Free radical polymerization of alkyl methacrylates with N,N-dimethylanilinium p-toluenesulfonate at above ambient temperature: a quasi-living system†
Atanu
Kotal
a,
Tapas K.
Paira
a,
Sanjib
Banerjee
a,
Chinmoy
Bhattacharya
b and
Tarun K.
Mandal
*a
aPolymer Science Unit, Indian Association for the Cultivation of Science, Jadavpur, Kolkata, 700032, India. E-mail: psutkm@iacs.res.in; Fax: (+91) 33-2473-2805
bAustin Paints and Chemicals, 3, Ambica Mukherjee Road, Kolkata, 700056, India
First published on 13th September 2010
Abstract
N,N-Dimethylanilinium p-toluenesulfonate (PTSA-DMA) has been successfully used as a versatile initiator for the quasi-living free radical polymerization of several alkyl methacrylate monomers such as methyl, ethyl, n-butyl, tert-butyl, and benzyl methacrylates (MMA, EMA, n-BuMA, t-BuMA and BzMA respectively) at 60 °C in dry THF. The initiator, PTSA-DMA was prepared by a simple complexation reaction of readily available p-toluenesulfonic acid (PTSA) and N,N-dimethylaniline (DMA). The yield of this polymerization system was moderate to good (60–75%). The produced poly(alkyl methacrylates) had narrow polydispersities (PDIs) (Mw/Mn = 1.16–1.45). Although, this polymerization follows first order kinetics but the obtained molecular weight remains almost unchanged with conversion. This polymerization proceeds through radical mechanism as confirmed by electron paramagnetic resonance spectroscopy. The ‘quasi-living’ nature of this polymerization was verified from the chain extension experiment as well as the successful synthesis of a block copolymer, poly(methyl methacrylate)-b-poly(methyl methacrylate-co-benzyl methacrylate), by the sequential addition of the respective monomers. The obtained block copolymer was characterized by 1H NMR, differential scanning calorimetric and atomic force microscopic studies.
Introduction
Over the past few decades, synthesis of poly(alkyl methacrylates) with controlled molecular weights and low polydispersities (PDIs) has attracted great attention among the polymer chemists belonging to the academic as well as the industrial community. Among the various polymerization techniques, controlled free radical polymerization is known to be one of the best techniques for the large-scale synthesis of poly(alkyl acrylates/alkyl methacrylates), as this polymerization can be carried out at room or high temperature and at less-stringent reaction condition compare to anionic polymerization.1–4 The discovery of nitroxide mediated stable free radical polymerization (NMP),1,5 radical addition fragmentation chain transfer polymerization (RAFT),3,6,7 atom transfer radical polymerization (ATRP)2,4,8–11 and degenerative chain transfer polymerization12,13 enabled tremendous development in the area of controlled living radical polymerization of alkyl acrylates/alkyl methacrylates in the past few decades. However, the development of new free radical initiators/initiator systems that are capable of producing polymers with controlled molecular weights with low polydispersity is still an emerging area of research compared to the use of conventional radical initiators such as benzoyl peroxide and azobisisobutyronitrile (AIBN), where there is poor control over polymerization14,15 in terms of polydispersity and livingness in the absence of chain transfer agents (such as dodecane thiol, butyl mercaptan and chloro alkyl alcohol) or stable free radical such as 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO).16–18In this context, herein, we report the use of N,N-dimethylanilinium p-toluenesulfonate (PTSA-DMA) as a versatile initiator for the free radical polymerization of several alkyl methacrylate monomers. In the literature, there are complementary reports regarding the ability of dimethylaniline (DMA) initiator to initiate polymerization of vinyl monomers.19–24 The binary systems consisting of N,N-dimethylaniline (DMA) and any one of the following compounds o-benzoic sulfimide,21,25 trichloroacetic acid,26p-tolylsulfonylacetic acid,24 benzoyl peroxide,19,27,28 benzyl chloride,22 and p-toluenesulfonyl chloride,29–31 have been used as thermal initiators for the free radical polymerization of several vinyl monomers. According to these reports, no polymerization took place when any one component of the binary systems was used alone. In contrast, Tsuda et al. have reported that DMA itself can act as an initiator for free radical polymerization of several vinyl monomers such as methyl methacrylate (MMA), methyl acrylate (MA) and methyl vinyl ketone.20,23 On the other hand, binary systems comprising p-toluenesulfonic acid (PTSA) and either N-acyloxyimides,32 tetramethyl-2-tetrazene,33 or N-chlorosuccinimide34 have also been utilized as an effective radical initiators for polymerization of vinyl monomers, but again there was no polymerization when either of the component was used alone. In our case, we also did not observe any significant polymer formation when PTSA or DMA was used alone under our reaction condition. But, the PTSA-DMA complex is shown to be an effective initiator for the polymerization of several alkyl methacrylate monomers to obtain poly(alkyl methacrylates) of narrow polydispersities (PDIs) (Mw/Mn = 1.16–1.45). This complex can be easily synthesized by simple mixing of readily available p-toluenesulfonic acid (PTSA) and N,N-dimethylaniline (DMA). It is shown that this polymerization is best at 60 °C in dry THF. It is also shown that the system is quasi-living in nature. Furthermore, the radical nature of this polymerization is established from electron paramagnetic resonance (EPR) spectroscopic study.
Experimental section
Materials
p-Toluenesulfonic acid (PTSA) and N,N-dimethyl aniline (DMA) were received from s d fine-Chem Limited, Mumbai. PTSA was recrystallized twice from dry benzene under inert atmosphere and dried under vacuum. DMA was distilled over KOH under reduced pressure just before use. Methyl methacrylate (MMA) (Burgoyne Urbidges & Co.), ethyl methacrylate (EMA) (Aldrich), and n-butyl methacrylate (n-BuMA) (Aldrich) were washed with 5 wt% NaOH to remove the inhibitor and then dried with calcium chloride (CaCl2) overnight and distilled over calcium hydride (CaH2) under reduced pressure prior to use. Benzyl methacrylate (BzMA) (Aldrich) and tert-butyl methacrylate (t-BuMA) (Aldrich) were passed through a 150 mesh activated alumina (Aldrich) column to remove inhibitor and then dried over anhydrous CaCl2 and CaH2 followed by distillation under reduced pressure before reaction. 1,4-Benzoquinone (FERAK, Germany), 4-methoxyphenol (Merck, Germany) and 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) was purchased from Aldrich and used as received.All the solvents were reagent grade and were purchased from Merck, India. Dichloromethane (DCM) and diethyl ether were dried with phosphorous pentoxide (P2O5) and sodium/benzophenone respectively and distilled just before use. Tetrahydrofuran (THF) was purified by refluxing over sodium/benzophenone and distilled prior to the polymerization.
Synthesis of N,N-dimethylanilinium p-toluenesulfonate (PTSA-DMA)
PTSA-DMA was synthesized by mixing the recrystallized PTSA (5 mmol) and distilled DMA (5 mmol) in 10 mL dry DCM followed by the drop wise addition of this mixture into magnetically stirred anhydrous diethyl ether (80 mL) under argon atmosphere following the method reported elsewhere.35 The as-prepared salt was then recrystallizing thrice from DCM–diethyl ether mixture followed by drying at room temperature in high vacuum and kept under argon atmosphere in a dark freezer for further use.Polymerization method
Polymerization was carried out in a specially designed borosil glass tube in dry THF under an argon atmosphere. The glass tube filled with argon was charged with the desired amount of purified PTSA-DMA salt and dry THF. Dry argon gas was bubbled through the mixture for 30 min to remove dissolved oxygen completely from the polymerization system. The tube containing the above mixture was then sealed by a high temperature silicon rubber septum. The reaction tube was then placed in a silicone oil bath thermostated at 60 °C and the reaction mixture was continuously magnetically stirred throughout the polymerization. After the equilibration of temperature, the required amount of distilled monomer was injected to the reaction mixture by a syringe (typically within 20 s to 40 s depending on the amount of monomer). Aliquots (∼2 mL) were taken from the reaction mixture time-to-time via syringe for determining conversion and molecular weight by GPC analysis. Polymerization was terminated by the addition of little methanol and then precipitated in large excess of hexane. Finally, the resultant polymer was dried under vacuum oven at 70 °C for 24 h and the yield was determined gravimetrically.Two-stage monomer addition/chain extension experiment
At first polymerization of a representative monomer, MMA (4.68 mmol) was carried out at 60 °C using PTSA-DMA initiator (0.0936 mmol) in a glass tube in dry THF (15 mL) under argon atmosphere for 36 h following the procedure described above. At this point, 2 mL of reaction mixture was withdrawn and precipitated in large excess of hexane (50 mL). At this point, the yield was 75% as determined gravimetrically. Degassed MMA (9.36 mmol) was then added to this reaction mixture that means a total of 10.53 mmol of MMA monomer was available for the polymerization at the second stage and continued polymerization for further 36 h. After polymerization, the reaction mixture was quenched by the addition of little methanol (1 mL) followed by the precipitation in large excess of hexane (150 mL) and the resultant precipitated poly(methyl methacrylate) (PMMA) was dried under vacuum at 70 °C for 24 h. The yield at the second stage was ∼90% as determined gravimetrically.Synthesis of block copolymer [poly(methyl methacrylate)-b-poly(benzyl methacrylate-co-methyl methacrylate), P(MMA)-b-P(BzMA-co-MMA)] by sequential addition of monomer
At first stage, MMA (4.68 mmol) was polymerized at 60 °C using PTSA-DMA initiator (0.0936 mmol) in a glass tube in dry THF (15 mL) under argon atmosphere for 36 h following the procedure described above. At this point, 2 mL reaction mixture was withdrawn and precipitated in large excess of hexane (50 mL) followed by drying under vacuum at 70 °C for 24 h. The yield was ∼75% as determined gravimetrically. 5.9 mmol of degassed BzMA monomer was then added to the reaction mixture that means 5.9 mmol of BzMA and 1.17 mmol of MMA monomers were available for the polymerization at the second stage and continued polymerization for further 36 h. After polymerization, the reaction mixture was quenched by the addition of little methanol (1 mL) followed by the precipitation in large excess of hexane (150 mL) and dried under vacuum at 70 °C for 24 h. The yield at the second stage was ∼92% as determined gravimetrically.Characterization
NMR spectra of the initiator and block copolymers [P(MMA)-b-P(BzMA-co-MMA)] were recorded in a 300 MHz Bruker DPX spectrometer from CDCl3 solution.Molecular weights and molecular weight distributions of the synthesized polymers and block copolymers were measured by size exclusion chromatography (SEC) using a Waters 1515 isocratic HPLC pump connected to three Waters Styragel HR1, HR3 and HR4 columns having effective separation of molecular weight ranges (100–5000), (500–30000) and (5000–500000) respectively. A Waters 2414 refractive index detector was used at room temperature (25 °C) for the above measurement. THF was used as eluent with a flow rate of 1 mL min−1 and narrow polystyrene standards having peak molecular weights (Mp) 3600, 8500, 19100, 43400, 76300, and 139400 were used for calibrating the GPC. The molecular weight of the polymer was determined by using Waters Breeze software.
The glass transition temperature (Tg) of the polymers was measured using a Perkin-Elmer Dimond DSC equipped with an intercooler at a scan rate of 20 °C min−1. For all DSC traces at least two cycles of heating and cooling was performed before taking the final heating scan.
AFM study of the block copolymer was carried on the thin film cast from THF solution of the block copolymer (2.5 mg mL−1) on a cleaned glass slide. The solvent was first evaporated at room temperature followed by drying the film at 130 °C for 1 h and subsequently quenched the film into liquid nitrogen. AFM images of the films were taken in a VEECO Digital Instrument CP-II microscope operating in tapping mode at room temperature.
For electron paramagnetic resonance (EPR) study, we performed the polymerization of MMA by PTSA-DMA in an EPR tube for 1 h under argon atmosphere at 60 °C, subsequently, the EPR tube was quenched in liquid nitrogen and the EPR spectrum was recorded in a JEOL JES-FA200 ESR spectrometer.
Results and discussion
The PTSA-DMA initiator was prepared simply by mixing the equimolar amounts of PTSA and DMA following the method reported elsewhere (see experimental section for details).35 The as-prepared salt was then recrystallized thrice from DCM–diethyl ether mixture. The purity of the PTSA-DMA salt was checked from its 1H NMR spectra by comparing the area of N-methyl proton peak (δ 3.20 ppm) of the cation with that of 4-methyl proton peak (δ 2.34 ppm) of the anion as shown in Fig. 1. The purified PTSA-DMA salt can be stored under argon atmosphere in a refrigerator under dark for months and subsequently can be used as an initiator for the polymerization of several alkyl methacrylates (AMAs) without the loss of its reactivity.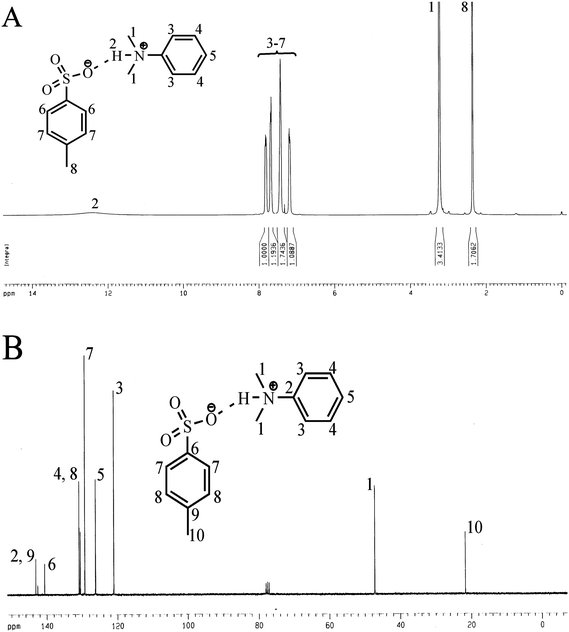 | ||
| Fig. 1 (A) 1H NMR and (B) 13C NMR spectra of the PTSA-DMA complex in CDCl3. | ||
To optimize the polymerization temperature, as a representative case, we performed polymerization of MMA ([M]0/[I]0 = 100) using PTSA-DMA initiator (1.02 × 10−4 mol) in THF (15 mL) as the solvent at different temperatures such as at 0, 40 and 60 °C. The polymerization did not proceed at all at 0 °C. Although, the polymerization took place at 40 °C, the yield was very low (∼25%) after 36 h. However, the polymerization proceeded smoothly at 60 °C with a yield of ∼60–75% after 36 h for most of the alkyl methacrylates (AMAs). Therefore, all the polymerizations were carried out at this temperature (60 °C).
To check the effect of solvent on this system, as a representative case, we conducted polymerization of MMA by PTSA-DMA complex in different solvents such as toluene, xylene, dioxane, anisole and acetonitrile other than THF (conditions: PTSA-DMA = 1.02 × 10−4 mol; [M]0/[I]0 = 100; solvent = 15 mL; T = 60 °C). It was observed that the polymerization occurred in most of these solvents except in acetonitrile. The yields of the polymerizations in xylene and dioxane are quite similar to that of THF (∼70%) after 36 h, but the yields in anisole and in toluene are ∼30% and ∼42% respectively. The PDIs of the obtained poly(methyl methacrylate)s (PMMAs) from different solvents are as follows: PDI = 1.22 (THF), PDI = 1.88 (dioxane), PDI = 1.42 (toluene), PDI = 1.45 (anisole) and PDI = 1.46 (xylene). As the PMMA obtained from THF gives lowest PDI, we have chosen THF as the solvent for polymerization of all the monomers.
Table 1 summarizes the results for the polymerizations of various AMA monomers such as MMA, EMA, n-BuMA, t-BuMA, and BzMA using this initiator. The molecular weight (Mn) and PDIs of the obtained poly(alkyl methacrylates) (PAMAs) were measured by GPC using THF as the eluent. The corresponding GPC traces of the PAMAs in the entry numbers 1–6 of Table 1 are shown in Fig. 2, which shows unimodal molecular weight distribution in all cases with relatively narrow polydispersities (PDIs) (<1.5). Table 1 also shows that the polymerization yields for all the monomers are moderately high (60–75%, as determined gravimetrically) after 36 h. It should be noted that among all the AMAs, MMA is polymerized most efficiently by this initiator. It should also be noted that the obtained PMMAs are of comparatively narrow PDIs (1.16–1.3) compared to other PAMAs. It is noteworthy that the obtained values of molecular weights (Mns) of poly(alkyl methacrylates) (PAMAs) from SEC measurement are “apparent” since the GPC instrument was calibrated using polystyrene standards (see column 5 of Table 1). Thus, the corrected Mn values of these PAMAs were calculated following the principle of universal calibration using the available Mark-Houwink parameters in the literature,36–39 which are provided in column 6 of Table 1. The data (entry numbers 1–3 of Table 1) also reveal that as the initial molar feed ratio of MMA to initiator increases, both the apparent and the corrected Mn of the obtained PMMA increases (see columns 5 and 6 of Table 1), which is usually expected for a controlled polymerization system. However, the initiator efficiency (f) is bit poor (<0.25) for all cases. The poor initiator efficiency might be arises due to the different possible structures present in the initiator. According to the literature report, PTSA-DMA salt exhibits three possible structures such as “corner”, “edge” and “axial” (see Scheme 1).35 Although according to them, the majority of the ion pairs exist as “corner” structure along with few percentage of “edge” structure.35 The presence of more than one kind of these initiating species in the polymerization media might also be responsible for such low initiator efficiencies.
| Entry No. | Monomer | [M]0/[I]0 | Yield (%) | M n (GPC) | M n (corrected) | M w/Mn (PDIs) | fc |
|---|---|---|---|---|---|---|---|
| a Conditions: initiator (I) = 1.02 × 10−4 mol; solvent = 15 mL (THF); T = 60 °C; polymerization time = 36 h. b M n (corrected) values were calculated using Mn (SEC) values and applying the principle of universal calibration using the available Mark-Houwink parameters in the literature36–39 at 30 °C in THF. c Initiator efficiency (f) = Mn,cal/Mn,SEC. | |||||||
| 1 | MMA | 50 | 75 | 26200 | 29760 | 1.16 | 0.14 |
| 2 | MMA | 100 | 75 | 40700 | 46270 | 1.22 | 0.18 |
| 3 | MMA | 200 | 75 | 59500 | 67700 | 1.30 | 0.24 |
| 4 | EMA | 85 | 60 | 78500 | 85100 | 1.22 | 0.07 |
| 5 | n-BuMA | 50 | 70 | 47600 | 29400 | 1.30 | 0.10 |
| 6 | t-BuMA | 50 | 70 | 36800 | 70460 | 1.43 | 0.13 |
| 7 | BzMA | 50 | 70 | 40500 | — | 1.45 | 0.15 |
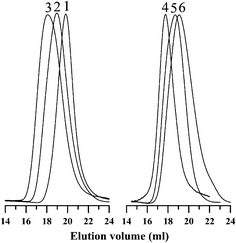 | ||
| Fig. 2 GPC traces of poly(alkyl methacrylate) samples of entry numbers 1–6 in Table 1. | ||
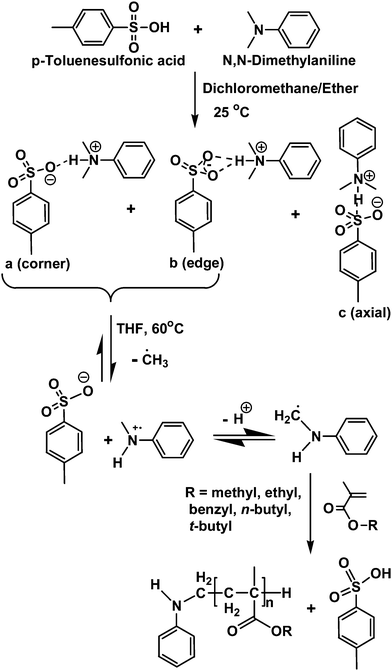 | ||
| Scheme 1 Plausible mechanistic pathway for the polymerization of alkyl methacrylates using PTSA-DMA initiator. | ||
To study the kinetics of this polymerization system, again, we have chosen the MMA over the other methacrylate monomers for polymerization using PTSA-DMA complex, as a representative case. Two sets of polymerization reactions, whose recipes are given in entries 2 and 3 of Table 1, were studied to find whether there is any effect of varying monomer to initiator ratios ([M]0/[I]0 = 100 and 200) on the kinetics of this polymerization. For this, known aliquots were withdrawn at different time intervals and the monomer-to-polymer conversion was determined gravimetrically (see details in the experimental section). The monomer conversion was plotted against time (Fig. 3A), indicating that the conversion increased with polymerization time. The trend of increase is similar in both the cases. As shown in Fig. 3A, 65–70% conversion was reached in 36 h of reaction, indicating that the polymerization was slow. This is may be because of poor initiator efficiency as mentioned above. Fig. 3B shows the semilogarithmic plot of monomer conversion versus time. It is clear that both the plots are linear and of same nature, indicating that this polymerization follows the first order kinetics. It should also be noted that the nature of the plot did not change due to change of used monomer to initiator ratios. Therefore, this polymerization is so well controlled that proceeds with constant active radical concentration, which is characteristic of a living polymerization system. But the molecular weight of the obtained PMMAs did not increase linearly with monomer conversion (see Fig. 3C) as expected for a living polymerization system. At this point, we do not know the exact reason behind this discrepancy. However, we were able to control the molecular weight by varying the initial molar feed ratio of MMA to initiator and the obtained polymers have narrow polydispersities (<1.5) (see columns 5 and 6 of Table 1). From the above results we may conclude that this polymerization system is quasi-living in nature. To check this issue further, we performed two-stage sequential monomer addition experiment as well as block copolymer synthesis experiment. The detailed results of these experiments will be discussed later in this section.
![(A) Conversion versus time, (B) kinetic plot of ln([M]0/[M]) versus time and (C) dependence of Mn and PDIs of the obtained PMMA on conversion of the MMA polymerization using PTSA-DMA initiator with various ratios of [M]0/[I]0 in THF. Conditions: PTSA-DMA = 1.02 × 10−4 mol, solvent (THF) = 15 mL, T = 60 °C.](/image/article/2010/PY/c0py00180e/c0py00180e-f3.gif) | ||
| Fig. 3 (A) Conversion versus time, (B) kinetic plot of ln([M]0/[M]) versus time and (C) dependence of Mn and PDIs of the obtained PMMA on conversion of the MMA polymerization using PTSA-DMA initiator with various ratios of [M]0/[I]0 in THF. Conditions: PTSA-DMA = 1.02 × 10−4 mol, solvent (THF) = 15 mL, T = 60 °C. | ||
To establish the mechanism of this polymerization system, we performed the following experiments. First of all, we observed that neither PTSA or DMA alone initiated polymerization under our reaction conditions. Also, the polymerization did not proceed at all in presence of a radical scavenger such as 1, 4-benzoquinone, 4-methoxyphenol and TEMPO under similar reaction conditions, which indicates that polymerization, may proceed through radical pathway. To confirm this, we have recorded an EPR spectrum of the polymerization solution (polymerization of MMA by PTSA-DMA), which clearly showed the radical signature at 313.285 mT (g = 2.06234) and at 323.182 mT (g = 1.99919) as depicted in Fig. 4. However, we are unable to identify the actual structure of radical species at this moment but the exhibition of characteristic radical signature at g ≈ 2.0 by this system as well as results of the controlled experiment mentioned above confirmed that this polymerization indeed proceed through radical pathway. From the above observations, we proposed the possible mechanism of this polymerization as depicted in Scheme 1, which was also proposed as the mechanism by other researchers for similar kinds of systems.20,28,30,34
![An EPR spectrum of the propagating radical corresponding to the polymerization of MMA by PTSA-DMA initiator [experimental conditions: temperature = −150 °C; microwave frequency = 9.042 (GHz), microwave power = 0.998 (mW), modulation amplitude = 800, modulation width = 1.40 mT].](/image/article/2010/PY/c0py00180e/c0py00180e-f4.gif) | ||
| Fig. 4 An EPR spectrum of the propagating radical corresponding to the polymerization of MMA by PTSA-DMA initiator [experimental conditions: temperature = −150 °C; microwave frequency = 9.042 (GHz), microwave power = 0.998 (mW), modulation amplitude = 800, modulation width = 1.40 mT]. | ||
Two-stage sequential monomer addition experiments for chain extension as well as the synthesis of block copolymers were carried out to check the living nature of the MMA polymerization. The GPC traces of the PMMAs obtained by chain extension experiment are shown in Fig. 5A. In the first stage, a conversion of 75% was reached after 36 h of reaction and in the second stage the conversion was 90%. The Mn and PDI values for the two stages are Mn (1st stage) = 27000, PDI = 1.2 and Mn (2nd stage) = 50900, PDI = 1.44. Both the GPC traces (Fig. 5Aa and 5Ab) show unimodal distribution along with a clear lateral shift of maxima towards higher molecular weight region on going from stage 1 to stage 2. Although, in the latter case, a slight broadening of the GPC trace was noticed in the lower molecular weight region (Fig. 5Ab). From the above results along with the kinetics data described earlier, we may conclude that this polymerization system is quasi living in nature.
![GPC traces of (A) PMMAs, obtained from the chain extension experiment [a: Mn (1st stage) = 27000, PDI = 1.2 and b: Mn (2nd stage) = 50900, PDI = 1.44] and (B) the block copolymer of PMMA and PBzMA [a: Mn (1st stage) = 28000, PDI = 1.2 and b: Mn (2nd stage) = 71200, PDI = 1.46].](/image/article/2010/PY/c0py00180e/c0py00180e-f5.gif) | ||
| Fig. 5 GPC traces of (A) PMMAs, obtained from the chain extension experiment [a: Mn (1st stage) = 27000, PDI = 1.2 and b: Mn (2nd stage) = 50900, PDI = 1.44] and (B) the block copolymer of PMMA and PBzMA [a: Mn (1st stage) = 28000, PDI = 1.2 and b: Mn (2nd stage) = 71200, PDI = 1.46]. | ||
For the block copolymer synthesis, at the first stage, MMA was polymerized for 36 h (conversion ∼75%) followed by the addition of benzyl methacrylate (BzMA) monomer to this reaction medium and continued polymerization for further 36 h. GPC traces (Fig. 5Ba and 5Bb) clearly reveal further growth of the PMMA chain with Mn (1st stage) = 28000, PDI = 1.2 and Mn (2nd stage) = 71200, PDI = 1.46. The further growth of the PMMA chain is indeed due to the second block formation. However, as expected considering the remaining MMA monomer in the reaction media, the second block is a statistical copolymer of PMMA and PBzMA. The mole percentage of PMMA present in the second block is calculated to be ∼16 (see ESI†). 1H NMR spectra of the resultant copolymer, poly(methyl methacrylate)-b-poly(benzyl methacrylate-co-methyl methacrylate), P(MMA)-b-P(BzMA-co-MMA) shows the characteristic resonance peak at δ 3.6 ppm for –OCH3 group of MMA unit and at δ 4.92 ppm for the –OCH2 group of the BzMA unit (see Fig. S1 in the ESI†). The molecular weight (Mn,cal ≈ 92000) of the block copolymer as measured from 1H NMR (see ESI for calculation†) correlates well with that measured by GPC (see Fig. 5Bb). Considering the area under these two peaks, the weight ratio of PMMA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PBzMA in the copolymer was found to be 1:3.35.
:PBzMA in the copolymer was found to be 1:3.35.
Furthermore, differential scanning calorimetric analysis of the synthesized block copolymer shows a single glass transition temperature [Tg (onset)] at 70.65 °C (see Fig. 6b for thermogram). This experimental [Tg (onset)] value is close to the predicted [Tg (onset)] = 68.6 °C, calculated using the Fox equation40 for the above-mentioned composition of the copolymer. The [Tg (onset)] values of the individual PMMA and PBzMA homopolymers (synthesized by PTSA-DMA initiator) used in the above equation, having almost same block length as in the copolymer, were measured to be 110.26 °C and 57.89 °C respectively (see Fig. 6d and 6a for thermograms). We are surprised with this result of the appearance of a single Tg in a block copolymer that is composed of two immiscible polymer blocks as the physical blend of this two blocks (PMMA and PBzMA) shows two separate Tgs (see Fig. 6c for the thermogram). Angot et al. also made a similar observation for their systems.41 The apparent miscibility between the two immiscible blocks (PMMA and PBzMA) is possibly due to the formation of phase domains of lower size by the respective block in the copolymer, which probably below the detection limit of phase size of any polymers by DSC instrument.
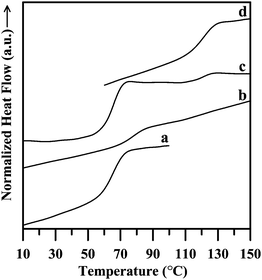 | ||
| Fig. 6 DSC thermograms of (a) neat PBzMA (Mn = 40500), (b) the synthesized block copolymer; P(MMA)-b-P(BzMA-co-MMA), (c) blends of PMMA (Mn = 28000) and PBzMA (Mn = 40500) with the same weight ratio as in the block copolymer (PMMA:PBzMA = 1:3.35), and (d) neat PMMA (Mn = 28000). | ||
To check this supposition, we have further examined the P(MMA)-b-P(BzMA-co-MMA) thin film via atomic force microscopy (AFM). The AFM image clearly shows two distinct phases where one phase, with spherical morphology (∼30–35 nm), is distributed throughout the matrix of other phase as shown in Fig. 7. Such small phase domains are beyond the limit of detection of DSC instrument and as a result only a single Tg was observed for the block copolymer. Thus, we may conclude that this initiator (PTSA-DMA) can be successfully used to prepare block copolymers and the system was found to behave like a quasi-living one.
 | ||
| Fig. 7 AFM study of the block copolymer P(MMA)-b-P(BzMA-co-MMA) thin film (a) topographic image and (b) phase image. The root mean square roughness is about 1.7 nm, illustrating the flatness of the film. | ||
Conclusions
In conclusion, successful polymerizations of several alkyl methacrylate monomers using N,N-dimethylanilinium p-toluenesulfonate (PTSA-DMA) as a versatile initiator in dry THF at 60 °C is described. This polymerization resulted in the formation of poly(alkyl methacrylates) of narrow polydispersities (PDIs) (Mw/Mn = 1.16–1.45). This polymerization followed first order kinetics but the obtained molecular weight remained almost unchanged with conversion. An EPR spectroscopic study showed that this polymerization proceeded through radical mechanism. The quasi-living nature of this polymerization system was established by the chain extension experiment as well as the successful synthesis of block copolymer of PMMA and PBzMA as confirmed from NMR, DSC and AFM study.Acknowledgements
T.K.P. and S.B. thank CSIR, India for the fellowship. This research was supported by the grants from CSIR, India. Thanks are also due to the partial support from the Nanoscience and Nanotechnology Initiatives, DST, India.Notes and references
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Acc. Chem. Res., 2008, 41, 1133 CrossRef CAS.
- M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963 CrossRef CAS.
- C. J. Hawker, Acc. Chem. Res., 1997, 30, 373 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079 CrossRef CAS.
- A. B. Lowe and C. L. McCormick, Prog. Polym. Sci., 2007, 32, 283 CrossRef CAS.
- T. E. Patten and K. Matyjaszewski, Acc. Chem. Res., 1999, 32, 895 CrossRef CAS.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689 CrossRef CAS.
- M. Ouchi, T. Terashima and M. Sawamoto, Acc. Chem. Res., 2008, 41, 1120 CrossRef CAS.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93 CrossRef CAS.
- B. M. Rosen and V. Percec, Chem. Rev., 2009, 109, 5069 CrossRef CAS.
- S. Yamago, Chem. Rev., 2009, 109, 5051 CrossRef CAS.
- G. C. Eastmond, in Encyclopedia of Polymer Science and Technology, ed. H. F. Mark, N. G. Gaylord and N. M. Bilkales, Wiley-Interscience, New York, 1964, vol. 7, pp. 361 Search PubMed.
- B. Yamada and P. B. Zetterlund, in Handbook of Radical Polymerization, ed. K. Matyjaszewski and T. P. Davis, Wiley-Interscience, 2002, pp. 117 Search PubMed.
- A. Fischer, A. Brembilla and P. Lochon, Eur. Polym. J., 2001, 37, 33 CrossRef CAS.
- José Luis de la Fuente and E. L. Madruga, Macromol. Chem. Phys., 2001, 202, 375 CrossRef.
- S. Sugihara, A. H. M. Radzi, S. Hori and I. Ikeda, Polym. Bull., 2007, 58, 873 CrossRef CAS.
- M. Imoto and S. Choe, J. Polym. Sci., 1955, 15, 485 CrossRef CAS.
- T. Ishida, S. Kondo and K. Tsuda, Makromol. Chem., 1977, 178, 3221 CrossRef CAS.
- J. Lal and R. Green, J. Polym. Sci., 1955, 18, 430 CrossRef.
- T. Otsu, S. Aoki and K. Itakura, J. Polym. Sci., Part A-1, 1970, 8, 445 CrossRef CAS.
- K. Tsuda, S. Kondo, K. Yamashita and K. Ito, Makromol. Chem., 1984, 185, 81 CrossRef CAS.
- R. Uehara, Bull. Chem. Soc. Jpn., 1960, 33, 698 CrossRef CAS.
- J. Lal, R. Green and S. Ellis, J. Polym. Sci., 1957, 24, 75 CrossRef CAS.
- R. Uehara, Bull. Chem. Soc. Jpn., 1958, 31, 685 CrossRef CAS.
- S. Z. Chen, Y. P. Bai and Z. L. Li, Chin. Chem. Lett., 2006, 17, 258 CAS.
- H. Yu, Y. Fang, L. Chen and S. Chen, Polym. Int., 2009, 58, 851 CrossRef CAS.
- T. Sato, M. Yoshioka and T. Otsu, Makromol. Chem., 1972, 153, 47 CrossRef CAS.
- H. Kothandaraman and N. Arumugasamy, Eur. Polym. J., 1984, 20, 1195 CrossRef CAS.
- H. Kothandaraman and N. Arumugasamy, J. Polym. Mater., 1984, 1, 141 Search PubMed.
- T. Sato, M. Metsugi and T. Otsu, J. Macromol. Sci., Part A: Pure Appl. Chem., 1981, 15, 347 Search PubMed.
- K. Sugiyama, J. Macromol. Sci., Part A: Pure Appl. Chem., 1979, 13, 13 Search PubMed.
- M. Ko, T. Sato and T. Otsu, Makromol. Chem., 1975, 176, 643 CrossRef CAS.
- T.-P. I and E. Grunwald, J. Am. Chem. Soc., 1976, 98, 1351 CrossRef CAS.
- S. Beuermann, D. A. Paquet Jr., J. H. McMinn and R. A. Hutchinson, Macromolecules, 1996, 29, 4206 CrossRef CAS.
- R. A. Hutchinson, S. Beuermann, D. A. Paquet Jr. and J. H. McMinn, Macromolecules, 1997, 30, 3490 CrossRef CAS.
- A. Karandinos, S. Nan, J. W. Mays and N. Hadjichristidis, Macromolecules, 1991, 24, 2007 CrossRef CAS.
- M. Kurata and Y. Tsunashima, in Polymer Handbook, ed. J. Brandrup, E. H. Immergut and E. A. Grulke, Wiley-Interscience, New York, 4th edn, 1999, pp. 1 Search PubMed.
- Fox equation: 1/Tg = (wa/Tg,a) + (wb/Tg,b), where wa and wb are the weight fractions and Tg,a and Tg,b are the respective glass transition temperatures.
- S. Angot, N. Ayres, S. A. F. Bon and D. M. Haddleton, Macromolecules, 2001, 34, 768 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra of the block copolymer synthesized by PTSA-DMA initiator. See DOI: 10.1039/c0py00180e |
| This journal is © The Royal Society of Chemistry 2010 |