048 points per spectrum, allowing all hyperfine splitting to be measured directly with an accuracy of better than 0.1 G. Spectra processing and simulation were performed with Bruker WIN-EPR and SimFonia Software. The g-factors values were determined using 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO) as reference (g = 2.0058).
NMR spectroscopy
NMR spectra were recorded at room temperature in benzene solutions in J. Young vacuum NMR tubes equipped with DMSO-d6 capillary as external standard, using Bruker Avance 300 and Bruker Avance 500 instruments. NMR shifts are given in ppm.2b: 1H NMR (benzene, DMSO-d6 capillary): δ 5.95 (4H, s), 4.91 (4H, m, JH–H = 5.9 Hz), 1.41 (36H, s), 1.30 (24H, m, JH–H = 5.9 Hz); 29Si NMR: δ 65.7 (d, JSi–P = 12.7 Hz); 31P NMR: δ 129.3; 13C NMR: 113.0, 65.4 (d, JC–P = 10.9 Hz), 51.6, 32.0, 24.8.
2c: 1H NMR (benzene, DMSO-d6 capillary): δ 5.71 (2H, s), 4.52 (4H, m, JH–P = 6.3 Hz, JH–H = 6.2 Hz), 1.41 (18H, s), 1.19 (24H, dd, JH–P = 2.0 Hz, JH–H = 6.2 Hz); 29Si NMR: δ −71.2 (t, JSi–P = 1.3 Hz); 31P NMR: δ 126.2; 13C NMR: δ 110.4, 65.5 (d, JC–P = 11.1 Hz), 51.3, 30.6, 24.5.
5b: 1H NMR (benzene, DMSO-d6 capillary): δ 4.94 (4H, m, JH–H = 6.0 Hz), 2.94 (8H, s), 1.39 (36H, s), 1.33 (24H, d, JH–H = 6.0 Hz); 29Si NMR: δ 67.9 (d, JSi–P 16.4 Hz); 31P NMR: δ 129.7; 13C NMR: 65.0 (d, JC–P = 10.9 Hz), 51.2, 44.1, 30.8, 24.8.
5c: 1H NMR (benzene, DMSO-d6 capillary): δ 4.55 (4H, m, JH–P = 6.0 Hz, JH–H = 6.3 Hz), 2.90 (4H, s), 1.66 (18H, s), 1.22 (24H, dd, JH–P = 5.2 Hz, JH–H = 6.3 Hz); 29Si NMR: δ −69.1 (t, JSi–P = 3.2 Hz); 31P NMR: δ 126.0; 13C NMR: δ 65.2 (d, JC–P = 10.8 Hz), 50.9, 41.5, 29.3, 25.0.
Computational methods
Geometry optimizations were performed at the DFT B3LYP/6-31+G(d) level. This level was demonstrated to be reliable in predicting ground state geometries of radicals.18 All structures were confirmed as local minima by calculating second order derivatives. Orbital energies were calculated for optimized geometries using the UHF method because KS energies tend to overestimate the experimental values. However, trends are consistent within the methods.19 These calculations were carried out using the Gaussian0320 series of programs.Isotropic hyperfine coupling constants and g-factors were calculated using the PBE0 functional21 in conjunction with an uncontracted TZVP22 basis set using the ORCA 2.6.35 software package developed by F. Neese.23 The PBE0/TZVP level produces more accurate predictions of hfc constants and g-factors.23b,24
Acknowledgements
We are grateful to Dr Miriam Karni for helpful discussions. This research was supported by the Israel Science Foundation, by the USA-Israel Binational Science Foundation and by the Minerva Foundation in Munich. B.T. is grateful to the Center for Absorption in Science, Israel, Ministry of Immigrant Absorption, State of Israel for support. At the University of Wisconsin this research was sponsored by the National Science Foundation. R. West thanks the WCU program through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology (R33-10082) for support.Notes and references
- (a) M. B. Smith and J. March, Advanced Organic Chemistry, Wiley-Interscience, New York, 5th edn, 2001, ch. 5 Search PubMed; (b) C. Wentrup, Reactive Molecules: The Neutral Reactive Intermediates in Organic Chemistry, Wiley-Interscience, New York, 1984, ch. 4 Search PubMed.
- For example: (a) D. G. Leopold, K. K. Murray and W. C. Lineberger, J. Chem. Phys., 1984, 81, 1048 CrossRef CAS; (b) R. Srinivas, D. K. Bohme and H. Schwarz, J. Phys. Chem., 1993, 97, 13643 CrossRef CAS; (c) G. A. McGibbon, C. A. Kingsmill and J. K. Terlouw, Chem. Phys. Lett., 1994, 222, 129 CrossRef CAS; (d) P. C. Burgers, G. A. McGibbon and J. K. Terlouw, Chem. Phys. Lett., 1994, 224, 539 CrossRef CAS; (e) G. A. McGibbon, P. C. Burgers and J. K. Terlouw, Int. J. Mass Spectrom. Ion Processes, 1994, 136, 191 CrossRef CAS.
- For example: (a) T. Drahnak, J. Michl and R. West, J. Am. Chem. Soc., 1979, 101, 5427 CrossRef CAS; (b) G. Maier, J. Glatthaar and H. P. Reisenauer, Chem. Ber., 1989, 122, 2403 CrossRef CAS; (c) G. R. Gillette, G. Noren and R. West, Organometallics, 1990, 9, 2925 CrossRef CAS; (d) M. Veith, E. Werle, R. Lisowski, R. Löppe and H. Schnöckel, Chem. Ber., 1992, 125, 1375 CrossRef CAS.
- (a) A. J. Arduengo III, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361 CrossRef; (b) N. Korotkikh, G. Raenko, T. Pekhtereva, O. Shvaika, A. Cowley and J. Jones, Russ. J. Org. Chem., 2006, 42, 1822 CrossRef CAS; (c) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122 CrossRef CAS; (d) M. Denk, R. Lennon, R. Hayashi, R. West, A. V. Belyakov, H. P. Verne, A. Haaland, M. Wagner and N. Metzler, J. Am. Chem. Soc., 1994, 116, 2691 CrossRef CAS; (e) M. Denk, J. C. Green, N. Metzler and M. Wagner, J. Chem. Soc., Dalton Trans., 1994, 2405 RSC; (f) R. West and M. Denk, Pure Appl. Chem., 1996, 68, 785 CrossRef CAS; (g) M. Denk, R. West, Hayashi, Y. Apelog, R. Paunz and M. Karni, in Organosilicon Chemistry II. From Molecules to Materials, ed. N. Auner and J. Weis, VCH, Weinheim, 1996, p. 251 Search PubMed; (h) W. A. Herrmann, M. Denk, J. Behm, W. Scherer, F.-R. Klingan, H. Bock, B. Solouki and M. Wagner, Angew. Chem., Int. Ed. Engl., 1992, 31, 1485 CrossRef; (i) T. Gans-Eichler, D. Gudat and M. Nieger, Angew. Chem., 2002, 114, 1966 CrossRef.
- (a) A. J. Arduengo III, H. V. Rasika Dias, D. A. Dixon, R. L. Harlow, W. T. Kloostera and T. F. Koetzle, J. Am. Chem. Soc., 1994, 116, 6812 CrossRef; (b) A. J. Arduengo III, D. A. Dixon, R. L. Harlow, K. K. Kumashiro, C. Lee, W. P. Power and K. W. Zilm, J. Am. Chem. Soc., 1994, 116, 6361 CrossRef CAS; (c) A. J. Arduengo III, H. Bock, H. Chen, M. Denk, D. A. Dixon, J. C. Green, W. A. Herrmann, N. L. Jones, M. Wagner and R. West, J. Am. Chem. Soc., 1994, 116, 6641 CrossRef; (d) D. A. Dixon and A. J. Arduengo III, J. Phys. Chem., 1991, 95, 4180 CrossRef CAS; (e) J. Cioslowski, Int. J. Quantum Chem., 1993, 48, 309 CrossRef; (f) C. Heinemann and W. Thiel, Chem. Phys. Lett., 1994, 217, 11 CrossRef CAS; (g) Y. Apeloig, M. Karni and T. Müller, in Organosilicon Chemistry II. From Molecules to Materials, ed. N. Auner and J. Weis, VCH, Weinheim, 1996, p. 263 Search PubMed; (h) L. Nyulászi, T. Krápáti and T. Veszprèmi, J. Am. Chem. Soc., 1994, 116, 7239 CrossRef CAS; (i) C. Heinemann, W. A. Herrmann and W. Thiel, J. Organomet. Chem., 1994, 475, 73 CrossRef CAS; (j) C. Heinemann, T. Müller, Y. Apeloig and H. Schwarz, J. Am. Chem. Soc., 1996, 118, 2023 CrossRef CAS; (k) C. Boehme and G. Frenking, J. Am. Chem. Soc., 1996, 118, 2039 CrossRef.
- (a) W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290 CrossRef CAS; (b) E. Peris and R. H. Crabtree, Coord. Chem. Rev., 2004, 248, 2239 CAS; (c) N-Heterocyclic Carbenes in Synthesis, ed. S. P. Nolan, Wiley-VCH, Weinheim, 2006 Search PubMed; (d) A. G. Avent, B. Gehrhus, P.B. Hitchcock, M. F. Lappert and H. Maciejewski, J. Organomet. Chem., 2003, 686, 321 CrossRef CAS; (e) M. Denk, R. K. Haashi and R. West, J. Chem. Soc., Chem. Commun., 1994, 33 RSC; (f) D. S. McGuinness, B. F. Yates and K. J. Cavell, Organometallics, 2002, 21, 5408 CrossRef CAS; (g) T. A. Schmedake, M. Haaf, B. J. Paradise, A. J. Millevolte, D. R. Powell and R. West, J. Organomet. Chem., 2001, 636, 17 CrossRef CAS; (h) C. Boehme and G. Frenking, Organometallics, 1998, 17, 5801 CrossRef CAS.
- (a) P. L. Arnold and S. T. Liddle, Chem. Commun., 2006, 3959 RSC; (b) S. Ishida, T. Iwamoto and M. Kira, J. Am. Chem. Soc., 2003, 125, 3212 CrossRef CAS; (c) M. Haaf, T. A. Schmedake, B. J. Paradise and R. West, Can. J. Chem., 2000, 78, 1526 CrossRef CAS; (d) M. P. Egorov, O. M. Nefedov, T.-S. Lin and P. P. Gaspar, Organometallics, 1995, 14, 1539 CrossRef CAS; (e) L. Pause, M. Robert, J. Heinicke and O. Kuhl, J. Chem. Soc., Perkin Trans. 2, 2001, 1383 RSC; (f) B. Gehrhus, P. B. Hitchcock, R. Pongtavornpinyo and L. Zhang, Dalton Trans., 2006, 1847 RSC; (g) B. Gehrhus, P. B. Hitchcock and L. Zhang, Angew. Chem., Int. Ed., 2004, 43, 1124 CrossRef CAS.
- (a) J. K. Kochi and P. J. Krusic, J. Am. Chem. Soc., 1969, 91, 3944 CrossRef CAS; (b) A. G. Davies, R. W. Dennis and B. P. Roberts, J. Chem. Soc., Perkin Trans. 2, 1974, 1101 RSC; (c) R. W. Dennis and B. P. Roberts, J. Chem. Soc., Perkin Trans. 2, 1975, 140 RSC; (d) A. G. Davies, D. Griller and B. P. Roberts, J. Organomet. Chem., 1972, 38, C8 CrossRef CAS; (e) P. J. Krusic, W. Mahler and J. K. Kochi, J. Am. Chem. Soc., 1972, 94, 6033 CrossRef CAS; (f) W. G. Bentrude and T. B. Min, J. Am. Chem. Soc., 1972, 94, 1025 CrossRef CAS.
- (a) A. J. Arduengo III, Acc. Chem. Res., 1999, 32, 913 CrossRef; (b) M. Haaf, T. A. Schmedake and R. West, Acc. Chem. Res., 2000, 33, 704 CrossRef CAS; (c) B. Gehrhus and M. F. Lappert, J. Organomet. Chem., 2001, 617–618, 209 CrossRef CAS; (d) N. J. Hill and R. West, J. Organomet. Chem., 2004, 689, 4165 CrossRef CAS; (e) S. E. Boganov, M. P. Egorov, V. I. Faustov and O. M. Nefedov, in The Chemistry of Organic Germanium, Tin and Lead Compounds, ed. Z. Rappoport, Wiley, New York, 2002, vol. 2, part 1, ch. 12 Search PubMed; (f) P. P. Gaspar and R. West, in The Chemistry of Organic Silicon Compounds, ed. Z. Rapport and Y. Apeloig, Wiley, New York, 1998, vol. 2, part 3, ch. 43 Search PubMed.
- (a) B. Tumanskii, P. Pine, Y. Apeloig, N. J. Hill and R. West, J. Am. Chem. Soc., 2004, 126, 7786 CrossRef CAS; (b) B. Tumanskii, P. Pine, Y. Apeloig, N. J. Hill and R. West, J. Am. Chem. Soc., 2005, 127, 8248 CrossRef CAS; (c) B. Tumanskii, D. Sheberla, G. Molev and Y. Apeloig, Angew. Chem., Int. Ed., 2007, 46, 7408 CrossRef CAS; (d) G. A. Abakumov, V. K. Cherkasov, A. V. Piskunov, I. A. Aivaz'yan and N. O. Druzhkov, Dokl. Chem., 2005, 404, 189 CrossRef CAS; (e) A. V. Piskunov, I. A. Aivaz'yan, V. K. Cherkasov and G. A. Abakumov, J. Organomet. Chem., 2006, 691, 1531 CrossRef CAS; (f) I. McKenzie, J. Brodovitch, P. W. Percival, T. Ramnial and J. A. C. Clybume, J. Am. Chem. Soc., 2003, 125, 11565 CrossRef CAS; (g) radical addition to in situ generated triplet diphenyl carbene was also studied: H. L. Casal, N. H. Werstiuk and J. C. Scaiano, J. Org. Chem., 1984, 49, 5214 Search PubMed; (h) T. Iwamoto, H. Masuda, S. Ishida, C. Kabuto and M. Kira, J. Am. Chem. Soc., 2003, 125, 9300 CrossRef CAS; (i) A. Naka, N. J. Hill and R. West, Organometallics, 2004, 23, 6330 CrossRef CAS; (j) I. L. Fedushkin, N. M. Khvoinova, A. Yu. Baurin, G. K. Fukin, V. K. Cherkasov and M. P. Bubnov, Inorg. Chem., 2004, 43, 7807 CrossRef CAS; (k) S.-H. Ueng, A. Solovyev, X. Yuan, S. J. Geib, L. Fensterbank, E. Lacote, M. Malacria, M. Newcomb, J. C. Walton and D. P. Curran, J. Am. Chem. Soc., 2009, 131, 11256 CrossRef CAS.
- (a) A. G. Davies, D. Griller and B. P. Roberts, J. Am. Chem. Soc., 1972, 94, 1782 CrossRef CAS; (b) D. Griller, K. Dimroth, T. M. Fyles and K. U. Ingold, J. Am. Chem. Soc., 1975, 97, 5526 CrossRef CAS; (c) D. Griller and K. U. Ingold, J. Am. Chem. Soc., 1973, 95, 6459 CrossRef CAS; (d) B. Tumanskii and O. Kalina, Radical Reaction of Fullerenes and their Derivatives, Kluwer Academic Publishers, Dordrecht, 2001 Search PubMed.
- Prolonged unfiltered UV-irradiation (20 min) leads to complex radical transformations and as a result to the accumulation of side products, leading to the central multiplet in Fig. 2a. We interpret the central multiplet as the superposition of two radicals, the adduct of benzyl radical with 3 [a(214N) = a(21H) = 5.7 G, aH(CH2) = 16.85 G, g = 2.0015] and an adduct of an O-centered radical [a(214N) = a(21H) = 5.8 G, a (73Ge) = 13.3 G, g = 2.0014]. For details of simulation of superposition of the two spectra and model experiments in benzene see Fig. S2‡.
- A. G. Davies, M. J. M. Parott and B. P. Roberts, J. Chem. Soc., Chem. Commun., 1974, 973 RSC.
- J. D. Cotton, C. S. Cundy, D. H. Harris, A. Huson, M. F. Lappert and P. W. Lednor, J. Chem. Soc., Chem. Commun., 1974, 651 RSC.
- A. Sekiguchi, T. Fukawa, M. Nakamoto, V. Ya. Lee and M. Ichinode, J. Am. Chem. Soc., 2002, 124, 9868.
- (a) J. A. Weil and J. Bolton, Electron Paramagnetic Resonance, John Wiley & Sons, Hoboken, New Jersey, 2nd edn, 2007 Search PubMed; (b) K. U. Ingold and J. C. Walton, Acc. Chem. Res., 1989, 22, 8 CrossRef CAS; (c) K. U. Ingold and J. C. Walton, J. Am. Chem. Soc., 1987, 109, 6937 CrossRef CAS; (d) J. C. Matasyoh, P. Schuler, H. B. Stegmann, J. L. Poyer, M. West and E. G. Janzen, Magn. Reson. Chem., 1996, 34, 351 CrossRef CAS.
- B. L. Tumanskii, R. G. Gasanov, M. V. Tsikalova, A. V. Usatov, E. V. Martynova and Yu. N. Novikov, Russ. Chem. Bull., 2004, 9, 53 Search PubMed.
- L. Hermosilla, P. Calle, J. M. Garcia de la Vega and C. Sieiro, J. Phys. Chem. A, 2005, 109, 7626–7635 CrossRef CAS.
- R. Stowasser and R. Hoffmann, J. Am. Chem. Soc., 1999, 121, 3414 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision D.02), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- A. Schafer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829 CrossRef CAS.
- (a) F. Neese, ORCA, version 2.6.35, University of Bonn, Germany, 2008 Search PubMed; (b) F. Neese, J. Chem. Phys., 2001, 115, 11080 CrossRef CAS.
- R. Improta and V. Barone, Chem. Rev., 2004, 104, 1231 CrossRef CAS.
Footnotes |
| † Dedicated to Professor Wataru Ando, a pioneer of silicon chemistry, on the occasion of his 75th birthday. |
| ‡ Electronic supplementary information (ESI) available: Cartesian coordinates and energies of stationary points in the calculations of 2a, 3a, 4a, 5a, 6a and Fig. S1–S7. See DOI: 10.1039/c0sc00143k |
| This journal is © The Royal Society of Chemistry 2010 |