Hydrophilization of poly(ether ether ketone) films by surface-initiated atom transfer radical polymerization
Charlotte Juel
Fristrup
,
Katja
Jankova
and
Søren
Hvilsted
*
Danish Polymer Centre, Department of Chemical and Biochemical Engineering, Technical University of Denmark, Building 227, DK-2800, Kgs. Lyngby, Denmark. E-mail: sh@kt.dtu.dk; Fax: +45 4588 2161; Tel: +45 4525 2965
First published on 13th September 2010
Abstract
Surface-Initiated Atom Transfer Radical Polymerization (SI-ATRP) has been exploited to hydrophilize PEEK. The ketone groups on the PEEK surface were reduced to hydroxyl groups which were converted to bromoisobutyrate initiating sites for SI-ATRP. The modification steps were followed by contact angle measurements and XPS. Moreover, ATR FTIR has been used to confirm the formation of initiating groups. Grafting of PEGMA from PEEK was performed in aqueous solution. The presence of the PPEGMA grafts on PEEK was revealed by the thermograms from TGA whereas investigations with AFM rejected changes in the surface topography. Two possible applications arose from the hydrophilization of PEEK, metal deposition and protein repellency. The performed modification allowed for successful electroless deposition and good adhesion of nickel as well as copper.
Introduction
Poly(ether ether ketone) (PEEK) can replace aluminium and other metals in medical, aerospace, electronics, and automotive applications. Surface modification of PEEK will increase the applicability of the material considerably. For the pharmaceutical industry non-fouling surfaces are desired whereas in other fields the demand for metallization is continuously increasing.In order to obtain good adhesion between metals and the polymer surfaces strong bonds are needed e.g. charge transfer at the polymer–metal interface. The most appropriate way to form a metal–polymer complex is a carboxyl containing polymer surface.1 Electroless metallisation of polymers is performed in aqueous solutions of the metal salts, containing numerous additives. Many factors will contribute to the adhesion of metals to polymers e.g. polymers with higher surface energy can easier be metallised. Conventionally, polymer surfaces are etched with chromic acid prior to electroless metal deposition. The etching results in hydrophilization of the polymer surface and some roughness is also introduced, which will support the adhesion of the metal layer to the polymer. Several steps are needed before the deposition of the metal e.g. exposure to a catalyst for the reduction of the metal ions. The reduction is performed to ensure that the metal is deposited on the surface in question and not on the walls of the metallisation bath or any other places. Palladium particles show the desired catalytic activity and they can be applied in two different ways which are acceptable for almost all polymers. In the cheapest and most common method Pd particles are mounted on the chromic acid pretreated polymer surface by reduction of colloid Pd. The other method is more exclusive and involves premixed palladium in the polymer. After reduction the metal (Cu, Ni, Au, etc.) sticks better to the Pd particles.2 Attempts to avoid treatment with chromic acid mainly rely on substitution with other acids, their mixtures with various organic or inorganic compounds, as well as laser treatment and plasma reactive ion etching.3 Therefore, a new method is presented here to hydrophilize PEEK avoiding hazardous chromic acid and its attendant effects.
During the last 30 years PEEK has been known for its biological applications which require surface modification to change the hydrophobicity of the material. Techniques like plasma treatment or deposition (mainly plasma spraying of Ti and/or thermal plasma coating of hydroxyapatite) as well as wet chemistry4–6 are commonly used.7 The most powerful method is coating of the hydrophobic PEEK with a hydrophilic polymer. In that way, an entire new surface layer is added to the substrate. The method can be divided into “grafting to” or “grafting from”. Where “grafting to” a polymer surface requires both an anchoring group on the surface as well as a reactive group on the incoming polymer, in addition to an effective coupling mechanism. Steric hindrance or shielding from the first reacted polymers onto a surface often prevents subsequent polymer grafting and leads to a low surface coverage. “Grafting from” a surface involves graft polymerization which is often accomplished by the use of electrons, UV or plasma treatment of the surface followed by radical polymerization of various monomers. In general, conventional radical polymerization will result in poor control of the new surface layer in terms of chemical functionality and morphology. Controlled radical polymerization techniques, especially ATRP,8 provide the best control, where “control” means the ability to shape the polymer architecture and to design linear, block, graft, star or dendritic forms in a predictable manner including total control over the chain length (molecular weight). In principle, the concept of grafting from a polymer also allows an initiator gradient, enabling a variation in the grafting density. ATRP normally also offers the possibility to select the most convenient and appropriate initiator for the monomer in question. However, in “grafting from” a polymer surface by SI-ATRP the largest challenge is often to generate surface anchored initiating sites.9 PEEK has ketone groups which after reduction can be utilized for anchoring of initiating groups for ATRP.10 Therefore, Surface-Initiated Atom Transfer Radical Polymerization (SI-ATRP) has been selected as the way to covalently graft hydrophilic polymer chains from the PEEK surface. Recently, SI-ATRP was applied to prepare conductive cotton yarns by grafting of poly(2-(methacryloyloxy)ethyl trimethylammonium chloride (PMETAC), followed by ion exchange with (NH4)2PdCl4.11 The palladium moieties are known to act as effective catalyst sites for the deposition of metals.12 The hydrophilic polymer grafts in this work were prepared from poly(ethylene glycol)methacrylate (PEGMA)13 as coatings of poly(ethylene glycol) (PEG) are known to be compatible with proteins due to their water solubility and hydrophilicity. The PEG surfaces are in a liquid-like state with polymer chains showing high flexibility or mobility (increasing mobility with chain length up to 100).14 Steric stabilization and chain mobility play important roles in inhibition of non-specific fouling on PEG surfaces. The PEG molecules have a large excluded volume in water which makes them very effective for the steric repulsion. Additionally, the high surface mobility of PEG chains prevents protein adsorption as the contact time is shortened.14,15 The size of the proteins relative to the distance between the PEG chains is also important for the efficiency of the coatings to inhibit fouling.16
In this work the surface of PEEK is functionalized by covalent bonding of hydrophilic polymer brushes of PEGMA from initiator-modified PEEK using SI-ATRP. Surface reduction of PEEK to form hydroxyl groups was performed prior to the attachment of 2-bromoisobutyrate initiating groups. Each modification step of PEEK as well as the polymer grafting was followed and confirmed by ATR FTIR, water contact angle (WCA) measurements, and Thermal Gravimetric Analysis (TGA). The surface topography was evaluated by Atomic Force Microscopy (AFM). X-Ray Photoelectron Spectroscopy (XPS) has been used to investigate the degree of functionalization.
Experimental
Materials and methods
Acetone (Riedel-de Haën) was distilled and stored over molecular sieves. Dimethylsulfoxide (DMSO, Fluka) was distilled over calcium hydride under reduced pressure and stored over molecular sieves. Tetrahydrofuran (THF, Sigma-Aldrich) and triethylamine (TEA, Riedel-de Haën) were distilled over calcium hydride. 2,2′-Bipyridine (Bipy, 99%), 2-bromoisobutyrylbromide (Br-iBuBr, 98%), copper chloride (CuCl, 99%), 4-dimethylaminopyridine (DMAP, 99%), methanol (analytical grade), poly(ethylene glycol)methacrylate (PEGMA, Mn ≈360), sodium chloride (NaCl), and tin(II) chloride dihydrate (SnCl2·2H2O, 98%) were used as supplied by Sigma-Aldrich. Cataposit 958, ethanol (99.9% vol, Kemetyl), hydrochloric acid (HCl), sodium borohydride (NaBH4, ≥96%, Fluka), and ultrapure water were used without further purification. A sheet of 1.4 m2 of APTIV 1000-750 from Mape Plastics was cut into smaller pieces. APTIV 1000-750 are calendared films made of poly(ether ether ketone) (PEEK). The thickness of the films was 750 µm.Attenuated Total Reflectance (ATR) Fourier Transform Infrared (FTIR) spectra were obtained using a Spectrum One spectrometer from Perkin Elmer which was equipped with a universal ATR sample accessory. WCA measurements were made on an OCA20 Contact Angle System from Dataphysics with a temperature controller. The temperature was set to 25 °C. The dynamic method called “sessile drop (needle in)” was used and the WCAs were computed using “Ellipse Fitting”. The measurements were made on three drops of deionized water at different spots and three values for both the advancing and receding angles were used to determine the average value. Thermal degradation was investigated by TGA performed with a TGA Q500 from TA Instruments recording the total weight loss of approx. 10–12 mg samples from room temperature to 800 °C at a rate of 10 °C min−1 in a nitrogen flow of 90 mL min−1. The atomic force microscope was a Nanosurf EasyScan 2 system which could make nanometre scale resolution measurements of topography. The maximum scan range was 110 µm and the maximum Z-range was 22 µm, with a resolution of approximately 2 nm in the XY directions and 0.3 nm in the Z direction. The advantage of this system was that the sample size could be unlimited. XPS analysis was performed on a Thermo Fisher Scientific K Alpha using monochromatized aluminium KR radiation in a 400 µm spot on the sample. Survey and high-resolution spectra were acquired and analyzed using the manufacturer's Advantage software package.
The samples were pretreated before metallisation in electroless nickel and copper baths. Pretreatment of the samples involved immersion in an activation solution at room temperature for 3 min. The activation solution had the following composition (per litre): 180 g NaCl, 120 mL concentrated HCl, 1 g SnCl2·2H2O, and 20 mL Cataposit 958 (colloidal tin–palladium solution). After neutralization in 10 wt% HCl at room temperature for 10 seconds, either Ni-deposition at 90 °C or Cu-deposition at 45 °C in industrial electroless nickel bath (containing Ni-acetate) or copper bath for 15 min is followed. Pull-off tests and adhesive tape tests were performed according to ISO and ASTM standards.17 A dolly having a diameter of 8.2 mm was glued to the metallised surface of PEEK-g-PPEGMA with fast-acting cyanoacrylate glue (Loctite 422 or 432).
Modification of the PEEK surfaces
The formation of hydroxyl groups (PEEK-OH) on the PEEK films was achieved by treatment with NaBH4 according to Noiset et al.5 The PEEK films were refluxed with acetone under nitrogen followed by drying under vacuum for 2 hours at 60 °C prior to the reduction of the ketone groups. After formation of the hydroxyl groups the PEEK-OH films were washed first by immersion in methanol for 15 min, then water for 10 min, 0.5 M HCl for 10 min, water for 10 min, and ethanol for 15 min. Subsequently, the films were dried under vacuum for about 3 hours at 60 °C.Initiating groups (PEEK-Br) were anchored chemically to the PEEK–OH films. 5 PEEK–OH films (1 × 2 cm each), DMAP (0.42 g, 3.5 mmol), TEA (2.91 mL, 21 mmol), and 100 mL of THF were added into a 250 mL round bottom flask. Br-iBuBr (2.65 mL, 21 mmol) in 25 mL of dried THF was slowly added under nitrogen blanket to the flask while the reaction mixture was kept at 0 °C. Afterwards the reaction mixture was allowed to reach room temperature. The reaction proceeded overnight (about 18 hours) before the films were removed. The PEEK-Br films were rinsed and stirred three times for 10 min in THF.
Polymerization of PEGMA from the surface of PEEK-Br was performed in aqueous media. 14 mL of ultrapure water and 8.4 mL of PEGMA were added to a Schlenk tube. Nitrogen was bubbled through the mixture for 15 min. 2 PEEK-Br films were added and the bubbling with nitrogen was proceeded for 10 min. The Schlenk tube was kept at 0 °C while CuCl (0.146 g, 1.47 mmol) and Bipy (0.390 g, 2.50 mmol) were added. The bubbling with nitrogen was continued for 15 min. The polymerization was carried out at 30 °C for 45 min. Afterwards the PEEK-g-PPEGMA films were immersed and stirred in water/methanol 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for 15 min and in water/ethanol 5:1 for 15 min.
:1 for 15 min and in water/ethanol 5:1 for 15 min.
Results and discussion
The surface of PEEK was functionalized by covalent bonding of hydrophilic polymer brushes of PEGMA from initiator-modified PEEK using SI-ATRP. Surface reduction of PEEK to form hydroxyl groups5 (Fig. 1) was performed prior to the attachment of 2-bromoisobutyrate initiating groups (Fig. 2). The reduction of the ketone groups with sodium borohydride in DMSO at 120 °C did not dissolve the films which makes the method very useful for implantable devices e.g. spinal implants, orthopedic bearing and hip stem material.7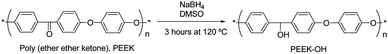 | ||
| Fig. 1 Surface activation of the PEEK films. | ||
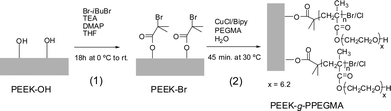 | ||
| Fig. 2 (1) Anchoring of the initiating groups on the hydroxyl-functionalized surface. (2) Grafting of PPEGMA brushes from the PEEK films using SI ATRP. | ||
SI-ATRP of the monomer PEGMA was performed in aqueous media in the presence of the catalyst system 2,2′-bipyridine and copper chloride. The same ATRP conditions have been used with the initiator, ethyl 2-bromoisobutyrate and without substrates. Moreover, water has been replaced with methanol and the homopolymerzations only resulted in a gel when performed in water but not in methanol. The gel formation strongly indicates that polymer chains have reacted with each other and formed crosslinks. Therefore, the PPEGMA grafts were presumably also crosslinked. Further characterization has only been performed on grafts prepared by SI-ATRP of PEGMA in water as metallisation of the PPEGMA grafts prepared in methanol did not result in uniform coverage.
ATR FTIR spectra (Fig. 3) were compared during the modification of PEEK. The formation of hydroxyl groups was observed as a C–O absorption band appearing at 1057 cm−1. The carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) absorption band from ester groups at 1736 cm−1 indicated the presence of initiating groups on the PEEK surface. This means that it was actually possible to observe absorption bands from the initiating groups on PEEK as opposed to unsuccessful attempts with many other substrates.18–20 For these polymeric substrates ATR FTIR spectroscopy has only been used for characterization of the polymer grafts as an indirect proof for the formation of the initiating groups. Grafting of PPEGMA from the surface resulted in an increase of the CO band (broad band at 1730 cm−1). ATR FTIR spectroscopy penetrates a few micrometres into the sample; therefore, the spectra of the PEEK-g-PPEGMA films contained absorption bands from both PPEGMA and the substrate. Since the ketones in PEEK are conjugated with two aromatic rings the absorption band for CO appears at 1647 cm−1. Moreover, the CC ring stretch absorptions from the aromatic rings occur in pairs at 1594 and 1487 cm−1. The spectra in Fig. 3 also display C–O–C stretching bands for the aryl ethers in PEEK at 1217, 1185, and 1157 cm−1. All spectra contained the characteristic absorption bands from PEEK which strongly suggest that only the PEEK surface has been modified.
O) absorption band from ester groups at 1736 cm−1 indicated the presence of initiating groups on the PEEK surface. This means that it was actually possible to observe absorption bands from the initiating groups on PEEK as opposed to unsuccessful attempts with many other substrates.18–20 For these polymeric substrates ATR FTIR spectroscopy has only been used for characterization of the polymer grafts as an indirect proof for the formation of the initiating groups. Grafting of PPEGMA from the surface resulted in an increase of the CO band (broad band at 1730 cm−1). ATR FTIR spectroscopy penetrates a few micrometres into the sample; therefore, the spectra of the PEEK-g-PPEGMA films contained absorption bands from both PPEGMA and the substrate. Since the ketones in PEEK are conjugated with two aromatic rings the absorption band for CO appears at 1647 cm−1. Moreover, the CC ring stretch absorptions from the aromatic rings occur in pairs at 1594 and 1487 cm−1. The spectra in Fig. 3 also display C–O–C stretching bands for the aryl ethers in PEEK at 1217, 1185, and 1157 cm−1. All spectra contained the characteristic absorption bands from PEEK which strongly suggest that only the PEEK surface has been modified.
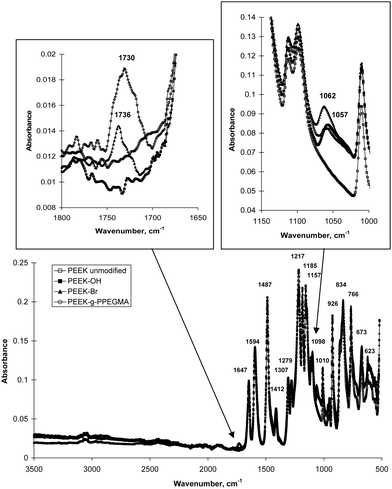 | ||
| Fig. 3 FTIR spectra of the unmodified and modified PEEK films. | ||
The advancing and receding WCAs decreased as the films were modified, reflecting the high hydrophilicity of the hydroxyl groups and PPEGMA (Table 1). For the unmodified PEEK and the PEEK with PPEGMA grafts the WCAs of the smooth and the rough side were compared. As expected it was observed that the contact angle hysteresis was larger for the rough side due to the enhanced surface roughness and possibly also surface heterogeneity. The rough side had uniform stripes from the calendaring process which may have caused a heterogeneous dispersion of the initiating groups and by that means the polymer grafts have been placed with some irregularities. Therefore, the WCAs for the smooth side should be compared in order to evaluate the influence of the modifications. The contact angle hysteresis was unchanged when the ketones on the surface were reduced to hydroxyl groups. However, the formation of initiating sites lowered the hysteresis as the surface became more hydrophobic. The grafting of PPEGMA from the surface increased the hysteresis due to hydrophilic ethylene glycol units in the side chains of PPEGMA. Thus, the WCA measurements seem to strongly support the chemical modifications.
| Material | S/R | WCA (adv.)/° | WCA (rec.)/° | Δ/° |
|---|---|---|---|---|
| PEEK | S | 99 ± 2 | 58 ± 4 | 41 ± 4 |
| PEEK | R | 103 ± 3 | 27 ± 4 | 76 ± 5 |
| PEEK-OH | S | 79 ± 5 | 38 ± 3 | 41 ± 6 |
| PEEK-Br | S | 92 ± 7 | 59 ± 5 | 33 ± 9 |
| PEEK-g-PPEGMA | S | 62 ± 1 | 25 ± 2 | 37 ± 2 |
| PEEK-g-PPEGMA | R | 68 ± 2 | 19 ± 4 | 49 ± 4 |
AFM analysis was additionally employed to evaluate the surface topography of the unmodified and the PPEGMA grafted PEEK surfaces as shown in Fig. 4. The determined roughness average (Ra) and root mean square roughness (Rq) of the rough side of the films are listed in Table 2. The measurements have been made on the rough side of the films as it was more uniform. Scratches on the smooth side of the film from the calendaring process or handling of the film made it impossible to obtain reliable values on that side. The AFM results showed that grafting of PPEGMA from the surface did not change the surface roughness significantly on the rough side. This was an important finding as surface roughness is expected to influence the adsorption of proteins. However, the smooth side will be of most interest for biological applications as it is known that even at the nanometre scale the roughness of the surface has a significant impact on protein adsorption. Thus more proteins will adsorb if the surface roughness is increased.21 The grafts are expected to be evenly distributed on both sides; therefore, a smooth PEEK surface without any scratches will presumably not be more rough after SI-ATRP of PEGMA.
 | ||
| Fig. 4 AFM images of the unmodified PEEK (left) and PEEK-g-PPEGMA (right). | ||
| Roughness | PEEK | PEEK-g-PPEGMA |
|---|---|---|
| R a/µm | 0.76 ± 0.12 | 0.79 ± 0.18 |
| R q/µm | 0.94 ± 0.14 | 1.0 ± 0.2 |
Thermal analysis made on unmodified PEEK, PEEK-g-PPEGMA, and PPEGMA homopolymer confirmed the presence of PPEGMA grafted from PEEK. The thermograms in Fig. 5 showed that the PPEGMA homopolymer started to decompose around 75 °C and total thermal decomposition was accomplished around 450 °C. On the other hand, the decomposition of PEEK begins well above 500 °C. Therefore, the 2–3% weight loss below 500 °C for the PEEK-g-PPEGMA film originated from PPEGMA. To our surprise the thermograms for PEEK and PEEK-g-PPEGMA cross at about 600 °C and we do not have a good explanation for this observation.
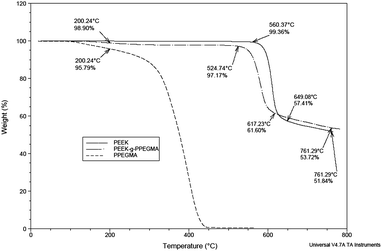 | ||
| Fig. 5 TGA of the unmodified PEEK, PPEGMA homopolymer, and PEEK-g-PPEGMA. | ||
XPS has been used to investigate the PEEK surface functionalization. Scan survey spectra were used to identify and quantify the elements in the modified PEEK samples. The results are collected in Table 3 where also the C/O ratio was calculated in order to follow the modification steps. When PPEGMA was grafted from the surface the C/O ratio was lowered as a consequence of the increased oxygen content in the grafts.
| Element (%) | PEEK | PEEK-OH | PEEK-Br | PEEK-g-PPEGMA | Calc. PPEGMA |
|---|---|---|---|---|---|
| C | 93.7 | 84.3 | 82.9 | 70.5 | 64 |
| O | 6.3 | 15.7 | 15.6 | 29.5 | 32 |
| Br | 1.6 | 4 | |||
| C/O ratio | 15.0 | 5.4 | 5.3 | 2.4 | 2 |
Chemical composition information was obtained from high resolution scans (Table 4). The ketones on the surface of the PEEK films were reduced to hydroxyl groups. Therefore, CO was not detected for PEEK-OH. This was previously also observed by Noiset et al.5 When the initiating sites were introduced OC was found due to the ester groups from Br-iBuBr. For the PPEGMA grafts the content of C–O increased as the PEG side chains contains ethers. XPS analysis of PEEK-g-PPEGMA confirmed that the PEEK surface was modified with PPEGMA as the measured chemical composition only resembles that of the PPEGMA brushes. Taken the penetration of the X-rays in XPS into account this implies that the PPEGMA layer is more than 10 nm.
| Binding Energy/eV | Attribution | PEEK (%) | PEEK-OH (%) | PEEK-Br (%) | PEEK-g-PPEGMA (%) |
|---|---|---|---|---|---|
| a The program was unable to resolve the peaks. | |||||
| 284.3–284.9 | C–C (C1s) | 90.3 | 77.4 | 72.0 | 42.5 |
| 285.3–286.5 | C–O (C1s) | 8.0 | 17.0 | 20.7 | 50.4 |
| 288.3–289.0 | CO (C1s) |
7.0 | |||
| <283.9 or >287.7 | Residual | 1.7 | 5.6 | 3.0 | |
| 530.8–531.9 | OC (O1s) |
82.3 | 50.8 | 3.5 | |
| 532.5 | O–H (O1s) | 77.4 | |||
| 532.6–533.5 | O–C (O1s) | 11.4 | 97.9 | 49.2 | 14.4 |
| <530.4 or >534.1 | Residual | 6.4 | 2.1 | 4.7 |
The hydrophilic PPEGMA grafts have two different applications as illustrated in Fig. 6. They can either be metallised or inherently used to avoid non-specific fouling. When PEEK-g-PPEGMA is pulled out of the metallisation bath the metal should be bound to the surface. On the other hand, proteins should not be adsorbed to the surface if it has been exposed to a protein solution.
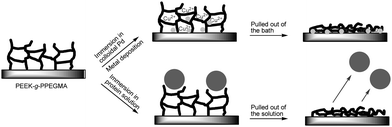 | ||
| Fig. 6 One functionalization with two different applications, electronics and medical. | ||
PEEK is highly inert and as many other polymers cannot be electrolessly metallised without a proper surface treatment.2 By the employed conditions described in Experimental section no metal deposition on the virgin surface of PEEK was achieved. However, metal films of both nickel and copper could be successfully deposited on the modified PEEK-g-PPEGMA. Optical microscope images at 40 times magnification (Fig. 7) showed a homogeneous rough surface, where the smooth surface is shiny, but has some cracks possibly from the scratches on the calendared PEEK.
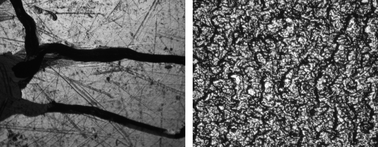 | ||
| Fig. 7 40 times magnification of PEEK-g-PPEGMA with nickel; (left) the smooth side of the film; (right) the rough side of the film. | ||
To evaluate how well the metal coating was bonded to the substrate a known simple adhesive tape-test was first performed. A pressure sensitive adhesive tape was applied to the surface, pressed well with fingers in order to adhere to the whole metal surface and then removed. If no metal was transferred to the adhesive tape, the adhesion of the metal layer was measured by the pull-off test. The metal film on the smooth surface can be removed by the adhesive tape-test, whereas the one on the rough surface withstand this test, and showed quite high adhesion as seen in Table 5. The bond failure for the Cu-deposition was mixed (both cohesive failure between the Cu-coating/PEEK-g-PPEGMA and adhesive failure Cu/glue). By performing the pull off test for PEEK-g-PPEGMA–Ni, the break was adhesive between the nickel and the glue. Therefore, the real pull off forces might be higher than the recorded forces listed in Table 5.
| Material | Failure | Force |
|---|---|---|
| PEEK-g-PPEGMA-Cu | Mixed | >4.4 MPa |
| PEEK-g-PPEGMA-Ni | Adhesive | >5.7 MPa |
In this way deposition of two important metals, nickel and copper, was achieved onto the prepared PEEK-g-PPEGMA surfaces. Moreover, this happens without use of the hazardous chromic acids, which are usually involved in the first treatment of polymer surfaces prior to metallisation. This process is schematically illustrated in Fig. 6. The metallisation takes place in two steps. Firstly, metal complexes are formed when colloidal palladium is deposited as the metal ions are trapped into the gelated polymer brush layer. Secondly, immersion in a copper or nickel bath resulted in complexation of copper or nickel with the ethylene oxide units of the PPEGMA. When pulled out of the solution and dried, the swelled layer of PPEGMA collapses and captures the metal. Since PPEGMA is covalently bound to the substrate, it provides good mechanical and chemical adhesion to the substrate.
Confocal microscopy has been applied to investigate whether proteins labeled with a fluorophore would adsorb to the PPEGMA surface. Unfortunately, PEEK exhibits autofluorescence at all the employed wavelengths (405, 445, 488, 555, and 639 nm) which makes it impossible to obtain good images of unmodified and modified PEEK exposed to fluorescenced proteins. However, PPEGMA grafted from both polymeric and metallic substrates using SI-ATRP has previously been reported as a non-fouling material.22
Conclusions
Polymer brushes of PPEGMA were grafted from PEEK films by use of surface-initiated ATRP. The water contact angles were lower for the modified PEEK films; thus hydrophilization of PEEK was achieved. AFM analyses showed that the surface modification did not change the surface roughness. The C/O ratio as well as data from the high resolution XPS confirmed the grafting from the PEEK films. The hydrophilicity of the material, PEEK-g-PPEGMA, makes it applicable for both metallisation and inhibition of non-specific fouling. Two metals, copper and nickel, were electroless deposited on modified PEEK surfaces with quite high adhesion. Pull-off forces higher than 4.4 MPa and 5.7 MPa were required to remove the deposited copper and nickel, respectively, from the modified PEEK surfaces.Acknowledgements
CJF acknowledges the Technical University of Denmark, Novo Nordisk A/S, and the Danish Agency for Science Technology and Innovation for financial support. The Ministry of Science Technology and Innovation is acknowledged for financial support through grant 61568. Furthermore, we thank Torben Tang for providing the electroless nickel and copper baths as well as fruitful discussions, Ilze Viskere for performing the metallization and pull off tests, Lene Hubert for the XPS results, and P. S. Ramanujam for the AFM analyses.References
- J. J. Pireaux, Synth. Met., 1994, 67, 39–46 CrossRef CAS.
- R. Suchentrunk and K. Heymann, Kunststoff-Metallisierung: Handbuch für Theorie und Praxis, Leuze, Saulgau/Württ, 1991 Search PubMed.
- A. Y. Yi, W. Lu, D. F. Farson and L. J. Lee, Adv. Polym. Technol., 2008, 27, 188–198 CrossRef CAS.
- (a) O. Noiset, Y. J. Schneider and J. Marchand-Brynaert, J. Biomater. Sci., Polym. Ed., 1999, 10, 657–677 CrossRef CAS; (b) O. Noiset, Y. J. Schneider and J. Marchand-Brynaert, J. Biomater. Sci., Polym. Ed., 2000, 11, 767–786 CrossRef CAS.
- O. Noiset, C. Henneuse, Y. J. Schneider and J. Marchand-Brynaert, Macromolecules, 1997, 30, 540–548 CrossRef CAS.
- (a) J. Marchand-Brynaert, G. Pantano and O. Noiset, Polymer, 1997, 38, 1387–1394 CrossRef CAS; (b) O. Noiset, Y. J. Schneider and J. Marchand-Brynaert, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 3779–3790 CrossRef CAS.
- S. M. Kurtz and J. N. Devine, Biomaterials, 2007, 28, 4845–4869 CrossRef CAS.
- (a) W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS; (b) N. M. L. Hansen, K. Jankova and S. Hvilsted, Eur. Polym. J., 2007, 32, 255–293 CrossRef; (c) J. A. Opsteen and J. C. M. van Hest, in Macromolecular Engineering, ed. K. Matyjaszewski, Y. Gnanou and L. Leibler, Wiley-VCH, Weinheim, 2007, vol. IV, ch. 16.4, pp. 2662–2671 Search PubMed; (d) N. V. Tsarevsky and K. Matyjaszewski, Chem. Rev., 2007, 107, 2270–2299 CrossRef CAS.
- (a) R. Barbey, L. Lavanant, D. Paripovic, N. Schüwer, C. Sugnaux, S. Tugulu and H.-A. Klok, Chem. Rev., 2009, 109, 5437–5527 CrossRef CAS; (b) C. J. Fristrup, K. Jankova and S. Hvilsted, Soft Matter, 2009, 5, 4623–4634 RSC; (c) D. Roy, M. Semsarilar, J. T. Guthrie and S. Perrier, Chem. Soc. Rev., 2009, 38, 1825–2148 RSC; (d) F. J. Xu, K. G. Neoh and E. T. Kang, Prog. Polym. Sci., 2009, 34, 719–761 CrossRef CAS.
- B. Yameen, M. Álvarez, O. Azzaroni, U. Jonas and W. Knoll, Langmuir, 2009, 25, 6214–6220 CrossRef CAS.
- X. Liu, H. Chang, Y. Li, W. T. S. Huck and Z. Zheng, ACS Appl. Mater. Interfaces, 2010, 2, 529–535 Search PubMed.
- P. C. Hidber, W. Helbig, E. Kim and G. M. Whitesides, Langmuir, 1996, 12, 1375–1380 CrossRef CAS.
- J. F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- J. H. Lee, H. B. Lee and J. D. Andrade, Prog. Polym. Sci., 1995, 20, 1043–1079 CrossRef CAS.
- D. L. Elbert and J. A. Hubbell, Annu. Rev. Mater. Sci., 1996, 26, 365–394 CrossRef CAS.
- M. Malmsten, K. Emoto and J. M. Van Alstine, J. Colloid Interface Sci., 1998, 202, 507–517 CrossRef CAS.
- ISO 4624 (2002) “Paints and Varnishes—Pull Off-Test for Adhesion” and ASTM D 4541-09(2009) “Standard Test Method for Pull-Off Strength of Coatings Using Portable Adhesion Testers”.
- A. Carlmark and E. Malmstrom, J. Am. Chem. Soc., 2002, 124, 900–901 CrossRef CAS.
- N. Singh, J. Wang, M. Ulbricht, S. R. Wickramasinghe and S. M. Husson, J. Membr. Sci., 2008, 309, 64–72 CrossRef CAS.
- J. Y. Huang, H. Murata, R. R. Koepsel, A. J. Russell and K. Matyjaszewski, Biomacromolecules, 2007, 8, 1396–1399 CrossRef CAS.
- K. Rechendorff, M. B. Hovgaard, M. Foss, V. P. Zhdanov and F. Besenbacher, Langmuir, 2006, 22, 10885–10888 CrossRef CAS.
- (a) F. X. Hu, K. G. Neoh, L. Cen and E. T. Kang, Biomacromolecules, 2006, 7, 809–816 CrossRef CAS; (b) B. S. Lee, Y. S. Chi, K. B. Lee, Y. G. Kim and I. S. Choi, Biomacromolecules, 2007, 8, 3922–3929 CrossRef CAS; (c) L. Li, G. P. Yan, J. Y. Wu, X. H. Yu and Q. Z. Guo, J. Macromol. Sci., Part A: Pure Appl. Chem., 2008, 45, 828–832 Search PubMed; (d) N. Singh, X. F. Cui, T. Boland and S. M. Husson, Biomaterials, 2007, 28, 763–771 CrossRef CAS; (e) S. Tugulu and H. A. Klok, Biomacromolecules, 2008, 9, 906–912 CrossRef CAS; (f) F. J. Xu, Y. L. Li, E. T. Kang and K. G. Neoh, Biomacromolecules, 2005, 6, 1759–1768 CrossRef CAS; (g) F. J. Xu, J. P. Zhao, E. T. Kang, K. G. Neoh and J. Li, Langmuir, 2007, 23, 8585–8592 CrossRef CAS; (h) F. J. Xu, S. P. Zhong, L. Y. L. Yung, E. T. Kang and K. G. Neoh, Biomacromolecules, 2004, 5, 2392–2403 CrossRef CAS; (i) G. Q. Zhai, E. T. Kang and K. G. Neoh, Macromolecules, 2004, 37, 7240–7249 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |