DOI:
10.1039/C0SC00202J
(Edge Article)
Chem. Sci., 2010,
1, 215-220
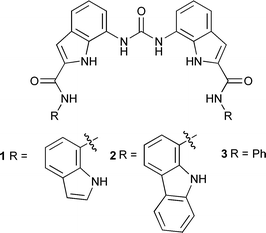
Acyclic indole and carbazole-based sulfate receptors†
Received
1st March 2010
, Accepted 8th April 2010
First published on
26th May 2010
Abstract
The anion complexation properties of a series of acyclic receptors consisting of diindolylurea groups appended with amide, amidoindole or amidocarbazole groups have been studied. The receptors selectively bind and encapsulate sulfate via either six or eight hydrogen bonds. Receptors containing eight hydrogen bond donors perturb the pKa of bound dihydrogen phosphate and bicarbonate to the extent that they are deprotonated by free anion in solution.
Introduction
The development of sulfate selective anion receptors is currently an area of intense interest due to the important roles this anion plays in biological systems and disease,1 in hydrometallurgy2 and as a pollutant.3 Tripodal tris-urea based systems have been developed by Custelcean and co-workers as selective receptors for SO42− with the goal of precipitating the anion from solution.4 These species form 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 receptor:sulfate complexes binding the anion via twelve NH⋯O hydrogen bonding interactions. Additionally Custelcean, Hay and co-workers have developed self-assembling urea containing cage systems to arrange six urea groups around sulfate.5 Sulfate has also been employed by Beer and Mullen to assemble a variety of interlocked structures.6 Macrocyclic sulfate receptors include Sessler et al.'s cyclo[8]pyrrole,7 Bowman-James et al.’s cyclic tetraamide/amine based receptor8 that forms a sandwich complex with SO42− and Kubik et al.'s cyclic peptide based molecular oysters.9
:1 receptor:sulfate complexes binding the anion via twelve NH⋯O hydrogen bonding interactions. Additionally Custelcean, Hay and co-workers have developed self-assembling urea containing cage systems to arrange six urea groups around sulfate.5 Sulfate has also been employed by Beer and Mullen to assemble a variety of interlocked structures.6 Macrocyclic sulfate receptors include Sessler et al.'s cyclo[8]pyrrole,7 Bowman-James et al.’s cyclic tetraamide/amine based receptor8 that forms a sandwich complex with SO42− and Kubik et al.'s cyclic peptide based molecular oysters.9
Despite the anion complexation properties of indole being recognised in biological systems,10 it was not until 200411 and 200512 that the first reports of the use of indole in synthetic anion receptors appeared. Since then indole has been employed in a number of receptor systems13 including a variety of foldamers reported by Jeong and co-workers that exhibit conformational changes in the presence of particular anions.14 Taking inspiration from this latter work, we decided to synthesise linear receptors that could wrap around tetrahedral oxo-anions such as sulfate but that would possess lower affinities for anions possessing other geometries.
We have previously shown that diindolylureas form 3:1 complexes with PO43− (binding the anion via twelve hydrogen bonds)15 in the solid state and in the process drive the deprotonation of the anion.16 In this edge article we report the synthesis of receptors 1 and 2 that contain pendant amido-indole and amido-carbazole groups and their interaction with a range of anionic guests. These compounds are designed to selectively bind sulfate over chloride (due to the large size of the binding site) or carboxylates (due to the carboxylate anion being saturated by four hydrogen bonds from the diindolylurea skeleton15). We compared the anion complexation properties of these species with compound 316 which contains pendant phenyl amides and a total of six hydrogen bond donor groups.
Results and discussion
Synthesis
7-Nitroindole-2-carboxylic acid was coupled to either 7-aminoindole or 1-aminocarbazole using pyBOP as an amide coupling reagent in DMF. The resulting amides were reduced from nitro- to amine derivatives using 10% Pd/C under a hydrogen atmosphere in DMF and the resulting amines coupled using triphosgene in a mixture of chloroform, DMF and saturated sodium bicarbonate solution to afford compounds 1 and 2 in 29% and 55% respective overall yields. Compound 3 was synthesised using literature methods.16
Initial anion complexation studies were conducted using single crystal X-ray diffraction. Crystals of the tetrabutylammonium benzoate complex of receptor 1 were obtained by slow evaporation from a DMSO solution containing excess tetrabutylammonium benzoate.‡ The structure (Fig. 1) reveals that receptor 1 binds three equivalents of benzoate in the solid state. The central diindolylurea group binds one benzoate anion via four hydrogen bonds N3⋯O6 2.719(4) Å; N4⋯O6 2.717(4) Å; N5⋯O7 2.820(4) Å; N6⋯O7 2.770(4) Å. The pendant amidoindole groups are oriented out of the cavity with each binding a single equivalent of benzoate via two or three hydrogen bonds N1⋯O4 2.870(4) Å; N1⋯O5 3.144(4) Å; N2⋯O5 2.811(4) Å and N7⋯O8 2.841(4); N8⋯O9 2.766(5) Å.
 |
| | Fig. 1 The X-ray crystal structure of 1·(TBA)3(C6H5CO2)·H2O. Tetrabutylammonium counter cations and water omitted for clarity. | |
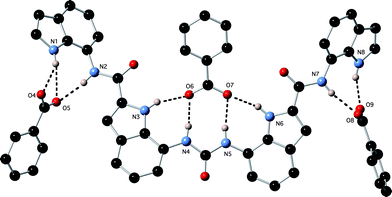
Crystals of the monohydrogen phosphate complex of receptor 1 were obtained by slow evaporation of a DMSO solution of the receptor in the presence of excess tetrabutylammonium dihydrogen phosphate.§ The structure (Fig. 2) shows the monohydrogen phosphate anion bound by nine hydrogen bonds from the receptor: N1⋯O7 2.970(8) Å; N1⋯O4 3.109(7) Å; N2⋯O4 2.790(8) Å; N3⋯O4 2.736(8) Å; N4⋯O5 2.818(8) Å; N5⋯O5 2.764(7) Å; N6⋯O6 2.815(7) Å; N7⋯O6 2.838(8) Å; N8⋯O6 2.782(7) Å. Additionally the monohydrogen phosphate dimerises in the solid state via two hydrogen bonds O7⋯O5i and O7i⋯O5 2.645(7) Å; the symmetry transformation −x + 1, −y + 1, −z generates atoms labelled (i). We have previously observed deprotonation of dihydrogenphosphate bound to neutral diindolylurea based receptors due to the multiple hydrogen bonding interactions lowering the pKa of the bound guest to the extent that it is deprotonated by free dihydrogen phosphate in solution and we suggest that the same process occurs here.16
 |
| | Fig. 2 Two views of the X-ray crystal structure of 1·(TBA)2(HPO4). Tetrabutylammonium counter cations have been omitted for clarity. | |
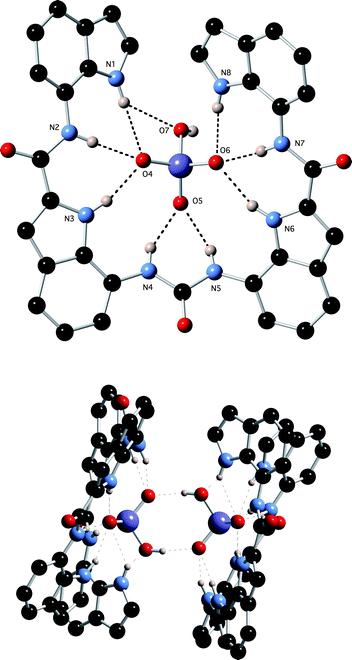
Crystals of the tetrabutylammonium sulfate complex of receptor 1 were obtained by slow evaporation of a DMSO solution of the receptor in the presence of excess tetrabutylammonium sulfate.17¶ The structure (Fig. 3) reveals that the anion is bound by eight hydrogen bonds from the receptor: N1⋯O6 2.927(4) Å; N2⋯O4 2.953(3) Å; N3⋯O4 2.798(3) Å; N4⋯O5 2.788(3) Å; N5⋯O5 2.896(3) Å; N6⋯O7 2.863(3) Å; N7⋯O7 2.896(3) Å; N8⋯O6 2.856(3) Å.
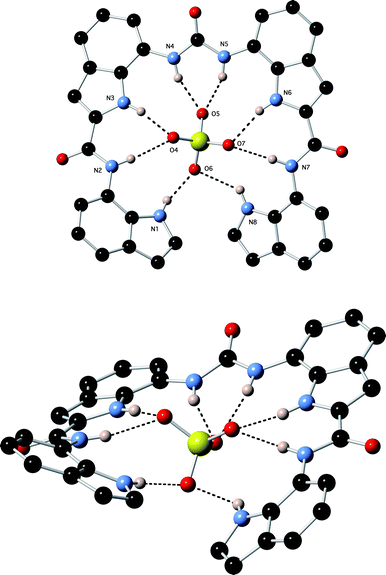 |
| | Fig. 3 Two views of the X-ray crystal structure of 1·(TBA)2SO4. Tetrabutylammonium counter cations have been omitted for clarity. | |
Solution studies
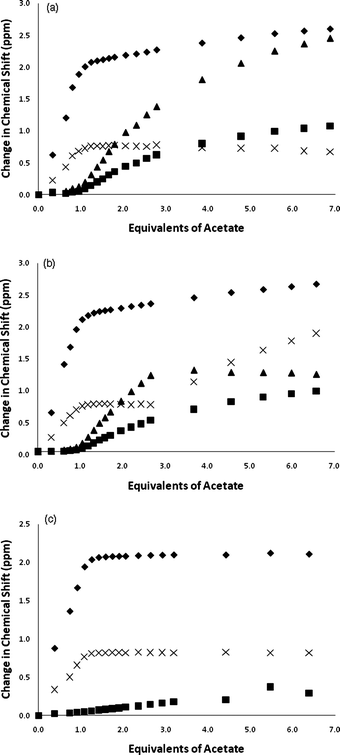
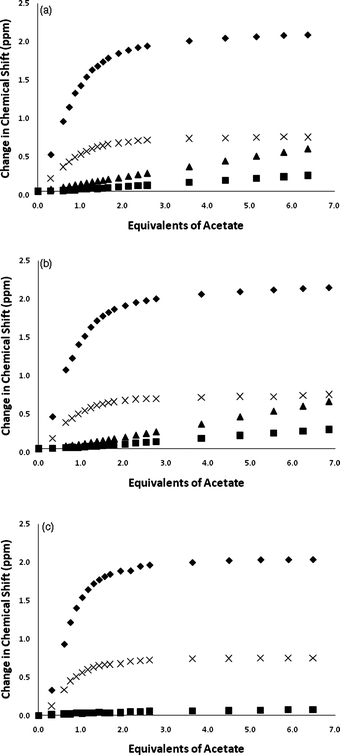
The solution anion binding properties of compounds 1 and 2 were studied by 1H NMR titration techniques in DMSO-d6/water mixtures and compared to those of compound 3. Initial 1H NMR titration studies were conducted in DMSO-d6/0.5% water. The NMR titrations of compounds 1–3 with acetate are shown in Fig. 4. The results show that the NH groups in the central diindolylurea unit in these three compounds bind the first equivalent of acetate added to solution. Further aliquots of acetate bind to the pendant amide or pendant amide and/or indole or carbazole groups (as shown in Scheme 1 for compound 1). This can be seen clearly in the NMR titration curves for compounds 1 and 2 wherein the pendant NH groups begin to shift downfield after the addition of 1.0 equivalents of acetate. In the case of compound 2, it appears that more complex equilibria are present as after the addition of three equivalents of acetate, the central indole groups begin to shift downfield again after having previously reached a plateau. This process does not occur in DMSO-d6/10% water (Fig. 5). Similar sequential binding of anions to simple urea-amide receptors containing multiple hydrogen bond donor groups has been observed previously by Gunnlaugsson and co-workers.18 Analogous titration experiments with compounds 1 and 2 in DMSO-d6/0.5% water with tetrabutylammonium benzoate resulted in precipitation.
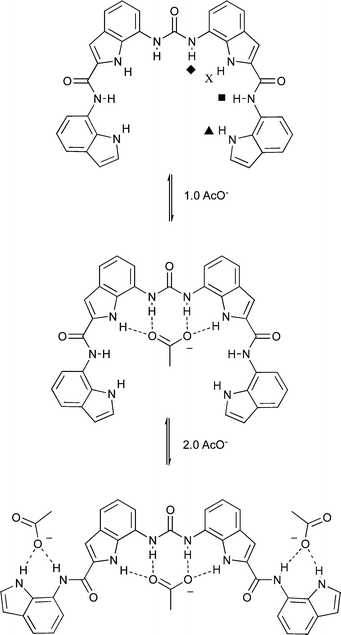 |
| | Scheme 1 Proposed solution equilibria of compound 1 with acetate in DMSO-d6/water solution. | |
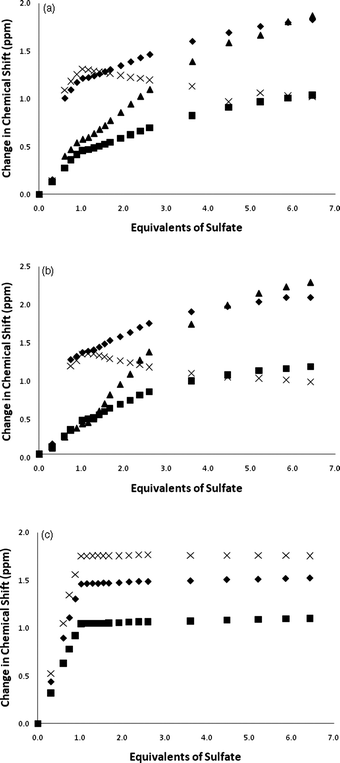
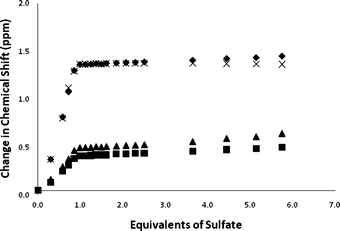
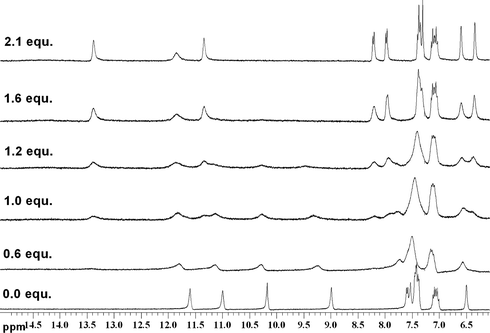
In contrast to the results found with acetate, addition of tetrabutylammonium sulfate causes all the NH groups in compounds 1, 2 and 3 to shift downfield upon addition of substoichiometric quantities of the anion. The NMR titration data conducted in DMSO-d6/0.5% water are shown in Fig. 6. Compound 3 binds sulfate strongly in 1:1 stoichiometry with a stability constant >104 M−1 under these conditions. Upon addition of sulfate to compounds 1 and 2, more complex equilibria are observed. The NMR titration curves show a discontinuity at one equivalent of sulfate which may be indicative of initial formation of a 1:1 complex followed by a conformational rearrangement and formation of higher order complexes. However, just as was observed with acetate, moving to DMSO-d6/10% water reduces the complexity of the equilibria in solution with predominant 1:1 complex formation and an apparent stability constant of >104 M−1 for sulfate with compound 2 under these competitive conditions (Fig. 7). Interestingly both compounds 1 and 3 crystallise over the course of about 20 min upon addition of tetrabutylammonium sulfate to DMSO-d6/10% water solutions of the receptors. This unfortunately precluded conducting NMR titrations under these conditions. The same process occurs more slowly with compound 2. The DMSO-d6/10% water solution of compound 1 after addition of 15 equivalents of tetrabutylammonium sulfate is shown in Fig. 8.
 |
| | Fig. 8 A view of the DMSO-d6/10% water solution of compound 1 in the presence of 15 equivalents of tetrabutylammonium sulfate. Crystals form approximately 20 min after the addition of sulfate. | |
By following the change in chemical shift of the urea NH groups in DMSO-d6/10% water it is possible to calculate apparent stability constants of 600 M−1 and 690 M−1 for the complexation of the first equivalent of acetate by compounds 1 and 2 (Table 1). Stability constants of 120 M−1 and 315 M−1 for compounds 1 and 2 with tetrabutylammonium dihydrogen phosphate in DMSO-d6/10% water were also measured. Addition of tetrabutylammonium chloride or hydrogen sulfate to both compounds in DMSO-d6/0.5% water resulted in only very small perturbations in the NMR spectra. Compound 3 binds dihydrogen phosphate with a stability constant of 107 M−1 in 0.5% water and acetate with a stability constant of 8460 M−1.16 In all cases these stability constants were calculated following the shift of the urea NH groups using the EQNMR computer program.19
Table 1 Stability constants (Ka/M−1) of compounds 1, 2 and 3 with a variety of putative anionic guestsa determined by 1H NMR titration techniques in DMSO-d6/10% water mixtures at 298 K following the urea NH group resonance.h Errors <15%
| Anion |
Compound |
|
1
|
2
|
3
|
|
Anions added as tetrabutylammonium salts except bicarbonate which was added as the tetraethylammonium salt.
Only small perturbations were observed in the 1H NMR spectrum upon addition of these anions.
K
1 values reported only.
Isotherm could not be fitted to a 1:1 or 1:2 binding model.
Deprotonation of bound guest was observed.
Peak broadening prevented a stability constant from being determined in this case (possibly due to deprotonation of the bound guest).
Data from ref. 16.
n.d. = not determined.
|
| Cl− |
—b |
—b |
n.d. |
| HSO4− |
—b |
—b |
n.d. |
| AcO− |
600c |
690c |
1422 |
| BzO− |
n.d. |
n.d. |
481 |
| H2PO4− |
120 |
315 |
—d |
| HCO3− |
—e |
—e |
—f |
In DMSO-d6/0.5% water, behaviour indicative of deprotonation of the bound anionic guest was observed with compounds 1 and 2 upon addition of dihydrogen phosphate and bicarbonate (with the latter anion added as the tetraethylammonium salt), i.e. new peaks appear in the NMR spectrum shifted significantly downfield from the free ligand due to formation of monohydrogen phosphate or carbonate complexes. This results from multiple hydrogen bonding interactions to the bound guest from the receptor, reducing the pKa of the anion, and subsequent deprotonation by free anion in solution.13 Deprotonation was confirmed by addition of one equivalent of the anionic guest followed by hydroxide which results in the appearance of the same new NMR resonances. This is shown in Fig. 9 for compound 1 and tetrabutylammonium dihydrogen phosphate. In 10% water this was observed only upon addition of HCO3− but not H2PO4−(see ESI for more details†).
Conclusions
Compounds 1–3 all bind sulfate strongly in DMSO-d6/0.5% water. Under these conditions compound 3 forms a 1:1 complex whilst the NMR titration evidence leads us to suggest that compounds 1 and 2 also form 1:1 complexes but that at higher sulfate concentrations there are multiple equilibria present. These additional processes do not occur in DMSO-d6/10% water to the same extent with predominant 1:1 complex formation. Hence by changing the solvent conditions from 0.5% water to 10% water we have reduced the complexity of the equilibria present. Compound 2 binds sulfate with a stability constant >104 M−1 under these conditions whilst compounds 1 and 3 crystallise upon addition of sulfate. It is interesting to note that compound 3 binds sulfate strongly despite having two fewer hydrogen bond donors than compounds 1 and 2. The receptors are also capable of perturbing the pKa of bound dihydrogen phosphate or bicarbonate to the extent that they are deprotonated by free anions in solution. The crystal structures show that compound 1 is capable of encapsulating tetrahedral oxo-anions via eight hydrogen bonds. We are currently exploring the extraction properties of this new family of sulfate selective anionophores. The results of these studies will be reported in due course.
Acknowledgements
PAG thanks the EPSRC for funding and for access to the crystallographic facilities at the University of Southampton.
Notes and references
- H. Xu, N. Sträter, W. Schröder, C. Böttcher, K. Ludwing and W. Saenger, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2003, 59, 815–822 CrossRef; J. W. Pflugrath and F. A. Quiocho, Nature, 1985, 314, 257–260 CrossRef CAS; J. W. Pflugrath and F. A. Quiocho, J. Mol. Biol., 1988, 200, 163–180 CAS; D. E. C. Cole, R. M. Hanning, S. H. Zlotkin and W. Balfe, Nephron, 1986, 44, 186–190 CrossRef CAS; R. M. Freeman and C. J. Richards, Kidney Int., 1979, 15, 167–175 CrossRef CAS.
- S. G. Galbraith, Q. Wang, L. Li, A. J. Blake, C. Wilson, S. R. Collinson, L. F. Lindoy, P. G. Plieger, M. Schroder and P. A. Tasker, Chem.–Eur. J., 2007, 13, 6091–6107 CrossRef CAS; S. G. Galbraith and P. A. Tasker, Supramol. Chem., 2005, 17, 191–207 CrossRef CAS; P. G. Plieger, P. A. Tasker and S. G. Galbraith, Dalton Trans., 2004, 313–318 RSC.
-
B. A. Moyer, L. H. Delmau, C. J. Fowler, A. Ruas, D. A. Bostick, J. L. Sessler, E. Katayev, G. D. Pantos, J. M. Llinares, Md. A. Hossain, S. O. Kang, and K. Bowman-James, in Advances in Inorganic Chemistry: Template Effects and Molecular Organisation, vol. 59, ed. R. van Eldik and K. Bowman-James, Academic Press, London, 2007, vol. 59, pp. 175–204 Search PubMed.
- R. Custelcean, B. A. Moyer and B. P. Hay, Chem. Commun., 2005, 5971–5973 RSC; R. Custelcean, P. Remy, Bonnesen, D. Jiang and B. A. Moyer, Angew. Chem., Int. Ed., 2008, 47, 1866–1870 CrossRef . This class of sulfate receptor has also been studied by other groups: B. Wu, J. J. Liang, J. Yang, C. D. Jia, X. J. Yang, H. R. Zhang, N. Tang and C. Janiak, Chem. Commun., 2008, 1762–1764 Search PubMed; F. Y. Zhuge, B. A. Wu, J. J. Liang, J. Yang, Y. Y. Liu, C. D. Jia, C. Janiak, N. Tang and X. J. Yang, Inorg. Chem., 2009, 48, 10249–10256 RSC.
- R. Custelcean, J. Barsano, P. V. Bonnesen, V. Kertesz and B. P. Hay, Angew. Chem., Int. Ed., 2009, 48, 4025–4029 CrossRef CAS.
- K. M. Mullen and P. D. Beer, Chem. Soc. Rev., 2009, 38, 1701–1713 RSC.
- L. R. Eller, M. Stepien, C. J. Fowler, J. T. Lee, J. L. Sessler and B. A. Moyer, J. Am. Chem. Soc., 2007, 129, 11020–11021 CrossRef CAS.
- Md. A. Hossain, J. M. Llinares, D. Powell and K. Bowman-James, Inorg. Chem., 2001, 40, 2936–2937 CrossRef CAS.
- S. Kubik, R. Kirchner, D. Nolting and J. Seidel, J. Am. Chem. Soc., 2002, 124, 12752–12760 CrossRef CAS; S. Otto and S. Kubik, J. Am. Chem. Soc., 2003, 125, 7804 CrossRef CAS; Z. Rodriguez-Docampo, S. I. Pascu, S. Kubik and S. Otto, J. Am. Chem. Soc., 2006, 128, 11206 CrossRef CAS.
- K. H. G. Verschueren, F. Seljee, H. J. Rozeboom, K. H. Kalk and B. W. Dijkstra, Nature, 1993, 363, 693–698 CrossRef CAS; J. J. He and F. A. Quiocho, Science, 1991, 251, 1479–1481 CrossRef CAS.
- M. J. Chmielewski, M. Charon and J. Jurczak, Org. Lett., 2004, 6, 3501–3504 CrossRef CAS; P. Piatek, V. M. Lynch and J. L. Sessler, J. Am. Chem. Soc., 2004, 126, 16073–16076 CrossRef CAS.
- D. Curiel, A. Cowley and P. D. Beer, Chem. Commun., 2005, 236–238 RSC; K.-J. Chang, D. Moon, M. S. Lah and K. S. Jeong, Angew. Chem., Int. Ed., 2005, 44, 7926–7929 CrossRef CAS.
- J.-m. Suk, M. K. Chae, N.-K. Kim, U.-l. Kim and K.-S. Jeong, Pure Appl. Chem., 2008, 80, 599–608 CrossRef CAS; P. A. Gale, Chem. Commun., 2008, 4525–4540 RSC; C. Caltagirone and P. A. Gale, Chem. Soc. Rev., 2009, 38, 520–563 RSC; P. A. Gale, S. E. García-Garrido and J. Garric, Chem. Soc. Rev., 2008, 37, 151–190 RSC.
- K.-J. Chang, B.-N. Kang, M.-H. Lee and K.-S. Jeong, J. Am. Chem. Soc., 2005, 127, 12214–12215 CrossRef CAS; J.-m. Suk and K.-S. Jeong, J. Am. Chem. Soc., 2008, 130, 11868–11869 CrossRef CAS; J. Kim, H. Juwarker, X. Liu, M. S. Lah and K. S. Jeong, Chem. Commun., 2010, 46, 764–766 RSC.
- C. Caltagirone, P. A. Gale, J. R. Hiscock, S. J. Brooks, M. B. Hursthouse and M. E. Light, Chem. Commun., 2008, 3007–3009 RSC; C. Caltagirone, J. R. Hiscock, M. B. Hursthouse, M. E. Light and P. A. Gale, Chem.–Eur. J., 2008, 14, 10236–10243 CrossRef CAS . For carbazole analogues see: J. R. Hiscock, C. Caltagirone, M. E. Light, M. B. Hursthouse and P. A. Gale, Org. Biomol. Chem., 2009, 7, 1781–1783 Search PubMed . For our earlier work on indoles see: G. W. Bates, P. A. Gale and M. E. Light, Chem. Commun., 2007, 2121–2123 RSC; G. W. Bates, Triyanti, M. E. Light, M. Albrecht and P. A. Gale, J. Org. Chem., 2007, 72, 8921–8927 Search PubMed . For related work by Caltagirone and co-workers see: C. Caltagirone, A. Mulas, F. Isaia, V. Lippolis, P. A. Gale and M. E. Light, Chem. Commun., 2009, 6279–6281 RSC.
- P. A. Gale, J. R. Hiscock, S. J. Moore, C. Caltagirone, M. B. Hursthouse and M. E. Light, Chem.–Asian J., 2010, 5, 555–561 CrossRef CAS.
- M. J. Chmielewski, L. Zhao, A. Brown, D. Curiel, M. R. Sambrook, A. L. Thompson, S. M. Santos, V. Felix, J. J. Davis and P. D. Beer, Chem. Commun., 2008, 3154–3156 RSC.
- C. M. G. dos Santos, T. McCabe, G. W. Watson, P. E. Kruger and T. Gunnlaugsson, J. Org. Chem., 2008, 73, 9235–9244 CrossRef CAS.
- M. J. Hynes, J. Chem. Soc., Dalton Trans., 1993, 311–312 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, characterisation data for compounds 1 and 2, NMR and titration data. CCDC reference numbers 767919–767921. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00202j |
| ‡ Crystal data for compound (1·(TBA)3(C6H5CO2)·H2O) C104H151N11O10, Mr = 1715.36, T = 120(2) K, triclinic, space group P1, a = 8.9320(1), b = 12.4500(2), c = 22.5830(4) Å, α = 93.6790(9)°, β = 100.5490(12)°, γ = 98.6400(11)°, V = 2429.96(6) Å3, ρc = 1.172 Mg m−3, μ = 0.075 mm−1, Z = 1, reflections collected: 38462, independent reflections: 8540 (Rint = 0.0637), final R indices [I > 2σI]: R1 = 0.0613, wR2 = 0.1183, R indices (all data): R1 = 0.0811, wR2 = 0.1288. |
| § Crystal data for compound (1·(TBA)2(HPO4)) C67H99N10O7P, Mr = 1187.53, T = 120(2) K, monoclinic, space group P21/n, a = 14.7860(6), b = 17.7836(6), c = 25.6491(10) Å, β = 103.512(2)°, V = 6557.7(4) Å3, ρc = 1.203 Mg m−3, μ = 0.102 mm−1, Z = 4, reflections collected: 63501, independent reflections: 11548 (Rint = 0.1856), final R indices [I > 2σI]: R1 = 0.1595, wR2 = 0.2338, R indices (all data): R1 = 0.2672, wR2 = 0.2775. |
¶ Crystal data for compound (1·(TBA)2(SO4)) C67H98N10O7S, Mr = 1187.61, T = 120(2) K, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 8.78020(10), b = 16.9156(4) c = 22.1638(5) Å, α = 88.1703(11)°, β = 85.7351(12)°, γ = 82.7653(13)°, V = 3255.72(11) Å3, ρc = 1.211 Mg m−3, μ = 0.110 mm−1, Z = 2, reflections collected: 61520, independent reflections: 11472 (Rint = 0.0907), final R indices [I > 2σI]: R1 = 0.0734, wR2 = 0.1616, R indices (all data): R1 = 0.1081, wR2 = 0.1822. , a = 8.78020(10), b = 16.9156(4) c = 22.1638(5) Å, α = 88.1703(11)°, β = 85.7351(12)°, γ = 82.7653(13)°, V = 3255.72(11) Å3, ρc = 1.211 Mg m−3, μ = 0.110 mm−1, Z = 2, reflections collected: 61520, independent reflections: 11472 (Rint = 0.0907), final R indices [I > 2σI]: R1 = 0.0734, wR2 = 0.1616, R indices (all data): R1 = 0.1081, wR2 = 0.1822. |
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.