Miktoarm core-crosslinked star copolymers with biologically active moieties on peripheries
Irakli
Javakhishvili
and
Søren
Hvilsted
*
Technical University of Denmark, Department of Chemical and Biochemical Engineering, Danish Polymer Centre, Building 227, DK-2800, Kgs. Lyngby, Denmark. E-mail: sh@kt.dtu.dk; Tel: +45 4525 2965
First published on 20th September 2010
Abstract
Miktoarm core-crosslinked star (CCS) copolymers featuring hydrophobic inner compartment based on poly(ε-caprolactone) (PCL), and hydrophilic multivalent exterior consisting of L-lysine dendritic wedges and estradiol or ferrocene moieties have been synthesized. Ring-opening polymerization (ROP) of ε-caprolactone (ε-CL) initiated by functional alcohols provides alkyne or azide end-capped linear PCL chains. Further derivatization of the hydroxyl chain ends of these hetero-telechelic macromolecules by methacrylic acid (MA), and subsequent Cu(I) mediated “click” coupling of terminal alkyne and azide groups with azide-functionalized dendritic wedge, 17α-ethynylestradiol and ethynylferrocene afford well-defined linear-dendritic and linear macromonomers (MMs), respectively. Atom transfer radical polymerization (ATRP) of the MMs together with a difunctional monomer 1,4-butanediol dimethacrylate (BDMA) results in miktoarm CCS copolymers. Elucidation of the structure and composition of the intermediate and final products demonstrates effectiveness, versatility, and high degree of control displayed by the proposed synthetic sequence.
Introduction
Unique globular shape, sophisticated macromolecular architecture providing core-shell microenvironment, and plethora of reactive functional groups of CCS polymers have bolstered their potential in myriads of applications ranging from polymer therapeutics and catalysis to membrane technology and lithography.1,2 Synthesis of CCS polymers by ATRP involves exploitation of macroinitiators (MIs) and/or MMs in combination with difunctional cross-linkers.1 Matyjaszewski and co-workers have prepared CCS polymer employing polystyrene (PS) MI and divinyl benzene (DVB) cross-linker.3 The authors pointed out that the method suffers from the consequences of inevitable radical termination reactions in the radical polymerization which results in partially functionalized MIs. Employing similar approach, Matyjaszewski and co-workers have designed surface functionalized CCS polymers based on poly(tert-butyl acrylate) MIs. Epoxy, cyano, hydroxyl, and amino groups have been incorporated at the end of the MI via functional ATRP initiators.4 While homoarm star polymers contain arms with similar molecular weight and same chemical composition, miktoarm stars are comprised of arms with diverse chemical compositions and/or molecular weights.1 Miktoarm CCS copolymer with degradable core have been synthesized via “in-out” method: poly(methyl methacrylate) (PMMA) MI has been reacted with the cross-linker containing degradable disulfide moiety, and thus obtained CCS polymer has been exploited as MI in subsequent ATRP of n-butyl acrylate (BA).5 Grafting the second monomer through the reactivation of the alkyl halide sites in the core of the star polymer features several obstacles such as star-star and intrastar couplings that broaden the molecular weight distribution (MWD), and low initiation efficiency of the star MI.5 Star polymers with hydrolysable cores prepared via group transfer polymerization of MMA have also been reported.6A characteristic trait of the star synthesis by cross-linking MI chains is star-star coupling reaction that takes place throughout the star formation process and broadens MWD.7 Star-star reaction occurs between the star cores where the number of initiating sites equals to the number of arms per star. Therefore, reduction of the ratio of initiating sites to arms per star could diminish the star-star coupling providing better control and improved consistency.7 In the quest of alleviating this intrinsic shortcoming, Matyjaszewski and co-workers have proposed preparation of CCS polymers by copolymerization of linear MM with cross-linker using low molecular weight ATRP initiator.7 Under these circumstances the number of initiating sites and arms are independently controlled allowing limiting of star-star coupling reactions resulting in narrow MWD. Employing this strategy, miktoarm star copolymers incorporating PBA and poly(ethylene glycol) (PEG) MMs have been prepared.8 Star congestion and obstruction of the initiating sites can be overpowered by addition of another portion of cross-linker and initiator. Thus, high yield and low polydispersity have been attained by stepwise addition of the cross-linker and initiator during cross-linking the mixture of the MMs.8
Miktoarm CCS copolymer bearing biocompatible PCL and PS arms have been developed.9 PCL MI possessing bromoisobutyryl end group has been reacted with DVB giving CCS polymer with relatively broad MWD (Mw/Mn > 2). PS arms have then been grown from the star PCL MI by ATRP of styrene. Besides controlled radical polymerization methods such as ATRP, ROP has also been exercised for preparation of CCS PCL.10 ROP of ε-CL affords MI for the ensuing cross-linking reaction via ROP of bislactone. This is a two-step one-pot approach which does not demand isolation and purification of the MI. However, adventitious water may produce α,ω-difunctional arms and facilitate star-star coupling leading to macrogelation.10 Wiltshire and Qiao have designed a range of degradable CCS polymers featuring core-degradable, arm-degradable, partially arm-degradable, and fully degradable stars.11 Dual initiator 2-hydroxyethyl 2-bromoisobutyrate has been used to prepare degradable PCL and nondegradable PS arms. Cross-linking of the mixture of PCL-Br and PMMA-Cl MIs provides partially degradable stars. Degradable core is formed by ROP of bislactone cross-linker, while DVB and ethylene glycol dimethacrylate (EGDMA) are employed in ATRP from the MI when nondegradable cores are desired.11 Since tuning coronal composition of CCS polymers is the handle for manipulating the physical properties of the stars, ways for tailoring the corona have been suggested.12 CCS polymers with degradable outer coronal layer, with initiating sites on the coronal surface, and with initiating groups in the core as well as on the peripheries have been prepared making use of the PCL building blocks amongst others.12
Hawker and co-workers have reported synthesis of CCS polymers with amine and hydroxyl surface functional groups employing functional initiators for nitroxide-mediated living free radical polymerization. The potential of star stabilized Pd nanoparticles in catalysis has also been demonstrated by exploiting them in hydrogenation and Heck reactions.13 Linear-dendritic MIs, carrying Fréchet type or polyester dendrons, have been utilized for preparation of CCS with dendron-decorated peripheries.14,15 Modification of the dendron functionalities results in dramatic changes in the morphologies produced by the star polymers.15
Potential application of star polymers in drug delivery stems from the inherent thermodynamic instability of classical amphiphilic copolymer micelles. The formation of classical micelles is thermodynamically favored only above critical micelle concentration (cmc). If the concentration of the copolymer drops below the cmc, the micelles tend to disassemble. Inasmuch as the delivery vehicle undergoes severe dilution upon injection into an animal or human, lysis of the micelles below the cmc impedes their application in vivo.16 Dendrimers, star and CCS polymers, which may serve as unimolecular reservoirs, may be the remedy in circumventing the problems posed by micelle disintegration upon dilution.16 Roovers and co-workers have prepared amphiphilic star polymer based on polyamidoamine dendritic core, which served as the macroinitiator for obtaining lipophilic PCL inner block, and hydrophilic PEG outer block. Thus, the arms of the star polymer are built by biocompatible and biodegradable blocks. The potential in encapsulation of hydrophobic anticancer drug etoposide has been demonstrated.16 Besides high and controllable drug loading and release capabilities, biocompatibility, and the capacity to compartmentalize various drugs, the polymeric micelles offer a handle for passive targeting due to enhanced permeability and retention (EPR) effect in tumors.17 By taking advantage of EPR effect highly selective delivery of the anticancer drugs can be attained.17 Core-crosslinked micelles as unimolecular containers offer all the above mentioned benefits of classical micelles, and provide bonus in terms of enhanced stability in biological fluids.17 Kissel and co-workers prepared core-crosslinked micelles by crosslinking di- and triblock copolymers PEG-b-PCL and PEG-b-PCL-b-PEG which incorporated a maleic acid residue at the end of PCL block.17 These micelles have been successfully loaded with anticancer drug paclitaxel. Core-crosslinked micelles have displayed higher loading capacity than the classical ones.17
Alongside passive targeting, delivery of drugs involving a specific molecular recognition plays an important role.18 Gadolinium based MRI contrast agent incorporating selective estrogen receptor modulator (SERM) which binds to the estrogen receptor (ER) with high affinity, and induces contrast enhancement in human breast cancer cells has been proposed.18 17β-Estradiol (E2) is the most potent estrogen that enters freely the cell and binds to nuclear estrogen receptors. Modification of E2 by introducing bulky substituents at the 17α position does not deteriorate its high affinity to ER.18 Thus, coupling of 17α-ethynylestradiol with suitable chelate for gadolinium affords ER targeting contrast agent.18 Since binding of E2 to ER results in conformational change initiating the transcription of various genes commonly upregulated in cancerous cells, ER are overexpressed in 75–80% of all breast cancers.19 ERs are located both intracellularly and on the cell membrane; antiestrogen compound tamoxifen competitively binds ER, and averts the recruitment of transcription cofactors.19 Gold nanoparticles decorated with tamoxifen-PEG-SH derivative have been synthesized in the attempt to enhance potency and selective intracellular delivery. The study suggests that the membrane localized ER may assist selective particle uptake.19
Kostarelos and co-workers have reported tumor growth delay by sixth generation cationic poly-L-lysine dendrimer (G6PLL).20 This has been ascribed to the systemic antiangiogenic activity of G6PLL. It has been shown that G6PLL, which possesses 64 peripheral amino groups, has the ability to accumulate and remain at the tumor site as the result of strong electrostatic interactions between the positively charged PLL dendrimer surface and the negatively charged, heparin-rich walls of the tumor neovasculature.20 The authors suggest that conjugation of these dendrimers with other therapeutic and diagnostic agents may provide a powerful tool in cancer therapy.20
In this paper, we describe the synthetic outline and structural characterization of PCL miktoarm CCS copolymers bearing multiple amine functional groups and targeting motifs on the periphery. The miktoarm CCS copolymers have been obtained through MM approach exploiting linear-dendritic (di-Boc-L-lysine)G2-b-PCL-methacrylate and linear E2-PCL-methacrylate or ferrocene-PCL-methacrylate. Synthesis of the MMs is the combination of ROP of ε-CL from a functional initiator, Mitsunobu coupling with MA, and Cu(I) catalyzed “click” reaction. Subsequent cross-linking of the mixtures of MMs with BDMA via ATRP affords miktoarm CCS copolymers. Unmasking of the amino groups of L-lysine dendritic wedge furnishes amphiphilic nanoparticles that may have potential in targeted drug delivery. Unlike classical micelles, our potential nanocarrier should exhibit robustness and versatility combining various exceedingly important features such as: positively charged hydrophilic surface, ER targeting vectors, covalently stabilized core, and hydrophobic and bioerodible interior. To the best our knowledge, this is a novel design based on elegant coupling of individual building blocks which may facilitate fine-tuning the composition and thus properties of these particular CCS copolymers.
Experimental
Materials and methods
ε-Caprolactone (ε-CL; Fluka, ≥99%) was dried over CaH2, and distilled under reduced pressure. 1,4-butanediol dimethacrylate (BDMA; Aldrich, 95%) was passed through a short basic Al2O3 column. Tetrahydrofuran (THF; Sigma-Aldrich, 99.9%), N,N-dimethylformamide (DMF; Fluka, 99.8%), toluene (Sigma-Aldrich, 99.9%), and triethylamine (TEA; Sigma-Aldrich, 99%) were dried over CaH2, and distilled under nitrogen flow. Diethyl ether (Sigma-Aldrich, 99.5%) was kept over molecular sieves. Tin octoate (Sn(Oct)2; Sigma, ∼95%) and 6-bromo-1-hexanol (Aldrich, 97%) were distilled under reduced pressure. Methanol (Sigma-Aldrich, 99.9%), heptane (Sigma-Aldrich, 99%), dichloromethane (DCM; Sigma-Aldrich, 99.8%), anisole (Aldrich, 99%), 5-hexyn-1-ol (Aldrich, 96%), L-lysine (Fluka, purum; crystallized; ≥98%), di-tert-butyl dicarbonate (Aldrich, 97%), N,N′-dicyclohexylcarbodiimide (DCC; Aldrich, 99%), 3-bromo-1,2-propanediol (Fluka, ≥96%), methacrylic acid (MA; Aldrich, 99%), diethyl azodicarboxylate (DEAD; Aldrich), triphenylphosphine (TPP; Fluka, ∼97%), sodium azide (Aldrich, 99%), 17α-ethynylestradiol (Sigma, 98%), ethynylferrocene (Aldrich, 97%), CuI (Aldrich, 98%), CuBr (Aldrich, 98%), 1,1,4,7,10,10-hexamethyltriethylenetetramine (HMTETA; Aldrich, 97%), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA; Aldrich, 99%), ethyl α-bromoisobutyrate (EBiB; Aldrich, 98%), and trifluoroacetic acid (TFA; Sigma-Aldrich, ≥98%) were used as received.Characterization by NMR spectroscopy was conducted on a Bruker 300 MHz spectrometer using CDCl3 or DMSO-d6 as solvents (both from Aldrich). The coupling constants are given in Hz. Molecular weights and polydispersity indices were estimated by size exclusion chromatography (SEC) using the following instruments: (a) Viscotek 200 instrument using two PLgel mixed-D columns (Polymer Laboratories (PL)), assembled in series, and a refractive index (RI) detector. SEC samples were run in THF at room temperature (1 mL min−1). (b) Viscotek GPCmax VE-2001 equipped with Viscotek TriSEC Model 302 triple detector array (RI detector, viscometer detector, and laser light scattering detector with the light wavelength of 670 nm, and measuring angles of 90° and 7°) and a Knauer K-2501 UV detector using two PLgel mixed-D columns (from PL). The samples were run in THF at 30 °C (1 mL min−1). Molecular weights were calculated using PS standards from PL. Thermogravimetric analysis (TGA) was carried out on a TGA Q500 instrument from 20–600 °C with a heating rate of 20 °C min−1 under nitrogen flow. Transmission electron microscopy (TEM) images have been acquired using Tecnai T20 instrument operated at 200 kV. The samples for TEM were prepared by evaporating droplets of the dilute solutions of the CCS polymers in THF, filtered through 0.45 μm Teflon filters, on lacey carbon coated nickel grids.
Synthesis
All reactions were carried out under nitrogen atmosphere.1 and 2 have been prepared according to the procedures described in ref. 21.
1: SECPSS: Mn = 4300 Da, Mw = 4700 Da, Mw/Mn = 1.09. Degree of polymerization (DP) has been estimated by 1H NMR to be 21, Mn = 2560 Da.
δ
H (300 MHz; CDCl3) 6.09 (1H, m, HCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CCH3), 5.55 (1H, m, HCHCCH3), 3.95–4.17 (44H, t, J1,3 6.7, (CH2)4CH2OC(O) and C(O)OCH2(CH2)3C
CCH3), 5.55 (1H, m, HCHCCH3), 3.95–4.17 (44H, t, J1,3 6.7, (CH2)4CH2OC(O) and C(O)OCH2(CH2)3C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH), 2.28 (t, J1,3 7.4, OC(O)CH2(CH2)4), 2.21 (m, CH2CCH) (2.16–2.42 corresponds to 44H), 1.89–2.00 (4H, m, CH2CCH, C(CH3)CH2), 1.50–1.89 (88H, m, OC(O)CH2CH2CH2CH2CH2 and C(O)OCH2CH2CH2CH2CCH), 1.20–1.50 (42H, m, OC(O)CH2CH2CH2CH2CH2).
CH), 2.28 (t, J1,3 7.4, OC(O)CH2(CH2)4), 2.21 (m, CH2CCH) (2.16–2.42 corresponds to 44H), 1.89–2.00 (4H, m, CH2CCH, C(CH3)CH2), 1.50–1.89 (88H, m, OC(O)CH2CH2CH2CH2CH2 and C(O)OCH2CH2CH2CH2CCH), 1.20–1.50 (42H, m, OC(O)CH2CH2CH2CH2CH2).
3: Cu(I) catalyzed “click” coupling of 1 and 2 has been carried out as reported in ref. 21. SECLS: Mn = 3200 Da, Mw = 3300 Da, Mw/Mn = 1.02. Mn(1H NMR) = 3340 Da.
δ
H (300 MHz; CDCl3) 7.40–7.80 (1H, br s, Ar), 6.07 (1H, m, HCHCCH3), 5.53 (1H, m, HCHCCH3), 5.10–5.50 (br, CHNHBoc and OCH2CHO), 3.93–5.00 (br m, CH2NHBoc, OCH2CHO, CHNHBoc, CH2Ar, (CH2)4CH2OC(O) and C(O)OCH2(CH2)3Ar) (3.93–5.50 corresponds to 55H), 2.97–3.23 (4H, br, CH2NHBoc), 2.60–2.86 (2H, br, C(O)O(CH2)3CH2Ar), 2.29 (42H, t, J1,3 7.5, OC(O)CH2(CH2)4), 1.92 (m, C(CH3)CH2), 1.53–1.97 (m, CH2CH2CHNHBoc, OC(O)CH2CH2CH2CH2CH2 and C(O)OCH2CH2CH2CH2Ar), 1.05–1.60 (m, BocNHCH2CH2CH2CH2CHNHBoc, C(CH3)3 and OC(O)CH2CH2CH2CH2CH2) (1.05–1.97 corresponds to 181H).
4: ROP of ε-CL initiated by 6-bromo-1-hexanol has been conducted as in ref. 22. SECPSS: 4m: Mn = 4300 Da, Mw = 4700 Da, Mw/Mn = 1.09; 4k: Mn = 3800 Da, Mw = 4200 Da, Mw/Mn = 1.09; 4p: Mn = 6000 Da, Mw = 6600 Da, Mw/Mn = 1.09. From 1H NMR the DP and number average molecular weight: m = 29, Mn = 3490 Da; k = 22, Mn = 2690 Da; p = 41, Mn = 4860 Da.
δ H (300 MHz; CDCl3) 4.04 (58H, t, J1,3 6.6, (CH2)4CH2OC(O) and C(O)OCH2(CH2)5Br), 3.62 (2H, t, J1,3 6.4, (CH2)4CH2OH), 3.39 (2H, t, J1,3 6.7, CH2Br), 2.29 (58H, t, J1,3 7.4, OC(O)CH2(CH2)4), 1.85 (2H, m, CH2CH2Br), 1.50–1.75 (118H, m, OC(O)CH2CH2CH2CH2CH2 and CH2CH2(CH2)4Br), 1.25–1.50 (62H, m, OC(O)CH2CH2CH2CH2CH2 and (CH2)2CH2CH2(CH2)2Br).
5: Mitsunobu coupling of 4 and methacrylic acid has been accomplished following the synthetic protocol in ref. 22. The esterified product has been precipitated twice from THF into cold MeOH to eliminate residue of TPP. SECPSS: Mn = 4600 Da, Mw = 5000 Da, Mw/Mn = 1.09.
δ
H (300 MHz; CDCl3) 6.08 (1H, m, HCHCCH3), 5.54 (1H, m, HCHCCH3), 4.13 (2H, m, CH2OC(O)C(CH3)CH2), 4.05 (58H, t, J1,3 6.6, (CH2)4CH2OC(O) and C(O)OCH2(CH2)5Br), 3.40 (2H, t, J1,3 6.7, CH2Br), 2.29 (58H, t, J1,3 7.5, OC(O)CH2(CH2)4), 1.93 (3H, m, OC(O)CH3CCH2), 1.85 (2H, m, CH2CH2Br), 1.50–1.78 (118H, m, OC(O)CH2CH2CH2CH2CH2 and CH2CH2(CH2)4Br), 1.25–1.50 (62H, m, OC(O)CH2CH2CH2CH2CH2 and (CH2)2CH2CH2(CH2)2Br).
6: 5 (2.54 g, 0.71 mmol), sodium azide (0.135 g, 2.07 mmol), and DMF (25 mL) were placed in a 50 mL round-bottom flask equipped with a magnetic stirring bar. The reaction was carried out at room temperature for 15 h. The product was precipitated into cold MeOH–H2O (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture, isolated by filtration, and dried in a vacuum oven at room temperature until constant weight. SECPSS: Mn = 4800 Da, Mw = 5200 Da, Mw/Mn = 1.1.
:1) mixture, isolated by filtration, and dried in a vacuum oven at room temperature until constant weight. SECPSS: Mn = 4800 Da, Mw = 5200 Da, Mw/Mn = 1.1.
δ
H (300 MHz; CDCl3) 6.08 (1H, m, HCHCCH3), 5.54 (1H, m, HCHCCH3), 4.13 (2H, m, CH2OC(O)C(CH3)CH2), 4.04 (58H, t, J1,3 6.6, (CH2)4CH2OC(O) and C(O)OCH2(CH2)5N3), 3.27 (2H, t, J1,3 6.8, CH2N3), 2.29 (58H, t, J1,3 7.4, OC(O)CH2(CH2)4), 1.93 (3H, m, OC(O)CH3CCH2), 1.50–1.78 (120H, m, OC(O)CH2CH2CH2CH2CH2, CH2CH2N3 and CH2CH2(CH2)4N3), 1.22–1.50 (62H, m, OC(O)CH2CH2CH2CH2CH2 and (CH2)2CH2CH2(CH2)2N3).
7: A 50 mL two-neck flask was charged with 6 (0.80 g, 0.23 mmol), 17α-ethynylestradiol (0.20 g, 0.67 mmol), CuI (0.042 g, 0.22 mmol), and THF (7.5 mL). The reaction mixture was stirred at room temperate until the polymer dissolved completely. Then TEA (0.13 mL, 0.93 mmol) was introduced. The reaction was conducted at 35 °C for 24 h in the dark. Then, the macromonomer was precipitated into a large excess of cold MeOH. The product was isolated by filtration, and dried in vacuo at room temperature until constant weight. SECPSS: Mn = 5100 Da, Mw = 5600 Da, Mw/Mn = 1.1.
δ
H (300 MHz; CDCl3) 7.41 (1H, s, Tr), 7.04 (1H, d, Jo 7.6, Ar), 6.55 (2H, br s, Ar), 6.08 (1H, s, HCHCCH3), 5.54 (1H, m, HCHCCH3), 5.35 (1H, br s, Ar–OH), 4.38 (2H, m, CH2Tr), 4.14 (2H, t, J1,3 6.6, CH2OC(O)C(CH3)CH2), 4.05 (58H, t, J1,3 6.7, (CH2)4CH2OC(O) and C(O)OCH2(CH2)5Tr), 2.80 (2H, m, CH2Ar), 2.30 (58H, t, J1,3 7.5, OC(O)CH2(CH2)4), 1.93 (3H, m, OC(O)CH3CCH2), 1.50–1.78 (m, OC(O)CH2CH2CH2CH2CH2 and CH2CH2CH2CH2CH2CH2Tr), 1.22–1.50 (m, OC(O)CH2CH2CH2CH2CH2 and (CH2)2CH2CH2(CH2)2Tr), 1.20–2.70 (signals from estradiol residue overlap with the resonances originating from the polymer backbone, and thus cannot be unambiguously assigned), 1.04 (3H, s, CH3C).
8: The synthetic protocol is same as for the preparation of 7. 6 (1.00 g, 0.28 mmol), ethynylferrocene (0.18 g, 0.86 mmol), CuI (0.055 g, 0.29 mmol), TEA (0.16 mL, 1.15 mmol) and THF (9.5 mL) were employed. SECPSS: Mn = 5200 Da, Mw = 5700 Da, Mw/Mn = 1.1.
δ
H (300 MHz; CDCl3) 7.44 (1H, s, Tr), 6.08 (1H, s, HCHCCH3), 5.54 (1H, m, HCHCCH3), 4.71 (2H, m, Cp), 4.36 (2H, m, CH2Tr), 4.28 (2H, m, Cp), 4.13 (2H, m, CH2OC(O)C(CH3)CH2), 4.05 (58H, t, J1,3 6.6, (CH2)4CH2OC(O) and C(O)OCH2(CH2)5Tr), 2.30 (58H, t, J1,3 7.5, OC(O)CH2(CH2)4), 1.93 (3H, s, OC(O)CH3CCH2), 1.50–1.78 (120H, m, OC(O)CH2CH2CH2CH2CH2 and CH2CH2CH2CH2CH2CH2Tr), 1.22–1.50 (62H, m, OC(O)CH2CH2CH2CH2CH2 and (CH2)2CH2CH2(CH2)2Tr).
Preparation of CCS polymers
:1) mixture at room temperature. The precipitate quickly sedimented, and the solvent mixture was removed by pipette. The product was isolated on filter paper, and washed with the fresh solvent mixture several times while applying vacuum with a water aspirator. It was then dried in a vacuum oven at room temperature until constant weight.
9: 6k (0.20 g, 0.074 mmol), CuBr (0.023 g, 0.16 mmol), toluene (5 mL), BDMA (0.25 mL, 1.13 mmol), HMTETA (90 μL, 0.33 mmol), and EBiB (16 μL, 0.11 mmol) were employed. The reaction was carried out for 72 h. SECLS: Mn = 1.13 × 106 Da, Mw = 1.61 × 106 Da, Mw/Mn = 1.43.
10: 3 (0.25 g, 0.075 mmol), 6p (0.51 g, 0.10 mmol), CuBr (0.059 g, 0.41 mmol), toluene (12 mL), BDMA (0.6 mL, 2.71 mmol), HMTETA (0.22 mL, 0.81 mmol), and EBiB (40 μL, 0.27 mmol) were employed. The reaction was carried out for 22.5 h. SECLS: Mn = 1.30 × 106 Da, Mw = 2.00 × 106 Da, Mw/Mn = 1.55.
11: A 50 mL two-neck flask was charged with 10 (0.2 g, 0.024 mmol of azide functional groups), ethynylferrocene (0.016 g, 0.076 mmol), CuI (0.015 g, 0.079 mmol), TEA (0.02 mL, 0.14 mmol) and THF (10.5 mL). The resulting slurry was stirred at 64 °C for 24 h. Then the product was precipitated into large excess of cold heptane. The solvent was decanted and the product was washed with heptane twice. It was then isolated on filter paper, and washed with the fresh solvent several times while applying vacuum with water aspirator. The product was dried in a vacuum oven at room temperature.
12: 3 (0.12 g, 0.036 mmol), 7 (0.25 g, 0.065 mmol), CuBr (0.034 g, 0.24 mmol), toluene (13.5 mL), BDMA (0.33 mL, 1.49 mmol), HMTETA (0.13 mL, 0.48 mmol), and EBiB (23 μL, 0.16 mmol) were employed. The reaction was carried out for 20.25 h. SECLS: Mn = 2.06 × 105 Da, Mw = 2.65 × 105 Da, Mw/Mn = 1.29.
13: 3 (0.12 g, 0.036 mmol), 8 (0.25 g, 0.067 mmol), CuBr (0.034 g, 0.24 mmol), toluene (13.5 mL), BDMA (0.35 mL, 1.58 mmol), HMTETA (0.13 mL, 0.48 mmol), and EBiB (23 μL, 0.16 mmol) were employed. The reaction was carried out for 16.5 h. SECLS: Mn = 8.5 × 104 Da, Mw = 1.16 × 105 Da, Mw/Mn = 1.37.
14: CCS polymer 12 (0.069 g) and DCM (3 mL) were placed in a 25 mL two-neck flask equipped with a magnetic stirring bar. The reaction mixture was stirred in the dark at room temperature until the polymer dissolved completely. The flask was immersed into an ice/water bath, and TFA (0.6 mL) was injected. The mixture was then stirred in the dark for 15 min on the ice/water bath, and for 2.5 h at room temperature. The product was recovered by precipitating into a large excess of cold heptane-Et2O (2:1) and isolating on filter paper. The polymer was washed several times with the fresh solvent mixture, and dried in a vacuum oven at room temperature.
Results and discussions
The linear-dendritic macromonomer (di-Boc-L-lysine)G2-b-PCL-methacrylate (MM 3) has been prepared according to the synthetic cascade depicted in Scheme 1.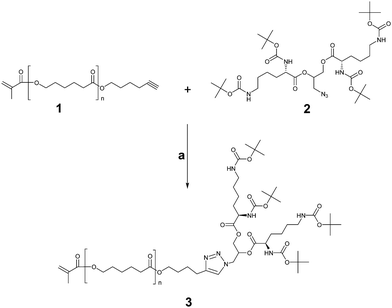 | ||
| Scheme 1 Synthetic pathway for preparation of linear-dendritic macromonomer. Reagents and conditions: (a) CuI/NEt3, 35 °C, THF. | ||
The synthetic procedures follow the algorithm which we have reported earlier.21 To the best of our knowledge, the macromonomer thus obtained has not been documented before. This synthetic layout bears significant benefits of exhaustive characterization of the individual building blocks 1 and 2. The Cu(I) catalyzed “click” coupling23 affords MM 3 with narrow molecular weight distribution, which implies that the goal product is obtained without any considerable side reactions. The 1H NMR spectroscopy confirms near to quantitative reaction (Fig. 1). The resonance signal originating from the methylene group next to the alkyne function (i) is shifted downfield, while the peak attributed to the acetylenic hydrogen disappears completely. The resonances o, p, and q that correspond to the methacrylic acid residue verify the preservation of the vinyl double bond which is the prerequisite for effectual participation of the macromonomer in subsequent polymerization reactions. The resonance h has been ascribed to the triazole proton. The heteronuclear single quantum coherence (HSQC) NMR experiment has revealed that h is the combination of two resonance signals that may be attributed to two regioisomers resulting from the “click” reaction. Formation of two regioisomers is not uncommon when conducting the reaction in THF.24 The number average molecular weight has been estimated by 1H NMR to be 3340 Da.
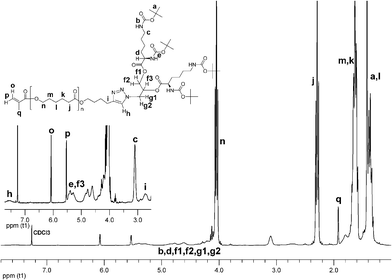 | ||
| Fig. 1 1H NMR spectrum of MM 3. | ||
Linear MMs N3-PCL-methacrylate (MM 6), E2-PCL-methacrylate (MM 7), and ferrocene-PCL-methacrylate (MM 8) have been synthesized according to Scheme 2. Synthetic routes leading to azide functional PCL include nucleophilic substitution of bromo end group of Br-PCL-OH by sodium azide25 and ROP of ε-CL using azide containing initiator.26 Since the latter carries the risk of reducing the azide functions by TPP in subsequent Mitsunobu coupling of N3-PCL-OH and MA,25 we follow the path involving the conversion of bromo end groups to corresponding azides after the esterification reaction (Scheme 2).
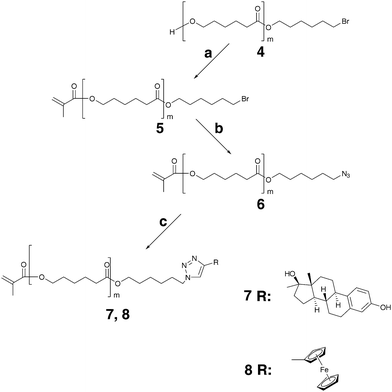 | ||
| Scheme 2 Synthetic pathway for preparation of linear macromonomers. Reagents and conditions: (a) MA, DEAD, TPP, THF; (b) NaN3, DMF; (c) 17α-ethynylestradiol or ethynylferrocene, CuI/NEt3, 35 °C, THF. | ||
ROP of ε-CL initiated by diethylaluminium 2-bromoethoxide and diethylaluminium 12-bromo-1-dodecyloxide in toluene at 25 °C has been carried out with excellent control.25 In a similar approach, we initially conducted the polymerization reaction in THF at 62 °C employing 2-bromoethanol and Sn(Oct)2 as initiator and catalyst, respectively. However, poor control over the reaction, translated into relatively high polydispersity index (PDI) of 1.24 and unsymmetrical SEC trace, was observed. The reaction rate was higher than in case of earlier reactions when different initiators were used.21,22 This may possibly be due to the inductive effects arising from the close proximity of Br and O in 2-bromoethanol. Indeed, when 6-bromo-1-hexanol was chosen as the initiator, the polymerization proceeded smoothly with SEC revealing monomodal, symmetrical trace and low PDI of 1.09. Br-PCL-OH with DP of 22, 29, and 41 has been obtained.
Esterification of the hydroxy chain end of 4 with MA was accomplished by Mitsunobu protocol as described earlier.22 Near to quantitative functionalization was attained as judged from 1H NMR data: resonance signal at 3.62 ppm corresponding to the methylene next to the hydroxy end group shifted completely at 4.13 ppm. After two-fold precipitation in MeOH, the product was free from residual TPP. This is important since TPP may lead to the reduction of azide groups in the reaction concomitant to the nucleophilic substitution of bromo chain ends by sodium azide. PDI remained same indicating preservation of the polyester backbone.
Conversion of bromo end groups into azide functions was first conducted as suggested by Teyssié and co-workers:25 Br-PCL-methacrylate 5 was reacted with 5 equivalents of NaN3 in DMF at 30 °C. The SEC chromatogram of the substituted product exhibited a small shoulder on the high molecular weight side. We reckoned that the appearance of the shoulder could be the result of the nucleophilic attack of the azide anion on the carbonyl carbons of the PCL backbone. Therefore, in the next trail 3 equivalents of NaN3 were utilized at room temperature in DMF. The shoulder, though diminished in intensity, was still present (PDI = 1.1). The NMR experiments suggested that the extent of the reaction was near to quantitative, and no fathomable side products were identified. This gave birth to the speculation that the azide groups were reacting with the methacrylic units even at room temperature. To ascertain or eliminate such a conjecture, NaN3 was reacted with Br-PCL-OH under identical conditions; the shoulder was no longer detected. Furthermore, MM 6 displayed a limited shelf-life at room temperature. After several days SEC trace with bimodal distribution was acquired. Triazolines, formed from 1,3-cycloaddition, were identified after careful investigation by 1H, COSY, and HSQC NMR experiments. Despite earlier reports regarding relative stability of methacrylic monomers bearing pendant azide functions at room temperature, which was attributed to the steric hindrance,27 our findings made it obvious that the uncatalyzed cycloaddition took place during the substitution reaction, though the share of this reaction was negligible, and then proceeded throughout the matrix of the isolated MM. This result is somewhat baffling if taken into consideration fairly low local concentration of the end functionalities in case of the MM with the molecular weight of almost 5 000 Da.
Cu(I) catalyzed azide-alkyne cycloaddition (CuAAC) between 6 and 17α-ethynylestradiol or ethynylferrocene28 provides MM 7 and MM 8, respectively (Scheme 2). To the best of our knowledge, participation of the azide end-capped PCL in CuAAC has been restricted to the synthesis of cyclic PCL.26,29 Neither has 17α-ethynylestradiol enjoyed extensive application in CuAAC not to mention two fairly recent reports about the synthesis of E2-peptoid30a and E2-cyclodextrin30b conjugates. Hence, MM 7 represents a novel macromolecular architecture – hormone-synthetic polymer conjugate – built via CuAAC and employed in ATRP. 6 and 17α-ethynylestradiol or ethynylferrocene were reacted under similar conditions as 1 and 2. However, 3 equivalents of the alkyne were taken to ensure complete conversion, and to avoid involvement of the methacrylic moieties in undesirable collateral reactions discussed above. Examination by SEC did not detect any chain scission or triazoline formation: PDI remained low (1.1), and the intensity of the shoulder did not increase. The 1H NMR spectrum of MM 7 displays all the characteristic resonances (Fig. 2).
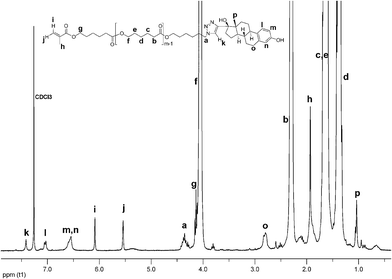 | ||
| Fig. 2 1H NMR spectrum of MM 7. | ||
The peak at 3.27 ppm arising from the methylene group next to the azide function has disappeared completely, and the new peak at 4.38 ppm attributed to the methylene group next to the triazole ring has emerged. This implies that the coupling was near to quantitative. Furthermore, resonances at 6.08 ppm, 5.54 ppm, and 1.93 ppm correspond to the methacrylic acid residue, and confirm the maintenance of the double bond. The triazole proton resonates at 7.41 ppm, while the signals designated as l, m, n, o, and p are ascribed to the E2 moiety. Investigation of MM 8 by SEC and NMR also rendered excellent results. While SEC trace convinced in the absence of chain scrambling and azide-methacrylate cycloaddition, the NMR experiment suggested almost complete functionalization (Fig. 3).
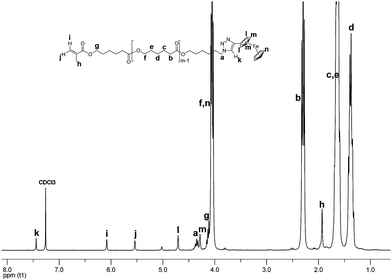 | ||
| Fig. 3 1H NMR spectrum of MM 8. | ||
Still oblivious of the fact that methacrylic double bond could react with azide end group even at room temperature; we attempted to prepare the first type of CCS polymer comprised solely of the MM 6. CCS polymers with peripheral alkyne functional groups have been reported earlier.31 If successful, our approach would generate aggregates with azide functionalities on the surface. Thus, MM 6k with the DP of 22 was cross-linked with BDMA via ATRP initiated by EBiB (Scheme 3).
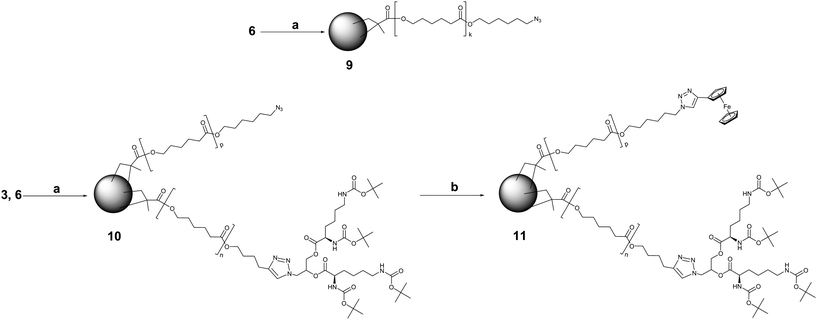 | ||
| Scheme 3 Synthesis and derivatization of CCS polymers possessing peripheral azide functions. Reagents and conditions: (a) BDMA, CuBr/HMTETA, EBiB, 50 °C, toluene; (b) ethynylferrocene, CuI/NEt3, 64 °C, THF. | ||
PCL MMs have been scarcely employed in controlled radical polymerization techniques.32 Therefore, it was necessary to adjust the reaction conditions in order to increase the MM conversion while maintaining narrow MWD. The MM conversion was roughly estimated from the peak intensities of RI detector signal on the SEC chromatogram. Initially the reaction was conducted in anisole, taking into consideration better solubility of copper(II) complex in polar solvents,4 at 50 °C employing CuBr/PMDETA catalytic complex and low initiator to MM molar ratio [MM 6k]0:[BDMA]0:[CuBr]0:[PMDETA]0:[EBiB]0 = 1:3:0.15:0.15:0.1. Only 3 equivalents of the cross-linker (BDMA) were employed to avoid premature gelation of the reaction mixture. Low MM conversion was achieved in a few hours, and it did not increase by extending the reaction time. This was ascribed to the rapid blocking of the initiating sites due to the core congestion, loss of the active alkyl bromide groups which has significant detrimental impact at such a low concentration of the initiating moieties, and catalyst deactivation since characteristic color change to bright green was observed. Since long reaction times encourage star-star couplings resulting in high PDI, it was desirable to increase the MM incorporation at the early stage of the polymerization. We improved the reaction conditions by changing the reaction medium to toluene in order to enhance the solubility of PCL, replacing PMDETA with HMTETA to augment the stability of the catalytic complex, and using the following initial molar ratios: [MM 6k]0:[BDMA]0:[CuBr]0:[HMTETA]0:[EBiB]0 = 1:15:2.25:4.5:1.5.
The large excess of the cross-linker, favoring gelation, was compensated by diluting the reaction mixture. Utilizing 1.5 equivalents of EBiB is in dissonance with the scheme proposed by Matyjaszewski suggesting decreasing the number of initiating groups to minimize the extent of star-star coupling.7,8 Thus, we anticipated reaching reasonable MM conversion on the expense of the MWD: we simply tried to escape wasting of the MMs 3, 6, 7, and 8, because their synthesis involves multiple steps. The CCS polymer was analyzed by SEC and NMR spectroscopy. The molecular weight characteristics were determined using light scattering detector. While PDI was fairly narrow (1.43), weight average molecular weight Mw was estimated to be 1.61 × 106 Da. Such a large value for Mw has not been reported before.3–5,7–12 We attribute the formation of such large CCS polymer to the star-star coupling throughout ATRP facilitated by relatively high concentration of the initiating sites, the cross-linker with soft aliphatic spacer,33 and azide-methacrylate cycloaddition reaction perceived at the later stage of our experiments. The 1H NMR spectroscopy data suggested the star formation, and preservation of the certain amount of peripheral azide functions (Fig. 4).
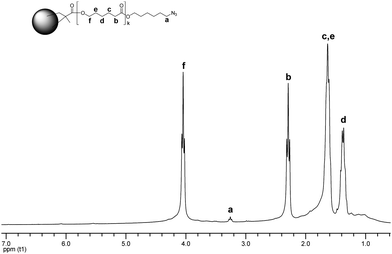 | ||
| Fig. 4 1H NMR spectrum of CCS 9. | ||
The resonances from the microgel core overlap with those from the PCL arms, and result in slight broadening at the baseline. The resonance signals arising from the unreacted double bonds are barely detected. The peak a corresponding to the methylene group next to the azide function of the arms has also been assigned.
The miktoarm CCS 10 was then prepared under similar reaction conditions: the mixture of the linear-dendritic MM 3 and the linear MM 6p was cross-linked with BDMA. The structure of the CCS polymer was elucidated by NMR spectroscopy. The 1H NMR experiment verified incorporation of both arms into the CCS (Fig. 5). Again, almost no unreacted methacrylic units were observed; resonances from the core were barely discernible. Signals from the methylene group next to the azide functionality (c), the methylene group adjacent to the Boc protected amine functions (b), and methyl groups of the Boc protection (a) have been identified together with the peaks characteristic to the PCL backbone.
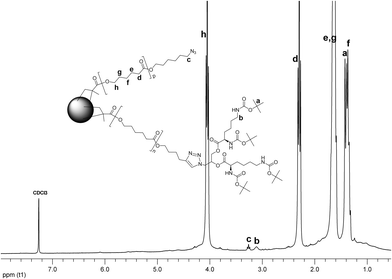 | ||
| Fig. 5 1H NMR spectrum of CCS 10. | ||
SEC trace was not symmetrical (Fig. 6), however it suggested that precipitation afforded the product free from the unreacted MMs. PDI of 1.55 and Mw of 2.00 × 106 Da were obtained using the light scattering detector and THF as eluent.
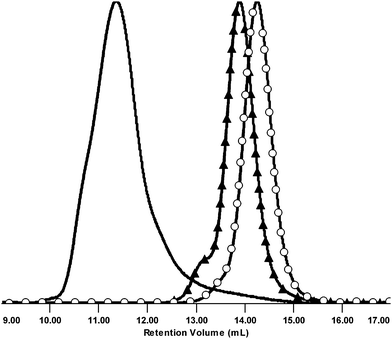 | ||
| Fig. 6 Normalized SEC RI detector signals of MM 3 (○), MM 6p (▲), and CCS 10. | ||
However, upon standing a few days, the CCS 10 was only partially soluble in THF and other common organic solvents. This implies that certain type of cross-linking reaction persisted in the isolated product. At this point the analysis of MM 6 unveiled azide-methacrylate cycloaddition. This reaction, provoking the star-star coupling, is responsible for such a large Mw, broad MWD, and loss of a certain amount of azide end groups. These results opted us for capping the MM 6 with the desirable moiety by CuAAC before employing it in ATRP with BDMA. Nevertheless, we decided to derivatize remaining accessible azide functions of CCS 10 with ethynylferrocene by conducting the CuAAC in a heterogeneous mixture (Scheme 3). Ferrocene would assume the role of stain in TEM investigations.
Three-fold excess of ethynylferrocene and CuI were used. The product was analyzed by NMR spectroscopy. The result, though inconclusive due to limited solubility of the CCS 11 in CDCl3, suggested that available azide groups had reacted since the resonance c (Fig. 5) disappeared, and peaks related to ferrocene were detected. The heterogeneous mixture of the CCS 11 in THF was filtered twice through 0.45 μm Teflon filter to remove the agglomerates of large sizes, and the nanoparticles in the filtrate were visualized by TEM (Fig. 7). Clusters of the nanoparticles in the range of 10–20 nm were observed. Clustering may be ascribed to the lack of repulsive interactions amongst the coronas of CCS copolymers. Evidently, ferrocene did not impart significant contrast.
 | ||
| Fig. 7 TEM image of CCS 11 obtained after removal of the insoluble matter. | ||
Being confronted by the destructive interference of azide-methacrylate cycloaddition with the cross-linking reaction, we utilized MMs 3, 7 and 8 in preparation of miktoarm CCS copolymers as shown in Scheme 4. Initially, we adopted the reaction conditions used for the cross-linking of azide end capped MMs.
 | ||
| Scheme 4 Synthesis and deprotection of miktoarm CCS copolymers bearing surficial E2 and ferrocene. Reagents and conditions: (a) BDMA, CuBr/HMTETA, EBiB, 50 °C, toluene; (b) TFA, CH2Cl2. | ||
As a result, ATRP of the MM 3, MM 7, and BDMA produced insoluble gel. Dilution of the reaction mixture was the only adjustment necessary to attain the formation of the goal product CCS 12. The molecular characteristics estimated by SEC (Fig. 8) include Mw = 2.65 × 105 Da and PDI = 1.29 (both were determined using the light scattering detector). The SEC analysis revealed that the product did not contain unreacted MMs. Fairly narrow MWD suggests that the ATRP was under control, and that the contribution from the star-star coupling was reduced significantly.
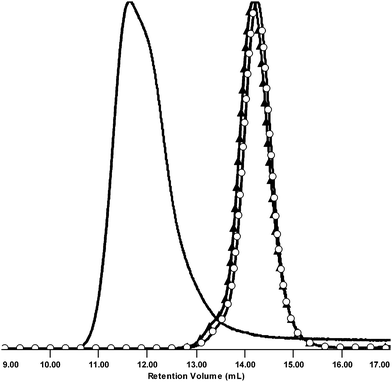 | ||
| Fig. 8 Normalized SEC RI detector signals of MM 3 (○), MM 7 (▲), and CCS 12. | ||
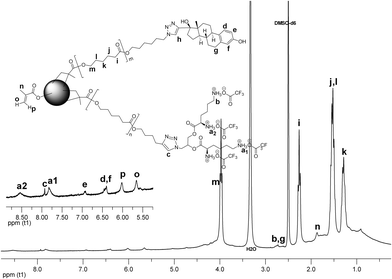
The structure of the CCS 12 was examined by 1H NMR spectroscopy (Fig. 9). The peaks corresponding to the PCL backbone overlap with the broad signals arising from the microgel core. This implies that the segmental mobility of the core has not been arrested completely. The resonances ascribed to the methyl group of the Boc protection (a), the methylene group neighboring the Boc protected amino group (b), and the triazole protons (c) of the MM 3 have been assigned. The MM 7 is represented by the signals attributed to the E2 moiety (d, e, f, g), and the triazole proton (h). Protons from the unreacted methacrylic units of the core resonate at 5.56 ppm and 6.09 ppm. Since the intensities of the signals originating from the L-lysine and E2 moieties are rather weak, the composition of the CCS 12 cannot be unambiguously estimated from the NMR data.
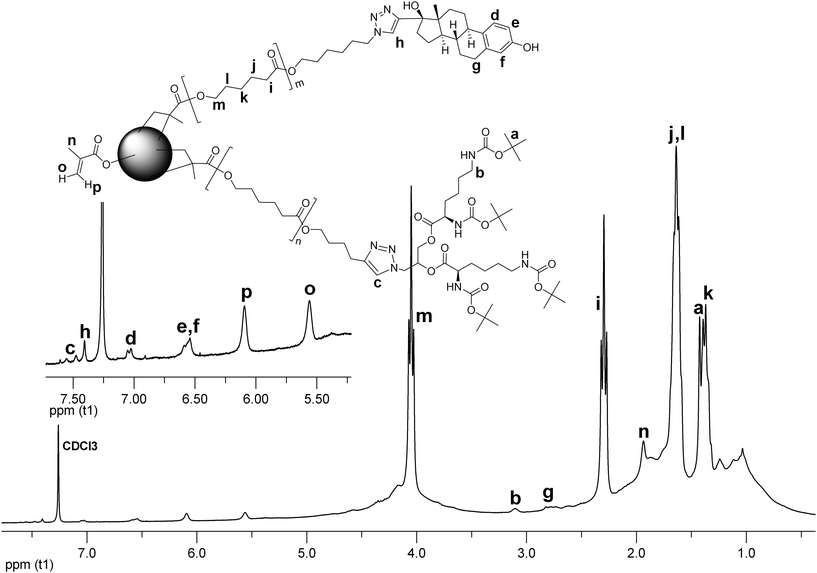 | ||
| Fig. 9 1H NMR spectrum of CCS 12. | ||
Therefore, TGA has been employed to gain insight about the ratio and the number of arms of the MMs. The overlay of TGA thermograms (Fig. 10) allows estimation of the weight fraction of the MM 3 as follows: the weight loss of 3.7% between 222 °C and 272 °C is attributed to the decomposition of the Boc protection groups, and corresponds to approximately 31% of the MM 3. About 26% weight loss in the temperature range 424–516 °C is ascribed to the decomposition of the core. Hence, the weight fraction of the MM 6 is roughly 43%. Obviously these results are crude estimates; nonetheless, they shed some light over the star composition. By making use of the following equation: 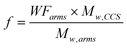 , where WFarms is the weight fraction of the arms determined by TGA, and Mw,CCS and Mw,arms are the weight average molecular weights of the CCS copolymer and the arms, respectively, it can be calculated that the CCS 12 consists of on average 24 (di-Boc-L-lysine)G2-b-PCL and 27 E2-PCL arms. The molar ratio of the incorporated arms is [E2-PCL]/[(di-Boc-L-lysine)G2-b-PCL] = 1.2, whereas in the initial mixture [MM 7]0/[MM 3]0 = 1.8. This rift between the initial and final compositions may be explained by higher reactivity of the MM 3 as a consequence of its lower molecular weight.11
, where WFarms is the weight fraction of the arms determined by TGA, and Mw,CCS and Mw,arms are the weight average molecular weights of the CCS copolymer and the arms, respectively, it can be calculated that the CCS 12 consists of on average 24 (di-Boc-L-lysine)G2-b-PCL and 27 E2-PCL arms. The molar ratio of the incorporated arms is [E2-PCL]/[(di-Boc-L-lysine)G2-b-PCL] = 1.2, whereas in the initial mixture [MM 7]0/[MM 3]0 = 1.8. This rift between the initial and final compositions may be explained by higher reactivity of the MM 3 as a consequence of its lower molecular weight.11
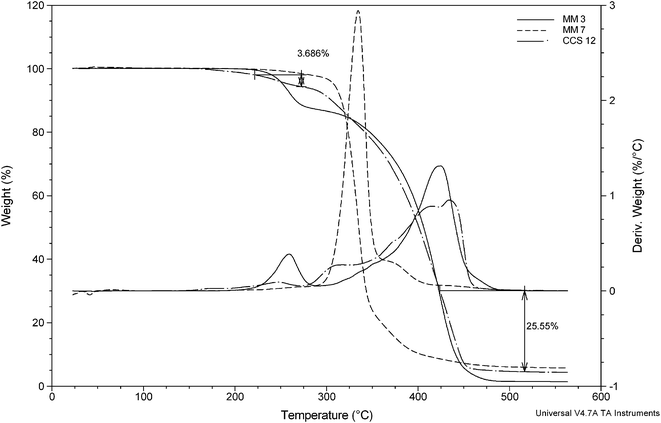 | ||
| Fig. 10 TGA thermograms of MM 3, MM 7, and CCS 12. | ||
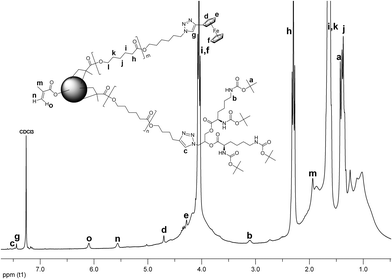
Deblocking of the amine functional groups of the L-lysine dendritic wedges was accomplished by treating the CCS 12 with TFA21 as shown in Scheme 4. The product CCS 14 was analyzed with 1H NMR spectroscopy using DMSO-d6 as solvent (Fig. 11). The resonance a arising from the tert-butyl groups disappeared completely (a refers to Fig. 9), while the broad peaks a1 and a2 related to free amine functions were detected. Moreover, the peak b has shifted upfield, and is now overlapped with the peak g ascribed to the E2 residue. Other resonances originating from the E2 unit have been identified as well. The signals from the core are somewhat weaker, perhaps due to the limited solubility of the interior of the CCS 14 in as polar a solvent as DMSO-d6.
 | ||
| Fig. 11 1H NMR spectrum of CCS 14. | ||
After successful preparation of the CCS 12, MM 3 and MM 8 were subjected to cross-linking with BDMA under similar conditions. Investigation of the product CCS 13 by 1H NMR spectroscopy validated integration of both arms in the star copolymer (Fig. 12).
 | ||
| Fig. 12 1H NMR spectrum of CCS 13. | ||
While the peaks c and g correspond to the triazole protons of MM 3 and MM 8 respectively, the signals a (tert-butyl) and b (CH2NHBoc) from the MM 3 and d and e (both related to Cp) from the MM 8 can be identified as well. SEC analysis (Fig. 13) provided the following characteristics: Mw = 1.16 × 105 Da and PDI = 1.37 (both values were obtained using the light scattering detector).
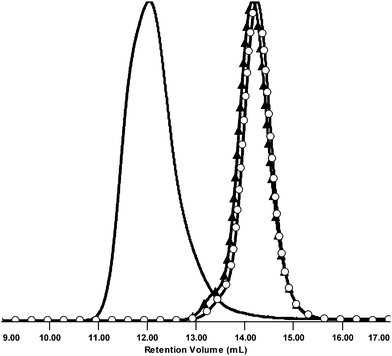 | ||
| Fig. 13 Normalized SEC RI detector signals of MM 3 (○), MM 8 (▲), and CCS 13. | ||
The composition of the CCS 13 was assessed employing the TGA. Taking the same approach as in the case of CCS 12, the following weight fractions have been estimated: 45% of the MM 3, 41% of the MM 8, and 14% of the core. Thus, the CCS 13 is comprised of 15 (di-Boc-L-lysine)G2-b-PCL and 12 ferrocene-PCL arms. TEM revealed clusters of the CCS 13 without additional staining (Fig. 14). However, low contrast and star-star overlaps do not allow precise evaluation of the size of these nanoparticles.
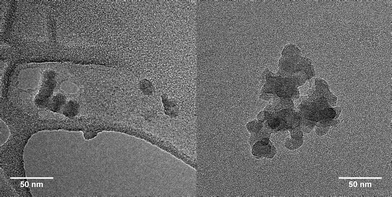 | ||
| Fig. 14 TEM images of CCS 13. | ||
Conclusions
In conclusion, we have synthesized miktoarm CCS copolymers consisting of linear-dendritic and linear arms. The cornerstone for preparation of the MMs is ROP of ε-CL by a functional initiator and quantitative functionalization of the hydroxyl chain end with methacrylic unit providing PCL MMs with terminal alkyne and azide groups. These hetero-telechelic building blocks elaborated by SEC and NMR spectroscopy exhibit narrow MWD and high degree of functionality, and represent flexible and versatile structural elements. A Cu(I) catalyzed “click” reaction has been employed as a robust and effectual tool for derivatization of these end functionalities with L-lysine dendron – bis-(di-Boc-L-lysine)-3-azido-1,2-propandiol, and 17α-ethynylestradiol and ethynylferrocene to obtain linear-dendritic and linear MMs, respectively. Cross-linking of the mixture of the MMs with BDMA through ATRP allows synthesis of the miktoarm CCS copolymers with fairly low PDI. Removal of the Boc protection affords novel nanoscale aggregates with hydrophilic, charged surface, targeting motifs, and hydrophobic core that may serve as excellent candidates for targeted drug delivery.Acknowledgements
The authors thank the Center for Electron Nanoscopy at Technical University of Denmark and Dr J. B. Wagner for acquiring the TEM images.Notes and references
- H. Gao and K. Matyjaszewski, Prog. Polym. Sci., 2009, 34, 317–350 CrossRef CAS.
- A. Blencowe, J. F. Tan, T. K. Goh and G. Qiao, Polymer, 2009, 50, 5–32 CrossRef CAS.
- J. Xia, X. Zhang and K. Matyjaszewski, Macromolecules, 1999, 32, 4482–4484 CrossRef CAS.
- X. Zhang, J. Xia and K. Matyjaszewski, Macromolecules, 2000, 33, 2340–2345 CrossRef CAS.
- H. Gao, N. V. Tsarevsky and K. Matyjaszewski, Macromolecules, 2005, 38, 5995–6004 CrossRef CAS.
- D. Kafouris, E. Themistou and C. S. Patrickios, Chem. Mater., 2006, 18, 85–93 CrossRef CAS.
- H. Gao, S. Ohno and K. Matyjaszewski, J. Am. Chem. Soc., 2006, 128, 15111–15113 CrossRef CAS.
- H. Gao and K. Matyjaszewski, Macromolecules, 2008, 41, 4250–4257 CrossRef CAS.
- J. Du and Y. Chen, Macromolecules, 2004, 37, 3588–3594 CrossRef CAS.
- J. T. Wiltshire and G. G. Qiao, Macromolecules, 2006, 39, 4282–4285 CrossRef CAS.
- J. T. Wiltshire and G. G. Qiao, Macromolecules, 2006, 39, 9018–9027 CrossRef CAS.
- J. T. Wiltshire and G. G. Qiao, Macromolecules, 2008, 41, 623–631 CrossRef CAS.
- A. W. Bosman, R. Vestberg, A. Heumann, J. M. J. Fréchet and C. J. Hawker, J. Am. Chem. Soc., 2003, 125, 715–728 CrossRef CAS.
- L. A. Connal, R. Vestberg, C. J. Hawker and G. G. Qiao, Macromolecules, 2007, 40, 7855–7863 CrossRef CAS.
- L. A. Connal, R. Vestberg, C. J. Hawker and G. G. Qiao, Adv. Funct. Mater., 2008, 18, 3706–3714 CrossRef CAS.
- F. Wang, T. K. Bronich, A. V. Kabanov, R. D. Rauh and J. Roovers, Bioconjugate Chem., 2005, 16, 397–405 CrossRef CAS.
- X. Shuai, T. Merdan, A. K. Schaper, F. Xi and T. Kissel, Bioconjugate Chem., 2004, 15, 441–448 CrossRef CAS.
- C. Gunanathan, A. Pais, E. Furman-Haran, D. Seger, E. Eyal, S. Mukhopadhyay, Y. Ben-David, G. Leitus, H. Cohen, A. Vilan, H. Degani and D. Milstein, Bioconjugate Chem., 2007, 18, 1361–1365 CrossRef CAS.
- E. C. Dreaden, S. C. Mwakwari, Q. H. Sodji, A. K. Oyelere and M. A. El-Sayed, Bioconjugate Chem., 2009, 20, 2247–2253 CrossRef CAS.
- K. T. Al-Jamal, W. T. Al-Jamal, S. Akerman, J. E. Podesta, A. Yilmazer, J. A. Turton, A. Bianco, N. Vargesson, C. Kanthou, A. T. Florence, G. M. Tozer and K. Kostarelos, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3966–3971 CrossRef.
- I. Javakhishvili, W. H. Binder, S. Tanner and S. Hvilsted, Polym. Chem., 2010, 1, 506–513 RSC.
- I. Javakhishvili and S. Hvilsted, Biomacromolecules, 2009, 10, 74–81 CrossRef CAS.
- (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS; (b) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS; (c) W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2008, 29, 952–981 CrossRef CAS.
- B. C. Englert, S. Bakbak and U. H. F. Bunz, Macromolecules, 2005, 38, 5868–5877 CrossRef CAS.
- Ph. Degée, Ph. Dubois, R. Jérôme and Ph. Teyssié, Macromolecules, 1992, 25, 4242–4248 CrossRef CAS.
- J. N. Hoskins and S. M. Grayson, Macromolecules, 2009, 42, 6406–6413 CrossRef CAS.
- (a) Y. Li, J. Yang and B. C. Benicewicz, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4300–4308 CrossRef CAS; (b) V. Ladmiral, T. M. Legge, Y. Zhao and S. Perrier, Macromolecules, 2008, 41, 6728–6732 CrossRef CAS; (c) G. Li, V. Zheng and R. Bai, Macromol. Rapid Commun., 2009, 30, 442–447 CrossRef CAS.
- J. P. Collman, N. K. Devaraj and C. E. D. Chidsey, Langmuir, 2004, 20, 1051–1053 CrossRef CAS.
- Ph. Lecomte, R. Riva, C. Jérôme and R. Jérôme, Macromol. Rapid Commun., 2008, 29, 982–997 CrossRef CAS.
- (a) J. M. Holub, M. J. Garabedian and K. Kirshenbaum, QSAR Comb. Sci., 2007, 26, 1175–1180 Search PubMed; (b) H.-Y. Kim, J. Sohn, G. T. Wijewickrama, P. Edirisinghe, T. Gherezghiher, M. Hemachandra, P.-Y. Lu, R. E. Chandrasena, M. E. Molloy, D. A. Tonetti and G. R. J. Thatcher, Bioorg. Med. Chem., 2010, 18, 809–821 CrossRef CAS.
- (a) J. T. Wiltshire and G. G. Qiao, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1485–1498 CrossRef CAS; (b) H. Durmaz, A. Dag, E. Erdogan, A. L. Demirel, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 99–108 CrossRef CAS.
- (a) C. J. Hawker, D. Mecerreyes, E. Elce, J. Dao, J. L. Hedrick, I. Barakat, Ph. Dubois, R. Jérôme and W. Volksen, Macromol. Chem. Phys., 1997, 198, 155–166 CrossRef CAS; (b) L. Mespouille, Ph. Degée and Ph. Dubois, Eur. Polym. J., 2005, 41, 1187–1195 CrossRef CAS.
- K.-Y. Baek, M. Kamigaito and M. Sawamoto, Macromolecules, 2001, 34, 215–221 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |