Dual hydrophilic polymers based on (meth)acrylic acid and poly(ethylene glycol) – synthesis and water uptake behavior†
Andreas
Krieg
ac,
Christian
Pietsch
ac,
Anja
Baumgaertel
ac,
Martin D.
Hager
ac,
C. Remzi
Becer
*abc and
Ulrich S.
Schubert
*abc
aLaboratory of Organic and Macromolecular Chemistry, Friedrich-Schiller-University Jena, Humboldtstrasse 10, 07743, Jena, Germany. E-mail: ulrich.schubert@uni-jena.de
bLaboratory of Macromolecular Chemistry and Nanoscience, Eindhoven University of Technology, PO Box 513, 5600, MB, Eindhoven, The Netherland. E-mail: c.r.becer@warwick.ac.uk
cDutch Polymer Institute (DPI), John F. Kennedylaan 2, 5612, AB, Eindhoven, The Netherlands
First published on 8th September 2010
Abstract
Synthesis and characterization of dual hydrophilic random and block copolymers of acrylic acid (AA) or methacrylic acid (MAA) with poly(ethylene glycol) (PEG) via different controlled radical polymerization techniques are discussed. Initially, reversible addition fragmentation chain transfer (RAFT) polymerization was employed to synthesize homo, random and block copolymers of AA and MAA in ethanol. The polymers were characterized in detail by means of size exclusion chromatography (SEC), 1H NMR spectroscopy, matrix assisted laser desorption ionization time of flight (MALDI-TOF) mass spectrometry as well as MALDI-TOF MS coupled with collision induced dissociation (CID) to identify the end groups and the repeating units. Following that, atom transfer radical polymerization (ATRP) and RAFT polymerization were employed for the preparation of block copolymers using a PEG macroinitiator and a PEG macro chain transfer agent. Moreover, graft copolymers that contain oligo(ethyleneglycol) pendant groups and AA or MAA have been prepared using the RAFT polymerization process. Additionally, selected homo or block copolymers were tested for their water-uptake properties using a thermal gravimetrical analyzer with a controlled humidity chamber. An advantageous behavior of the copolymers compared to the related homopolymers was reached with the obtained ability to absorb moisture over the complete humidity range as well as to a very high absolute water uptake.
Introduction
Water-soluble polymers with various architectures are of great interest in the field of polymer science due to their wide range of possible applications, i.e. in drug-delivery systems, dispersing agents and absorbent materials.1–4 The most widely used structures to construct dual hydrophilic polymers are based on acrylic acid (AA), methacrylic acid (MAA) and poly(ethylene glycol) (PEG). AA and MAA can be polymerized by controlled radical polymerization techniques whereas PEG can be obtained by ionic polymerization.5–7 Various polymeric architectures can be constructed using these monomers or polymers. However, the synthesis and characterization of well-defined water soluble polymers requires not only dedicated reaction conditions and catalysts but also suitable analytical instruments to precisely assign their structures.There are two main approaches which are possible for obtaining polymers of desired architectures. The first possibility is the reaction of end group or side group functionalized polymers with the second block or functional side group to form block or graft copolymers, respectively. The reactions which are nowadays employed in this route are mainly found among the so called “click” reactions.8–10 The other possibility is the construction of the desired compositions and architectures directly by the polymerization process itself. Therefore, macroinitiators, macromonomers or macro chain transfer agents (macro-CTA) have to be prepared according to the targeted polymeric structures.11 In the present study, the latter approach was applied to obtain linear as well as graft block and random copolymers.
Controlled radical polymerization (CRP) techniques provide enormous possibilities for synthesizing well-defined polymers with controlled architectures and molar masses. For instance, reversible addition fragmentation chain transfer (RAFT) polymerization allows the use of acidic monomers and also the use of polar solvents like ethanol. Therefore, this technique is the most widely employed method to prepare water soluble polymers.12 However, the CTA has to be carefully selected depending on the nature of the monomer.13–15
Alternatively, transition metal-catalyzed controlled radical polymerization techniques provide good control over the polymerization of several monomers.16 Unfortunately, these techniques, namely atom transfer radical polymerization (ATRP) and single electron transfer controlled radical polymerization (SET-LRP), are based on the oxidation reduction equilibrium of the transition metal and ligand complex which can be disturbed in the presence of acidic monomers.17–19 As a consequence, protected monomers have to be used during the polymerization.
While actual studies are, for example, investigating the morphology of dual hydrophilic copolymers, e.g. P(AA-co-EG) nanofibers, like recently reported by Charleux et al.,20 the present contribution focuses on structural investigation and the study of the water uptake behavior.
Water uptake properties of polymers are relatively rarely investigated, but represent a very important polymer characteristic and play a crucial role in several applications of polymers such as personal care products,21–23 coatings, composite materials, cement,24 membranes and sensors,25 agriculture products,26–30 biomedical materials,31–33 insulation of underwater cables34 or recreational activities (e.g. artificial snow). Researchers have focused on the modification and optimization of polymers in terms of water absorbency, absorption–desorption rates and gel strength (in cross-linked systems).35 Moisture uptake can be measured directly from water or from a humid atmosphere. However, most of the reported research has focused on the investigation of cross-linked polymer systems (super absorbent polymers) in direct contact with water. Other measurements require the use of conditioned desiccators and several days to weeks of measurement time until the samples are saturated with water molecules. Alternatively, water uptake properties of polymeric materials can be measured by a thermal gravimetric analyzer with a controlled humidity chamber. This technique requires a very small amount of sample (a few milligrams) and the measurement can be performed in any form, such as in powder, liquid or crosslinked gel.
Different strategies to synthesize dual hydrophilic polymers in various architectures were studied, as illustrated in Scheme 1. Homopolymers, block and random copolymers of AA and MAA have been prepared using RAFT polymerization. Moreover, graft copolymers were prepared using oligo(ethylene glycol) acrylate (OEGA) and AA or MAA. Furthermore, macroinitiator and macro chain transfer agents were synthesized in order to use those in the block copolymerization. ATRP and RAFT polymerization were employed to synthesize PEG-b-AA and PEG-b-MAA block copolymers. Finally, these polymers are tested for their water uptake properties.
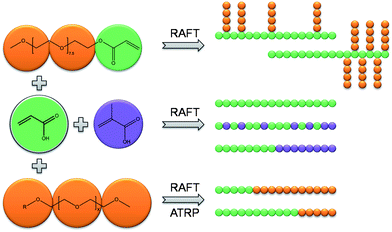 | ||
| Scheme 1 A schematic representation of the monomers used and the block and random copolymer architectures obtained by different synthetic approaches. | ||
Experimental section
Materials
Poly(ethylene glycol) monomethyl ether (mPEG1k: CH3-PEG22-OH, Mn = 1000 g mol−1, PDI = 1.07 and mPEG2k: CH3-PEG44-OH, Mn = 2000 g mol−1, PDI = 1.08) were purchased from Sigma-Aldrich. The monomers acrylic acid (AA), methacrylic acid (MAA), tert-butyl acrylate (tBuA) and oligo(ethylene glycol) acrylate (Mn = 480 g mol−1) (OEGA480) were purchased from Sigma-Aldrich and passed through a column with inhibitor remover by Sigma-Aldrich before using. CuBr, N,N′,N′,N′′,N′′-pentamethyl diethylene triamine (PMDETA) and ethyl 2-bromo-isobutyrate (EtBriB) were purchased from Sigma-Aldrich and used as received without further purification. 2-(Butylthiocarbonothioylthio) propanoic acid (BPTC) and 2-cyano-2-butyldithiobenzoate (CBDB) were synthesized according to the literature and used without further purification.36,37 Azobis(isobutyronitrile) (AIBN) was received from Sigma-Aldrich and recrystallized from methanol before using. Other solvents and reagents were used as received without further purification.Instrumentation
1H NMR spectra were recorded in CDCl3 or DMSO-d6 on a Bruker AC 300 MHz using the residual solvent resonance as an internal standard. Size exclusion chromatography (SEC) was performed on a Shimadzu system equipped with a SCL-10A system controller, a LC-10AD pump, a RID-10A refractive index detector and both a PSS Gram30 and a PSS Gram1000 column in series, whereby N,N-dimethylacetamide (DMAc) with 5 mmol lithium chloride (LiCl) was used as an eluent at 1 mL min−1 flow rate and the column oven was set to 60 °C. The system was calibrated with polystyrene (370 g mol−1–67![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 g mol−1) and poly(methyl methacrylate) (2000 g mol−1–88000 g mol−1) standards, respectively. For the measurement of the matrix assisted laser desorption/ionization (MALDI) spectra an Ultraflex III TOF/TOF (Bruker Daltonics, Bremen, Germany) instrument was used. The instrument was equipped with a Nd:YAG laser and a collision cell. All spectra were measured in the positive reflector or linear mode. The instrument was calibrated prior to each measurement with an external PMMA standard from PSS Polymer Standards Services GmbH (Mainz, Germany). Monomer conversions were determined by gas chromatography (GC) using anisole as internal standard. The number-average molar mass (Mn) and the polydispersity index (PDI) were determined by size-exclusion chromatography (SEC) using chloroform or N,N-dimethyl acetamide (DMAc) as solvents depending on the solubility behavior of the samples. The water-uptake measurements of the polymers were investigated on a Q5000 SA thermogravimetric analyzer from TA Instruments containing a microbalance in which the sample and reference pans were enclosed in a humidity and temperature controlled chamber. The temperature in the Q5000 SA was controlled by Peltier elements. Dried N2 gas flow (200 mL min−1) was split into two parts, of which one part was wetted by passing it through a water-saturated chamber. The desired relative humidity (RH) for the measurements could subsequently be obtained by mixing proper proportions (regulated by mass-flow controllers) of dry and wet stream.
500 g mol−1) and poly(methyl methacrylate) (2000 g mol−1–88000 g mol−1) standards, respectively. For the measurement of the matrix assisted laser desorption/ionization (MALDI) spectra an Ultraflex III TOF/TOF (Bruker Daltonics, Bremen, Germany) instrument was used. The instrument was equipped with a Nd:YAG laser and a collision cell. All spectra were measured in the positive reflector or linear mode. The instrument was calibrated prior to each measurement with an external PMMA standard from PSS Polymer Standards Services GmbH (Mainz, Germany). Monomer conversions were determined by gas chromatography (GC) using anisole as internal standard. The number-average molar mass (Mn) and the polydispersity index (PDI) were determined by size-exclusion chromatography (SEC) using chloroform or N,N-dimethyl acetamide (DMAc) as solvents depending on the solubility behavior of the samples. The water-uptake measurements of the polymers were investigated on a Q5000 SA thermogravimetric analyzer from TA Instruments containing a microbalance in which the sample and reference pans were enclosed in a humidity and temperature controlled chamber. The temperature in the Q5000 SA was controlled by Peltier elements. Dried N2 gas flow (200 mL min−1) was split into two parts, of which one part was wetted by passing it through a water-saturated chamber. The desired relative humidity (RH) for the measurements could subsequently be obtained by mixing proper proportions (regulated by mass-flow controllers) of dry and wet stream.
Synthesis
:PAA:AIBN = 5:1:0.25) and the solution was further heated for 6 h. The polymer was isolated by precipitation into cold ethyl acetate and washed with diethyl ether to remove the residual monomer and solvent. The polymer was dried until the mass of the sample was constant.
1H NMR (300 MHz, CDCl3) δ = 4.31 (m, CH2–OCO), 3.63 (m, O–CH2), 3.36 (m, CH3–O), 1.92 (s, (CH3)2CBr) ppm.
1H NMR (300 MHz, CDCl3) δ = 4.73 (CH–SCS2), 4.20 (CH2–OCO), 3.55 (CH2–O), 3.28 (CH3–O), 3.26 (CH2–SCS2), 1.59 (CH2(CH2)2), 1.50 (CH3–CH), 1.33 (CH2–CH3), 0.84 (CH2–CH3) ppm.
Results and discussion
The synthesis of polymers based on AA or MAA via CRP techniques can be challenging due to the acidic nature, as described previously. Therefore, the RAFT polymerization was preferred over ATRP to obtain the desired polymers. However, a dedicated CTA is necessary to successfully polymerize both acrylic and also methacrylic monomers. CBDB is an adequate CTA to reach control in the present case and allowed the preparation of the desired random and block copolymers of AA and MAA. The obtained SEC results of homopolymers, random- and block copolymers of AA and MAA are shown in Fig. 1. | ||
| Fig. 1 SEC traces of PAA (top left), P(MAA-r-AA) (top right) and P(MAA-b-AA) (bottom left). | ||
The relatively similar structures of AA and MAA raise the question about the possibilities to determine the compositions of the obtained polymers. An indirect approach is the calculation of molar mass and polymer composition in correlation to the monomer conversion during the polymerization which can be determined by GC. On the other hand, 1H NMR spectroscopy allows to distinguish between both compounds in the final polymer; thereby a selective and quantitative analysis of the real polymer composition can be performed (Fig. 2).
 | ||
| Fig. 2 1H NMR spectra (300 MHz, CDCl3) of P(MAA) (top) and P(MAA-co-AA) (bottom) with the corresponding schematic representation of the polymer structures. | ||
The conversion of the monomers were calculated using both GC and 1H NMR spectroscopy. This allows the direct calculation of the polymer composition. The comparison of the obtained values using both approaches shows a rather good agreement in most cases. It has to be considered that possible solvent residues, in particular those with alkyl functionalities, can lead to an overestimation of the MAA content using the 1H NMR technique.
The versatility of the RAFT technique allowed the synthesis of various polymers with different compositions. A comparison of the prepared polymers including their characteristic data obtained by several analytical techniques, i.e. GC, SEC, and 1H NMR spectroscopy, is provided in Table 1.
| Run | MAA | AA | MAAa | AAa | M n,theo | M n,SEC | PDIc | MAA:AA |
MAA : AAd |
|---|---|---|---|---|---|---|---|---|---|
| (feed) | (feed) | (conv.) | (conv.) | /g mol−1 | /g mol−1 | (GC) | (1H NMR) | ||
| a Conversion values were determined by GC. b Calculated according to formula (Mn(theo.) = ([M]/[CTA] × conv. × MMonomer) + MCTA). c Calculated according to PS standards. d The ratios were calculated from the corresponding peaks shown in Fig. 2. | |||||||||
| H1 | — | 30 | — | 0.84 | 2053 | 4300 | 1.19 | 0:1 |
0:1 |
| H2 | 25 | — | 0.54 | — | 1396 | 3600 | 1.31 | 1:0 |
1:0 |
| R 1 | 20 | 48 | 0.96 | 0.60 | 3960 | 7900 | 1.26 | 0.39:0.61 |
0.40:0.60 |
| R 2 | 25 | 25 | 0.87 | 0.55 | 3099 | 6400 | 1.25 | 0.61:0.39 |
0.60:0.40 |
| R3 | 11 | 67 | 0.88 | 0.43 | 3146 | 6900 | 1.21 | 0.26:0.74 |
0.33:0.67 |
| B1 | 18 | 35 | 0.99 | 0.50 | 3027 | 6000 | 1.34 | 0.50:0.50 |
0.71:0.29 |
| B2 | 20 | 25 | 0.95 | 0.45 | 2679 | 4300 | 1.39 | 0.63:0.37 |
0.67:0.33 |
| B3 | 20 | 60 | 0.96 | 0.3 | 3182 | 7300 | 1.28 | 0.53:0.47 |
0.55:0.44 |
SEC is a widely used technique in polymer analysis. The obvious discrepancy between the theoretical molar masses and the experimental values in the current case is caused by the significantly larger hydrodynamic volume of the polymers prepared in comparison to the polymer standards used for the calibration of the SEC (PS calibration). A route to circumvent this problem is the use of a direct molar mass determination technique, i.e. MALDI-TOF mass spectrometry. Since the exact determination of polymeric structures is one of the most important goals in polymer research, this technique represents a very powerful tool. The combination of a mass spectrometric analysis of a polymer, providing a molar mass distribution, with the subsequent fragmentation of single and distinct macromolecules offers a great possibility to analyze polymers down to their precise structure and composition. The prerequisite for such a tandem mass spectrometric analysis is the selective admittance of already desorbed and ionized molecules into a gas filled collision cell. Further fragmentation occurs inside, which is kinetically induced by the collision gas (argon or nitrogen). Fig. 3 shows the analysis of the selected polymers by MALDI-TOF MS and tandem-MS technique. However, besides the stated advantages, one should be aware of potential disadvantages of the MALDI-TOF MS technique. Due to its rather harsh ionization method, fragmentations can occur during the measurement process itself leading to fragments not representing the real polymer structure. Furthermore, possible ionization biases can make an accurate molar mass determination difficult. MS techniques applying softer ionization methods, e.g. electron spray ionization (ESI), can add further insights.38
 | ||
| Fig. 3 Representative MALDI-TOF mass spectra of PAA (H1) (top left), P(MAA-r-AA) (R3) (bottom left) and P(MAA-b-AA) (B3) (bottom right). Tandem MS spectrum of PAA (top right). | ||
The MALDI-TOF mass spectrum of the homopolymer allows the assignment of molecular structures corresponding to the obtained distributions. The distance between the signals of each distribution corresponds to the molar mass of the monomer unit. The main distribution represents the expected structure plus the charge carrying sodium ion. The three minor signal distributions are probably caused by fragmentations during the MALDI measurement itself. One of these fractions can for example be assigned to the expected structure after the loss of the n-butyl substituent from the end-group.
Furthermore, tandem mass spectrometry is possible providing an even deeper insight into the polymeric structure. The major signals in the high molar mass region can be assigned to the desired structure after losing H2O (−18) and CO2 (−44) due to anhydrite formation, decarboxylation reactions and combinations of both.39 The two important signals marked as −I and −E represent the polymer after loss of the initiator group (−I) or the chain transfer agent end group (−E). The Δ72 distribution in the middle mass region represents the different chain lengths, which can be formed by fragmentation of different amounts of repeating units of AA.
The clear structure of the homopolymer mass spectra is lost in the case of the random and block copolymers. The large amount of possible monomer combinations and, thereby, the wide range of resulting molar masses leads to complex spectra. Single signal assignments are no longer possible.
The resulting Mn values correspond to the expected molar masses calculated from the monomer. In case of the block copolymer, residual homopolymer of the unconverted first block is visible on the low molar mass shoulder of the distribution. However, it is not possible to draw any quantitative conclusions concerning the amount of remaining homopolymer from the MALDI-TOF mass spectra.
Dual hydrophilic block copolymers composed of EG and AA are a versatile polymer class suitable for many applications, e.g. to control structure and size of mineral particles.40 The synthetic approach mainly utilized towards these polymers up to now is the synthesis of a macro-initiator suitable for ATRP by an esterification reaction. Subsequently a protected acrylic monomer, e.g. t-butyl acrylate (t-BuA), is polymerized onto this macroinitiator, providing the desired poly(EG-b-AA) after a deprotection step.
A second possibility, with the advantage of avoiding the deprotection step, is the synthesis of a macro chain transfer agent applicable for a RAFT polymerization of acrylic acid. A visualization of both synthetic approaches is provided in Scheme 2. In the present study, both methods were applied to obtain P(EO-b-AA) block copolymers. In Table 2, the characteristic data for all polymers prepared using both approaches are summarized.
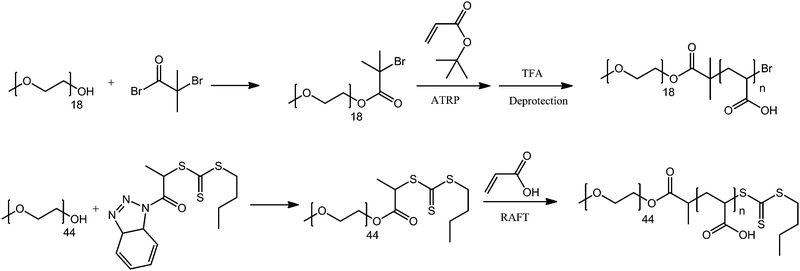 | ||
| Scheme 2 A schematic representation of the ATRP macroinitiator approach (top) and the RAFT macro chain transfer agent approach (bottom) towards P(EG-b-AA) block copolymers. | ||
| Type | M n | PDIa | M p | M n | Comp.d |
|---|---|---|---|---|---|
| /g mol−1 | /g mol−1 | /g mol−1 | EG:A. |
||
| a Determined by SEC analysis according to PS standards. b Determined by MALDI-TOF/MS analysis. c Determined by 1H NMR measurements. d The ratios were calculated from the corresponding peaks. | |||||
| mPEG1k | 1700 | 1.06 | 1000 | — | — |
| mPEG-Br | 2100 | 1.07 | 1040 | — | 1:0 |
| P(EG-b-tBuA) | 4100 | 1.09 | 5050 | 5250 | 0.32:0.68 |
| P(EG-b-AA) | 8500 | 1.22 | 3100 | 3390 | 0.33:0.67 |
| mPEG2k | 1950 | 1:0 |
|||
| mPEG-CTA | 2900 | 1.08 | 2000 | 2130 | 1:0 |
| P(EG-b-AA) | 12000 |
1.27 | 5300 | 5480 | 0.49:0.51 |
The macroinitiator synthesis as well as the polymerization and deprotection steps could be followed via MALDI-TOF mass spectrometry. The resulting spectra including the assigned structures are provided exemplarily for the ATRP approach in Fig. 4.
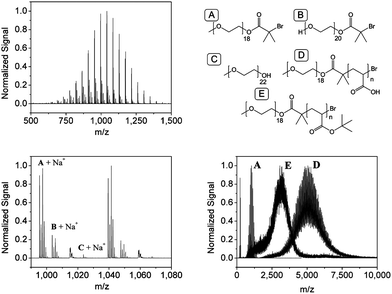 | ||
| Fig. 4 MALDI-TOF mass spectrum of the PEG macroinitiator (top left), its magnification (bottom left) and an overlay of the MALDI-TOF mass spectra of macro-initiator, P(EG-b-tBuA) and P(EG-b-AA) (bottom right) with a schematic representation of the assigned structures (top right). | ||
The main distribution could be assigned to the desired product plus a sodium ion. Also the second largest distribution represents an acceptable product with the only difference being a hydroxyl end-group at the second chain-end of the macroinitiator. Only a very small educt specific distribution is observed. An overlay of the MALDI-TOF mass spectra of the macroinitiator and its block copolymer with tBuA before and AA after deprotection illustrates the controlled synthesis of the block copolymer as well as its subsequent deprotection.
The approach via the RAFT-polymerization enabled the synthesis of comparably well-defined polymers. The MALDI-TOF mass spectra are provided in Fig. 5. The starting material and product resulted in the same molar mass and are thereby not distinguishable by MALDI-TOF/MS. Nevertheless, the desired structure could be proven by tandem MS analysis leading to a MS spectrum of the product which is different to mPEG-OH.
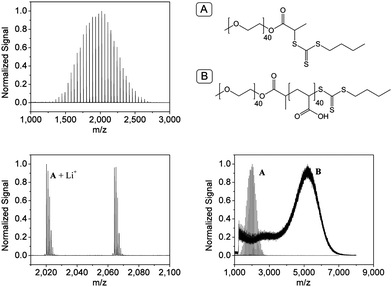 | ||
| Fig. 5 MALDI-TOF mass spectrum of the mPEG macro-CTA (top left), magnification of the main distribution (bottom left) and an overlay of the MALDI-TOF mass spectra of the macro chain transfer agent and the prepared P(EG-b-AA) (bottom right) with a schematic representation of the assigned structures (top right). | ||
The SEC analysis revealed once more the difference in hydrodynamic volume between polymers containing acrylic acid and the usual standard polymers used to calibrate the SEC. Fig. 6 visualizes the SEC results obtained for the P(EG-b-AA) polymers in comparison to their precursors prepared by the ATRP and the RAFT approach. To obtain comparable results PS-calibrations were used to calculate molar masses and PDI values. Nevertheless nearly quantitative conversions between different states of the block copolymer synthesis could be observed using SEC analysis.
 | ||
Fig. 6 Overlay of SEC traces of polymers obtained from the ATRP approach (P(EG-b-AA) (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ), P(EG-b-tBuA) ( ), P(EG-b-tBuA) ( ), mPEG macroinitiator ( ), mPEG macroinitiator ( )) (left) and the ones obtained during the RAFT approach (P(EG-b-AA) () and mPEG macro-CTA ( )) (left) and the ones obtained during the RAFT approach (P(EG-b-AA) () and mPEG macro-CTA ( )) (right). )) (right). | ||
The routes described led to linear block copolymers P(EG-b-AA). The copolymerization of AA and oligo(ethylene glycol) acrylate (Mn = 480 g mol−1) (OEGA480) will lead to graft block and random copolymers. The chosen technique to obtain these polymers was the RAFT polymerization using BPTC as CTA and AIBN as radical source.
The resulting polymers including characterizing data obtained by SEC and 1H NMR measurements are summarized in Table 3.
| Sample | Feed | M n (theor.)a | M n (SEC) | PDIb | Comp.c |
|---|---|---|---|---|---|
| /g mol−1 | /g mol−1 | (SEC) | (1H NMR) | ||
| a Calculated according to formula (Mn(theor.) = ([M]/[CTA] × conv. × MMonomer) + MCTA). b Calculated according to PS standards. c The ratios were calculated from the corresponding peaks shown in Fig. 7. | |||||
| POEGA480 | 0/8 | 3800 | 3000 | 1.15 | — |
| R4 | 35/5 | 4800 | 6400 | 1.28 | 0.81:0.19 |
| R5 | 45/5 | 5700 | 7100 | 1.21 | — |
| R6 | 70/5 | 7700 | 8100 | 1.38 | 0.91:0.09 |
| B4 | 35/5 | 4800 | 6000 | 1.20 | 0.87:0.13 |
| B5 | 45/5 | 5700 | 6600 | 1.25 | — |
| B6 | 70/5 | 7700 | 11 000 | 1.20 | 0.93:0.07 |
The specific separated signals obtained by 1H NMR spectroscopy enabled the determination of the relative and even the absolute polymer composition as seen in Fig. 7. The accuracy of the determination of the absolute composition is of course dependent on a quantitative end-group functionalization. Analysis via SEC revealed narrow molar mass distributions which led to different molar mass values due to the discrepancy in the hydrodynamic volume relative to the polystyrene standards used for calibration.
 | ||
| Fig. 7 1H NMR spectrum for P(AA-co-OEGA480) with the corresponding schematic representation of the structure and the signal assignments (300 MHz, DMSO-d6). | ||
To investigate the water uptake behavior of the prepared polymers, thermogravimetric analysis of dried polymer samples were performed under changing humidity. In previous studies the water uptake of several linear polymers including PEG, PAA and poly(sodium acrylate) was investigated and reported.4 These measurements showed the remarkable difference of the absorption behavior of pure PAA and the sodium salt of PAA as well as PEG. The highest water uptake at 90% relative humidity was observed for the PAA sodium salt (88%) and PEG (73%) whereas pure PAA could take up only 33%. In difference to the other two polymers, PAA absorbs water during the whole range of air humidity (10 to 90%) while PEG and PAA sodium salt do not absorb relevant amounts of water before a humidity level of at least 40% (PAA sodium salt) or even 70% (PEG) is reached.
In the present work the water uptake of P(MAA-b-AA) copolymers and P(EG-b-AA) was investigated. The results are shown in Fig. 8. While the ratio of MAA and AA in the corresponding block copolymers seems to have only a small influence, the total water uptake is surprisingly high in particular for B3 (59% at 90% humidity).
 | ||
| Fig. 8 Water uptake measurements of P(MAA-b-AA) with varying composition and P(EG-b-AA). | ||
An interesting result was observed in case of the P(EG40-b-AA42) block copolymer. The high water uptake of PEG at high humidity levels could be combined with the early starting uptake of water of PAA at lower humidity levels. Therefore, the resulting combined water uptake behavior could overcome the disadvantages of each homopolymer leading to the ability to absorb moisture over the complete humidity range as well as to a very high absolute water uptake level.
Conclusions
Dual hydrophilic copolymers consisting of acrylic acid and methacrylic acid could be synthesized successfully. Random as well as block copolymers were prepared by controlled radical polymerization, i.e. the RAFT technique. The materials obtained were characterized by multiple analytical techniques like SEC, MALDI-TOF mass spectrometry, 1H NMR spectroscopy, and water uptake measurements.Well-defined dual hydrophilic linear and graft block copolymers based on acrylic acid and poly(ethylene glycol) could be prepared by RAFT and ATRP. The synthesized precursors, i.e. macroinitiator and macro chain transfer agent, and the final block copolymers were characterized by SEC analysis, 1H NMR spectroscopy, and MALDI-TOF mass spectrometry. Additionally, the successful synthesis of well-defined block and random copolymers consisting of acrylic acid and oligo(ethylene glycol) acrylate led to dual hydrophilic copolymers of branched architecture. The synthesized polymers were characterized by 1H NMR spectroscopy and size exclusion chromatography (SEC).
During further investigations of the water uptake behavior of several selected polymers, an interesting water absorbing hybrid behavior could be observed for the P(EG-b-AA) block copolymer. The disadvantage of PEG, which is not able to absorb moisture below 70% humidity in a larger amount, could be overcome while keeping the final absolute water uptake at a high level of above 70%.
Acknowledgements
The authors thank the Dutch Polymer Institute (DPI) and the Thueringer Kultusministerium for financial support. The help from Juergen Vitz performing the water uptake measurements is greatly acknowledged.References
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967 CrossRef CAS.
- M. P. Patel, M. B. Johnstone, F. J. Hughes and M. Braden, Biomaterials, 2001, 22, 81 CrossRef CAS.
- Y. Oda, S. Kanaoka and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1207 CrossRef CAS.
- H. M. L. Thijs, C. R. Becer, C. Guerrero-Sanchez, D. Fournier, R. Hoogenboom and U. S. Schubert, J. Mater. Chem., 2007, 17, 4864 RSC.
- J. J. Kiefer, P. Somasundaran and K. P. Ananthapadmanabhan, Langmuir, 1993, 9, 1187 CrossRef CAS.
- S. J. Hou, E. L. Chaikof, D. Taton and Y. Gnanou, Macromolecules, 2003, 36, 3874 CrossRef CAS.
- C. R. Becer, S. Hahn, M. W. M. Fijten, H. M. L. Thijs, R. Hoogenboom and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7138 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900 CrossRef CAS.
- J. F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402 CrossRef CAS.
- A. B. Lowe and C. L. McCormick, Prog. Polym. Sci., 2007, 32, 283 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559 CrossRef CAS.
- M. Benaglia, M. Chen, Y. K. Chong, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2009, 42, 9384 CrossRef CAS.
- M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963 CrossRef CAS.
- B. M. Rosen and V. Percec, Chem. Rev., 2009, 109, 5069 CrossRef CAS.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS.
- S. Boissé, J. Rieger, K. Belal, A. Di-Cicco, P. Beaunier, M.-H. Li and B. Charleux, Chem. Commun., 2010, 46, 1950 RSC.
- U. I. Sires and S. B. Mallory, Postgrad. Med., 1995, 98, 79 Search PubMed.
- M. Gourmand and J. M. Corpart, Actual. Chim., 1999, 11, 46 Search PubMed.
- F. L. Buchholz, S. R. Pesce and C. L. Powell, J. Appl. Polym. Sci., 2005, 98, 2493 CrossRef CAS.
- O. M. Jensen and P. F. Hansen, Cem. Concr. Res., 2001, 31, 647 CrossRef CAS.
- D. Gao, R. B. Heimann, J. Lerchner, J. Seidel and G. Wolf, J. Mater. Sci., 2001, 36, 4567 CrossRef CAS.
- M. P. Raju and K. M. Raju, J. Polym. Mater., 2001, 18, 149 Search PubMed.
- S. J. Kohls, D. D. Baker, D. A. Kremer and J. O. Dawson, Plant Soil, 1999, 214, 105 CrossRef CAS.
- K. M. Raju and M. P. Raju, Polym. Int., 2001, 50, 946 CrossRef CAS.
- P. Chen, W. Zhang, W. Luo and Y. J. Fang, J. Appl. Polym. Sci., 2004, 93, 1748 CrossRef CAS.
- K. S. Kazanskii and S. A. Dubrovskii, Adv. Polym. Sci., 1992, 104, 97 CAS.
- L. C. Doug and A. S. Hoffman, J. Controlled Release, 1991, 15, 141 CrossRef.
- P. Colombo, Adv. Drug Delivery Rev., 1993, 11, 37 CrossRef CAS.
- P. Kuzma, A. J. Moo-Young, D. Moro, H. Quandt, C. W. Bardin and P. H. Schlegel, Macromol. Symp., 1996, 109, 15 CAS.
- F. L. Buchholz and N. A. Peppas, ACS Symp. Ser., 1994, 573, 128.
- M. Bakass, A. Mokhlisse and M. Lallemant, J. Appl. Polym. Sci., 2001, 82, 1541 CrossRef CAS.
- J.-S. Song and M. A. Winnik, Macromolecules, 2006, 39, 8318 CrossRef CAS.
- C. J. Ferguson, R. J. Hughes, D. Nguyen, B. T. T. Pham, R. G. Gilbert, A. K. Serelis, C. H. Such and B. S. Hawkett, Macromolecules, 2005, 38, 2191 CrossRef CAS.
- T. Gruendling, S. Weidner, J. Falkenhagen and C. Barner-Kowollik, Polym. Chem., 2010, 1, 599 RSC.
- R. Giordanengo, S. Viel, B. A. Breton, A. Thévand and L. Charlesa, J. Am. Soc. Mass Spectrom., 2009, 20, 25 CrossRef CAS.
- M. Sedlák, M. Antonietti and H. Coelfen, Macromol. Chem. Phys., 1998, 199, 247 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Comparison of measured and calculated isotopic patterns for MS-MS spectra. See DOI: 10.1039/c0py00156b |
| This journal is © The Royal Society of Chemistry 2010 |