A spoonful of sugar: the application of glycopolymers in therapeutics
Sebastian G.
Spain
a and
Neil R.
Cameron
*b
aSchool of Pharmacy, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: sebastian.spain@nottingham.ac.uk; Tel: +44 (0)115 846 6241
bDepartment of Chemistry, University of Durham, University Science Laboratories, South Road, Durham, DH1 3LE, UK. E-mail: n.r.cameron@durham.ac.uk; Tel: +44 (0)191 3342008
First published on 9th July 2010
Abstract
Glycopolymers, synthetic polymers displaying carbohydrate moieties, have been linked to many potential applications at the biology–chemistry interface. One area that holds particular promise is the employment of glycopolymers as vehicles for therapeutics or as therapeutics themselves. This review summarises some of the more prominent examples as well as those in the early stages of development.
 Sebastian G. Spain | Sebastian Spain was born in Gloucestershire, England, in 1981. He studied for an MChem degree at the University of Durham, graduating in July 2004. Subsequently, he remained at Durham where he undertook a PhD on the controlled synthesis and properties of glycopolymers under the supervision of Prof. Neil Cameron. Between March 2008 and February 2010 he was a postdoctoral research assistant for Prof. Len Seymour in the Department of Clinical Pharmacology, University of Oxford, investigating the polymeric modification of viruses for systemic gene delivery. Since March 2010 he has been a postdoctoral fellow for Prof. Cameron Alexander in the School of Pharmacy, University of Nottingham, developing self-assembling polymer–DNA hybrids as potential drug-delivery vehicles. |
 Neil R. Cameron | Prof. Neil Cameron undertook his BSc and PhD at the University of Strathclyde in Glasgow. Following two post-doctoral periods, he was appointed as a Lecturer in the Department of Chemistry at Durham University in 1997. He was promoted in 2005 to Reader and in 2008 he was appointed Professor of Bioactive Chemistry in the same department. His research is focused on the preparation of bioactive and/or bio-inspired macromolecules, including bioactive glycopolymers. His research to date has led to >90 publications and he has given >90 invited lectures. He was awarded the 2003 Young Researchers' Medal from the Macro Group UK and held a Durham University Christopherson/Knott Fellowship for 2008–09. |
Introduction
Glycopolymers,1–3 synthetic polymers featuring pendant and/or terminal carbohydrate moieties, have been of particular interest to the field of drug delivery and therapeutics. This interest is derived from the complex roles that carbohydrates play in vivo, particularly in recognition events with carbohydrate-binding proteins known as lectins.4,5 The interaction between lectins and carbohydrates is weak; dissociation constants, Kd, are typically 10−3–10−6 M, but may be greatly enhanced, in a non-statistical manner, through multivalency. This phenomenon has become known as the clusterglycoside effect.6 As polymers are, typically, multivalent by definition, they provide simple methodologies for accessing this effect. The ubiquitous nature of lectins within recognition and binding events suggests great potential for their exploitation as drug targets and at least one review on the subject has already been published.7 Additionally, glycopolymers provide easily synthesisable analogues of naturally occurring polysaccharides. Several targets for glycopolymeric drugs (some lectin-based and some not) have been identified, including influenza, Alzheimer's disease and some cancers. Herein, we review some of the glycopolymeric drugs8 and drug-delivery systems9–11 developed to date.Glycopolymeric drugs
Influenza hæmagglutinin and neuraminidase inhibitors
One class of disease-carrying agent that has received considerable attention as a target for glycopolymeric treatments are the influenza viruses. Considering their abundance and the number of fatalities that these viruses can cause—tens of thousands of deaths are attributed to influenza each year in the USA alone12—the search for effective treatments is unsurprising.Influenza infection is a multistep process: initially, the virus binds to N-acetyl neuraminic acid residues on the target cellvia lectin structures known as hæmagglutinin (HA) fingers on its surface membrane and then enters the cellviaendocytosis. The virus membrane then fuses with the endosome releasing a complex of RNA and proteins into the cytoplasm. These are transported into the nucleus and the process of virus replication begins.13 If initial binding of the virus to the cell can be prevented, subsequent uptake and replication can also be halted. A molecule that could block HA binding efficiently could prove a useful prophylactic during influenza breakouts, such as the recent H1N1 pandemic. Influenza hæmagglutinin, like most lectins, has a shallow binding site and its interaction with monovalent sialosides is typically weak (Kd ≈ 2 mM),14 thus multivalent ligands should provide improved avidity. Additionally, the virus surface presents a neuraminidase (NA) enzyme and consequently hæmagglutinin inhibitors need to be stable with respect to neuraminidase action.15,16
The first example of an influenza hæmagglutinin inhibitor (HAI) based around a glycopolymer was reported by Bovin et al. in 1990. Polymeric sialosides of varying carbohydrate densities were synthesised by the reaction between poly[4-nitrophenylacrylate] with monosialosides with amino-terminated linkers (Fig. 1). As would be expected, little or no inhibition was seen for monovalent sialosides, β-sialosides, or polymers carrying low quantities of α-sialoside residues (∼5%). Increasing the sialoside density from 10 to 30% indicated a maximum in inhibition at 20%, with 30% having a lower inhibitory effect than the 10% sialylated polymer.17
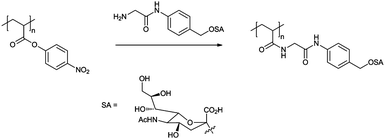 | ||
| Fig. 1 Synthesis of polymeric multivalent sialosides as used by Bovin et al.17 | ||
The main contributors to the field of glycopolymeric HAIs are Whitesides and collaborators. Throughout the 1990s Whitesides et al. published several studies on the inhibition activity of polymeric sialosides synthesised by both polymerisation of sialylated monomers and post-polymerisation functionalisation of reactive polymers.18–23
Initial studies involved copolymers of acrylamido α-O-sialoside (1, Fig. 2) with various N-substituted acrylamides. As Bovin et al. had described previously,17 a maximum level of inhibition was observed at intermediate levels of sialylation. This was rationalised by the competition between cooperative and efficient binding of the sialic acid groups: at low SA levels the groups are well separated and thus binding of one residue does not increase the likelihood of a subsequent group's binding; with high SA levels, binding may be limited as the steric bulk of the groups overcrowd one another. It was also noted that bulky or charged groups on the comonomer tended to reduce the binding efficiency and, in turn, inhibition.19,23 Although copolymers of 1 resulted in highly effective HAIs (inhibition constants (KiHAI) were typically 104–105 fold greater than monomeric equivalents on a per sugar basis), the O-linked SA made them susceptible to cleavage by neuraminidases. In order to alleviate this problem, acrylamido α-C-sialoside 2 was synthesised and copolymerised with acrylamide. Polymers with C-linked SA groups were found to have a maximum inhibitory effect comparable to that of their O-linked equivalents and had a far greater effect at low SA concentrations, probably due to their ability to interact with neuraminidase without deactivation.21 Despite high levels of inhibition displayed by polymers synthesised from sialylated monomers, they were still less efficient than either non-polymeric synthetic HAIs, such as sialylated liposomes,24 or naturally occurring HAIs, such as equine α2-macroglobulin,25KiHAI ≈ 100–200 nM. Consequently, Whitesides et al. turned their attention to the sialylation of reactive polymer backbones. The reasons for this are three-fold: firstly, due to differing monomer reactivities, it is unlikely that sialosides will be statistically placed along a polymer based upon feed ratio. If the comonomer is of higher reactivity it is likely that, especially at the low feed level of SA monomers, the result would be gradient copolymers. Secondly, as overcrowding was considered to be responsible for the reduction in activity of polymers containing greater quantities of SA, steric interactions should reduce over-functionalisation. Finally, polymers may be more directly compared; a single batch of a precursor polymer results in all derivative polymers having the same polydispersity and degree of polymerisation.
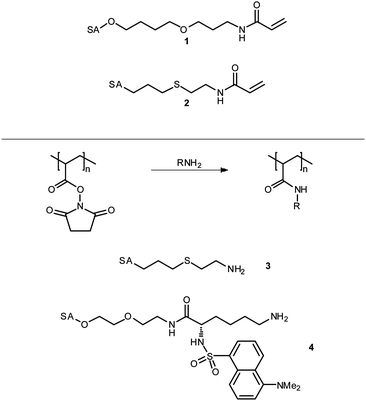 | ||
| Fig. 2 Sialic acid derived monomers and amines as used by Whitesides et al. in the synthesis of glycopolymeric influenza hæmagglutinin inhibitors. See Fig. 1 for structure of sialic acid (SA). | ||
The precursor polymers of choice were those featuring activated esters,26,27 such as poly[N-(acryloyloxy) succinimide] (pNAS) or poly[acrylic anhydride] (pAAn), which were reacted with amino-terminated sialosides including 3 and 4. pNAS was treated with a varying number of equivalents of 3 from 0.2–1.2 with respect to the number of succinimide groups; the level of sialoside incorporation was found to correlate directly with the number of sialoside equivalents up the maximum value of 1. After reaction of the sialoside, any remaining succinimide groups were functionalised by addition of an excess of a second amine or ammonia to yield copolymers of various N-substituted acrylamides. As was seen with the previous polymers, addition of charged groups had a negative effect on the inhibition, particularly positive charges where a singly positively charged side group has a more detrimental effect than a triply negatively charged side group; neutral, polar side groups also reduced inhibition with increasing steric bulk. Hydrophobic side groups increased or decreased inhibition depending on the steric bulk. Benzylamine, for example, was found to improve efficacy as its level of incorporation was increased; presumably through increased hydrophobic–hydrophobic interactions between the polymer and virus surface. Conversely, hexylamine resulted in reduced inhibition.20 Overall polymers with sub-nanomolar values of KiHAI could be produced. Polymers synthesised by similar methods from pAAn gave similar results.18 It was also determined that a synergistic treatment combining C-sialoside–acrylamide copolymers and low molecular weight monomeric neuraminidase inhibitors resulted in even greater inhibition of hæmagglutination. Although the mechanism of this synergy was not confirmed it is thought that the NA inhibitor displaces the polymer from the NA sites either allowing more SA residues to bind to HA sites or increasing the overall steric bulk of the polymer around the virus.28
In addition to hæmagglutinins, the neuraminidases are also targets for influenza treatment; in fact, the currently preferred influenza antivirals, such as oseltamivir (Tamiflu®, Hoffman-La Roche) and zanamivir (Relenza®, GlaxoSmithKline), act as transition state analogues of sialic acid cleavage.29 The presence of NAs on influenza at first seems counterproductive for the virus; NAs cleave sialic acid residues which would, in effect, reduce the chance of viral binding to the cell surface. In fact, the neuraminidases are essential for spread of the virus and infection of further host cells. Once a replicated virus has matured and budded from the cell, it can once again bind to the cell surfacevia the hæmagglutinin molecules. The neuraminidase cleaves the cell surfacesialic acid groups, releasing the new virus.16 Multivalent sialosides that are resistant to NAs have the potential of binding to these receptors and preventing the release of the virus from the host cell and therefore limiting the infection.
Linhardt et al. synthesised C-linked glycopolymer 5 (Fig. 3) and tested its ability to inhibit neuraminidase from Clostridium perfringens, a common bacterium. Compound 5 was synthesised by enzymatic polymerisation of the aromatic monomer by soybean peroxidase in the presence of hydrogen peroxide. Compound 5 was seen to inhibit neuraminidase 10-fold greater than monomeric equivalents.30 Matsuoka et al. synthesised 6 as a copolymer by radical polymerisation of an acetate-protected vinyl precursor with vinyl acetate; after treatment with NaOH, sialylated poly[vinyl alcohol] was isolated. In preliminary tests, polymers were shown to have an inhibitory effect against influenza neuraminidases in the millimolar range.31 Dendritic sialosides synthesised by the same group display similar levels of inhibition.32
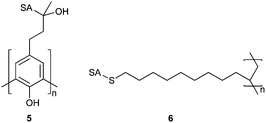 | ||
| Fig. 3 Polymeric neuraminidase inhibitors as synthesised by Linhardt et al. (5) and Matsuoka et al. (6). SA = α-sialoside, structure given in Fig. 1. | ||
Human immunodeficiency virus
Human immunodeficiency virus (HIV) infection is now a major international pandemic and is estimated to have killed 25 million people through progression to acquired immunodeficiency syndrome (AIDS). It is estimated that between 30 and 36 million people are currently infected with HIV and between 2.2 and 3.2 million people were infected in 2007.33 One area of interest for the development of improved HIV treatments is the use of anionic polysaccharides which have been shown to prevent HIV binding to the CD-4 receptor, and thus its entrance to the host cell, in vitro.34 Yoshida et al. synthesised methacrylate polymer 7 (Fig. 4) with maltoheptaose pendant groups by polymerisation of the peracetylated monomer followed by deacetylation and sulfation with either piperidine-N-sulfonic acid or SO3–DMF complex. The polymers were assayed for their ability to inhibit the infection of MT-4 cells by HIV. HIV inhibition was seen to increase with increasing polymer chain length and degree of sulfation for homopolymers but was poor compared to naturally derived polysaccharides such as dextran and curdlan sulfates. Copolymers with methyl methacrylate (MMA) increased in inhibitory effect as the number of maltoheptaose groups was reduced. At approximately 80% MMA the copolymers displayed inhibition in the same order of magnitude as the polysaccharides. Although the level of inhibition is still 2 orders of magnitude worse than that of azidothymidine, a common anti-retroviral, these materials display reduced cytotoxicity in comparison and may have potential in the future.35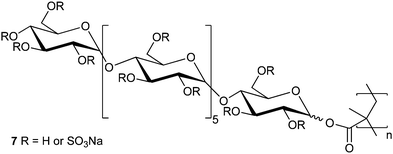 | ||
| Fig. 4 Sulfated maltoheptaose derived methacrylate glycopolymers as synthesised by Yoshida et al. | ||
Alzheimer's disease
Alzheimer's disease (AD) is a neurodegenerative disorder that is the leading cause of dementia of which there are an estimated 24 million sufferers worldwide, a figure that is expected to rise to over 80 million by 2040.36 Alzheimer's disease is characterised pathologically by the accumulation of senile plaques, insoluble aggregates of misfolded amyloid β-peptide (Aβ). One of the current hypotheses for the progression of AD is that these plaques,37 or soluble precursors as small as dimers,38–40 are neurotoxic resulting in atrophy of brain tissue; Aβ itself is considered harmless. Prevention of amyloid-β aggregation may slow, or stop, brain degeneration. Although the underlying cause and mechanism of Aβ misfolding and plaque formation are still the subject of debate there have been several reports of glycoconjugates, such as gangliosides and glycosaminoglycans accelerating aggregation of Aβ, potentially by acting as templates on which the process may occur.41–45 In an attempt to understand further the interaction between glycosaminoglycans and Aβ, Miura et al. synthesised glycopolymeric mimics by copolymerisation of glycomonomers 8 and 9 with acrylamide (Fig. 5). In contradiction of the expected result, polymers containing relatively small quantities of monomer 9 (10–30%) were seen to reduce the level of Aβ aggregation and the morphology of those aggregates that did form; no appreciable effect was observed for polymers of 8. Cytotoxicity of Aβ to HeLa cells was found to be significantly reduced by addition of a copolymer containing 11% of 9, presumably through reduced Aβ aggregation; the polymer itself was found to be non-cytotoxic.46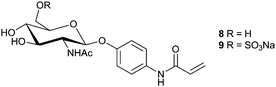 | ||
| Fig. 5 Glucosamine based glycomonomers as synthesised by Miura et al.47 | ||
Glycopolymeric drug-delivery
One of the greatest problems facing pharmaceutical development is the production of an efficacious drug that does not produce undesirable side effects, patient death being the least desirable of all. Side effects are usually the product of a drug having little or no selectivity with regard to its site of action or poor pharmacokinetics, where the drug is cleared from the body too quickly requiring either larger doses or regular administration in order to keep the treatment at an adequate level. Poor pharmacokinetics are a particular problem with anti-tumour drugs which, by their very nature, are usually cytotoxic and thus doses must be accurately controlled in order to destroy the tumour without disrupting healthy cells. The importance of dosage is clear when one considers methotrexate, a widely used anti-tumour drug, the use of which often requires the subsequent administration of a ‘rescue drug’, folinic acid or its salts, to prevent methotrexate toxicity.47 Another major obstacle in drug delivery is the blood–brain barrier (BBB), which is remarkably efficient at preventing the diffusion of molecules into the brain. The walls of the majority of the capillaries in the body consist of endothelial cells with small pores between, known as fenestræ, which allow small molecules to diffuse between the bloodstream and surrounding tissues and vice versa. The capillary walls of the blood brain barrier have no such fenestræ and consequently transport through the BBB must be via the lipid membrane or some form of transport protocol. This is well demonstrated in Fig. 6 which shows a full body radiogram of a mouse 30 min after injection with radiolabelled histamine; the histamine enters all organs of the body except the brain and spinal column.48Glycosylation has recently been shown to be effective at enabling peptides,49proteins50,51 and nanoparticles52 to cross the BBB. Currently this area is poorly understood and the mechanism of transport is unknown. Consequently such methodologies may not be considered generally applicable but may provide a route for allowing delivery to the brain. In order to combat the problems outlined above, methodologies for site-specific delivery of drugs are required.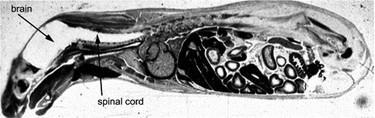 | ||
| Fig. 6 Autoradiogram of an adult mouse 30 min after intravenous injection of radiolabelled histamine. The dark regions show where the histamine is located—none is detected in the brain and spinal cord regions. Reprinted from W. M. Pardridge, The Blood–Brain Barrier: Bottleneck in Brain Drug Development, NeuroRx, 2005, 2, 3–14, Copyright (2005), with permission from The American Society for Experimental NeuroTherapeutics, Inc. Published by Elsevier Inc. | ||
Carbohydrate targeting
In the mid-1970s Ringsdorf introduced a simple model for the polymeric delivery of drugs, a so-called ‘magic bullet’ method. The Ringsdorf model is remarkably simple. Attached to a polymer backbone are three types of group: a targeting moiety, a solubilising moiety and, via a cleavable linker, the drug to be delivered (Fig. 7).53 In the case of glycopolymers the carbohydrate moieties may act as both the targeting vector, specific to a lectin on the surface of the tissue to be treated, as well as aiding solubility. There are now approximately 20 drugs either on the market or in clinical trials based upon this type of model.54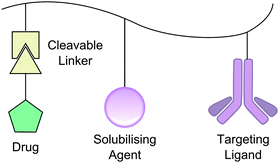
An example of such a treatment has been demonstrated by Hashida et al. in mice. The K vitamins are a family of hydrophobic molecules required for the synthesis of the proteins involved in blood coagulation.55 The denotation of ‘K’ vitamins derives from the German naming koagulations vitamin,56 consequently vitamin K deficiency may lead to hæmorrhaging. It is common for expectant mothers and newborns to be administered vitamin K as a prophylactic but this has found controversy due to its administration having been weakly linked to childhood cancers and other side effects.57,58 The majority of coagulating proteins are synthesised in the liver and thus targeted delivery of vitamin K to the liver may allow suitable prophylaxis with reduction of potential side effects. Hepatic (liver) cells are known to express the galactoside-binding asialoglycoprotein receptor (ASGPR) on their surface; on binding, the galactoside-conjugate is internalised by the cell.59 Hashida et al. synthesised terpolymers based upon a poly[L-glutamic acid] (PLGA) backbone (Fig. 8) by reaction with ethylenediamine followed by 2-imino-2-methoxyethyl 1-thiogalactoside to produce galactosylated PLGA. In turn, this was reacted with vitamin K5, a synthetic K vitamin analogue, to yield a galactosyl–PLGA–vitamin K conjugate. The anti-hæmorrhagic effect of such polymers was determined in mice models by comparison of the prothrombin time, a measure of coagulation efficiency, after systemic treatment with warfarin. As expected, in all cases prothrombin time was increased for warfarin treated mice compared with untreated. Warfarin treated mice that received intravenous (IV), unconjugated vitamin K only had a statistically significant reduction in prothrombin time 4 h after treatment, those receiving IV galactose–PLGA–K conjugate had a significant reduction at 2, 3 and 4 h time points.60
![Anti-hæmorrhagic, poly[l-glutamic acid] based terpolymers as synthesised by Hashida et al.](/image/article/2011/PY/c0py00149j/c0py00149j-f8.gif) | ||
| Fig. 8 Anti-hæmorrhagic, poly[L-glutamic acid] based terpolymers as synthesised by Hashida et al. | ||
Similarly Fleming et al. have targeted boar spermatozoa, which are known to display a galactose-binding lectin with great similarity to the hepatic ASGPR, with galactosyl polymersin vitro. They synthesised terpolymers of 2-(β-D-galactosyloxy)ethyl methacrylate, 2-(dimethylamino)ethyl methacrylate (DMAEMA) and a methacrylate featuring an α-tocopherol functionality. The resulting polymers were incubated with spermatozoa in an attempt to deliver the α-tocopherol, an antioxidant, to the cells to reduce oxidative damage during storage. Although the polymers appeared to have some protective effect to confirm their entrance into the cells, rather than acting as an extra-cellular protectant, the α-tocopherol monomer was replaced by a fluorescent monomer, hostasol methacrylate, and the polymer inside the cell visualised by confocal microscopy (Fig. 9).61
![Confocal micrograph of boar spermatozoa after incubation with a poly[GalEMA-DMAEMA-hostasol methacrylate] terpolymer. Image provided by the author.](/image/article/2011/PY/c0py00149j/c0py00149j-f9.gif) | ||
| Fig. 9 Confocal micrograph of boar spermatozoa after incubation with a poly[GalEMA-DMAEMA-hostasol methacrylate] terpolymer. Image provided by the author. | ||
PK2: glycopolymers in clinical trials
The field of polymeric vectors for drug delivery has been dominated by work of Duncan, Kopeček and Seymour. The most well known of their polymers—PK1, an untargeted doxorubicin conjugate, has been evaluated in Phase II clinical trials for breast, lung and colorectal cancers.62 Typically, they synthesised copolymers of N-(2-hydroxypropyl) methacrylamide (HPMA) and a second monomer featuring an active ester, such as a p-nitrophenyl ester, as a pendant group; the active ester was linked to the polymerisable moiety by a peptide linker, Gly-Gly or Gly-Phe-Leu-Gly for example. These polymers were then reacted with the ‘drug’ to be delivered and a glycosamine in order to provide targeting. Initial studies used model drug compounds such as tyrosinamide which were radiolabelled to allow easy detection of their relative biodistribution. The radiolabelled polymers were administered intravenously to rats and blood radioactivity levels were measured at regular intervals. After a set time (1 or 5 h) the rats were sacrificed and radioactivity levels of individual organs assayed. When the polymers bore galactosamine moieties, 90% clearance of the polymer from the bloodstream was seen within 1 hour and nearly 70% was seen to be present in the liver. Polymers featuring gluco- or mannosamine moieties, or control polymers with a simple amine, were seen to be cleared from the bloodstream more slowly with ∼10% accumulating in the liver. For all polymers, after 5 h ≥ 80% of radioactivity was to be found in the urine and faeces.63,64A polymer based upon this model, PK2 (FCE28069, Fig. 10), with the majority of tyrosinamide replaced with the anti-tumour drug doxorubicin (DOX) has been evaluated in a Phase I clinical trial for hepatoma. Trace levels of tyrosinamide were maintained to facilitate radiolabelling and subsequent imaging. Doxorubicin is a highly effective chemotherapy drug, however, off-target cardiotoxicity limits its use.65,66 A preclinical study in a rat model was used to determine the level of cardiotoxicity cf. free DOX. PK2 and free DOX were administered by both IV and intraperitoneal (IP) injection. Acute and cardiovascular toxicities were monitored by weight loss and cardiac output respectively. Animals receiving free DOX IV displayed acute toxicity at doses greater than 2 mg kg−1, with significant (>20%) weight loss 8–12 weeks after administration. By comparison, animals treated with PK2 at up to 12 mg kg−1 (DOX equivalent) were seen to gain weight, albeit at a reduced rate compared to the saline control. IP administration was considerably less toxic for both PK2 and free DOX, with no mean weight loss after 12 weeks. Animals administered 12 and 18 mg kg−1DOX equivalents of PK2 gained weight at a comparable rate to the saline controls. Cardiotoxicity, measured by relative cardiac output, was found to be insignificant in IV doses of PK2 up to 12 mg kg−1 and free DOX at 2 mg kg−1. 3 mg kg−1DOX yielded significant decreases in cardiac output (>35%, p < 0.0005) after 12 weeks. No animals survived to the 12 week end-point when administered 4 mg kg−1DOX IV. Overall survival curves showed that all animals receiving IV PK2 survived to 12 weeks post-administration. IP administration was, again, seen to be far less toxic with only doses of 5 and 6 mg kg−1 of DOX or 36 mg kg−1PK2 resulting in less than 80% survival at 12 weeks.67
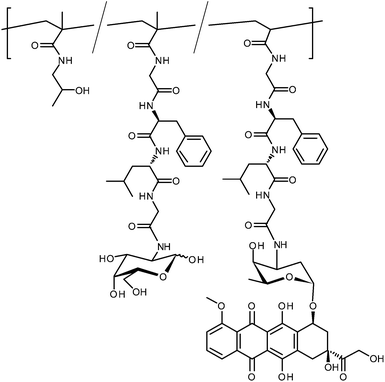 | ||
| Fig. 10 Structure of doxorubicin-conjugated polymer PK2. The trace tyrosinamide modifications are omitted for clarity. | ||
PK2 was assessed in a Phase I clinical trial involving patients with confirmed primary or secondary solid hepatic tumours. Patients were administered PK2 IV at 3 week intervals for a maximum of 6 treatment cycles. Doses administered ranged from 20–160 mg m−2DOX equivalent. Blood and urine samples were collected at various times up to 8 days after each treatment. Blood and urine samples were analysed to determine the level of polymer-bound and free doxorubicin and metabolites; distribution of the polymer was determined by full body imaging of 123I using single photon emission computed tomography. The galactosamine–polymer–doxorubicin conjugate was seen to be rapidly cleared from the bloodstream with 15–20% of the administered dose accumulating in the liver after 24 h. A control polymer, identical except for the absence of galactosamine residues, was seen to remain in the bloodstream for longer and was found to have a general body distribution with no organ specificity. Despite a large accumulation of the polymer in the liver the majority was found in healthy hepatic cells rather than in the tumours themselves, although the accumulation was still significant compared to background. The reduced uptake to cancerous cells was rationalised by the reduced levels of ASGPR that hepatoma cells are known to express compared to their healthy counterparts.68–71 The increased uptake compared to other tissues may instead be due to the enhanced permeability and retention effect and not a result of lectin targeting.72
Glycosylated micelles
Heldrick et al. recently described glycopolymeric micelles assembled from amphiphilic, glycosylated polycarbonates (Fig. 11). The micelles were loaded with DOX and its delivery to HepG2 and HEK293 cell lines in vitro was studied using flow cytometry. In the HEK293 cells (ASGPR negative) no difference was seen between free DOX and either of the glycosyl micellar formulations. In HepG2 cells (ASGPR positive) a three-fold increase in uptake was observed for the galactosyl formulation over the glucosyl formulation and free DOX. The galactose-specific targeting was confirmed by a dose-responsive reduction in uptake when cells were pre-treated with asialofetuin, a galactose-presenting protein.73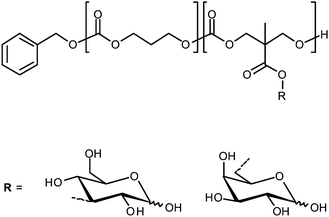 | ||
| Fig. 11 Amphiphilic polycarbonate block copolymers as used by Hedrick et al. for the delivery of doxorubicin. | ||
Glycopolymeric chaperones
The difficulties in effective drug-delivery are apparent when the payload to be delivered is a protein or nucleic acid for which the immune system is a veritable minefield. Ideally, a suitable drug-delivery system for such molecules would provide protection from degradation and a means of targeting the payload to a specific site. The simplest method of introducing a gene or other biomolecule into a cell is direct microinjection but this is hardly practical for the treatment of large multicellular organisms such as humans. The use of viruses, modified to carry the nucleic acid sequence of choice, allows efficient delivery but involves the use of potentially pathogenic precursors and nucleic acid strand length may be limited. Non-viral delivery vectors have the potential to replace viral vectors with non-immunogenic, low cost and easily produced materials.74The use of glycopolymers as molecular chaperones for proteins has been demonstrated by Chaikof et al. They utilised cyanoxyl-mediated polymerisation75,76 for the synthesis of several biomimetic glycopolymer species from alkenyl, acryloyl and acrylamido glycomonomers, often in their sulfated form (Fig. 12).75,77–79 Several of these polymers were tested with respect to their ability to act as mimics of heparan sulfates.78In vivo, fibroblast growth factor 2 (FGF-2) is bound by heparan sulfate, an anionic polysaccharide, which acts as a molecular chaperone protecting FGF-2 from deactivation and facilitating its binding to FGF receptor-1 (FGFR-1).80 Binding assays found that polymers featuring N-acetylglucosamine residues did bind, but weakly compared to heparan sulfate, the linker length was seen to have little effect.78 Sulfated sugars bound more strongly than their non-sulfated equivalents, particularly for polymers featuring pendant lactose groups. Further investigation into the FGF-2 chaperone role of glycopolymers featuring lactose sulfate residues found that low molecular weight (∼10 kDa) polymers containing ca. 10% of the glycomonomer were nearly as effective as heparin in dimerising FGF-2 and binding it to FGFR-1. The chaperone qualities of the polymer were also demonstrated by the extra stability that it gave FGF-2 with respect to degradation by acid, heat and trypsin.81 The glycosyl functionalised polymers were also compared to heparin in anticoagulant activity assays. Polymers featuring monosaccharides had no anticoagulant activity, those featuring lactosyl groups displayed interesting anticoagulant activity dependent on polymer composition and functionalisation. Non-sulfated polymers, like the monosaccharide polymers, had no activity, sulfated polymers were active but homopolymers less so than copolymers with acrylamide.
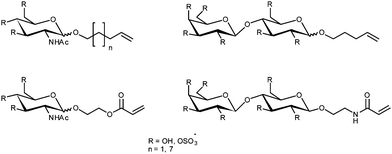 | ||
| Fig. 12 Glycosylated monomers as used by Chaikof et al. for the synthesis of heparin mimics viacyanoxyl-mediated polymerisation. | ||
Glycohydrogels
Glycohydrogels, chemically or physically crosslinked polymeric materials, have been linked to a variety of therapeutic and drug-delivery applications. Nakamae et al. synthesised glycohydrogels by polymerising the glycosylated monomer 2-(β-D-glucosyloxy)ethyl methacrylate in the presence of a divinyl crosslinker and the lectin concanavalin A (Con A). In addition to the chemical crosslinking provided by the divinyl monomer, physical crosslinking is provided by Con A. The presence of Con A leads to gels displaying sensitivity to glucose (and mannose) with swelling increasing with glucose concentration as this displaces the polymeric ligand from the Con A binding site resulting in reduced crosslinking. Consequently, such materials display promise as either glucose sensors or as drug-delivery devices for insulin as increased glucose levels would promote the release of the drug.82,83Wang et al. have produced glycohydrogels for the treatment of norovirus infection, one of the main causes of gastroenteritis. Noroviruses have been demonstrated to bind to the surface glycans on erythrocytes that determine the blood group with a particular affinity to the B group epitope. They synthesised the monomer 1-acrylamido-3,6-dioxa-8-octyl-O-(α-L-fucopyranosyl)-(12)-O-(α-D-galactopyranosyl)-(13)-β-D-galactoside (10) and subsequently polymerised it in the presence of acrylamide, diallyldimethylammonium chloride and N,N′-methylene bisacrylamide to yield hydrogels of the type shown in Fig. 13. The entrapment ability of the glycohydrogels with respect to norovirus was determined using recombinant virus-like particles, virus particles that carry no payload, and ELISA assays. Glycohydrogels were seen to reduce dramatically the solution virus concentration compared to hydrogels prepared in the absence of the glycosylated monomer.84
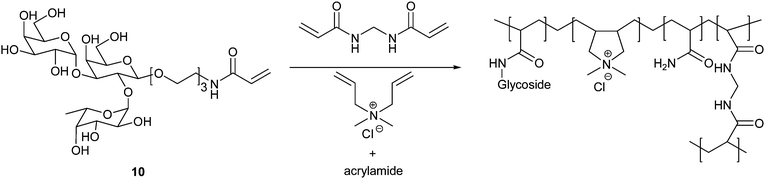 | ||
| Fig. 13 Synthesis of glycohydrogels capable of binding norovirus as synthesised by Wang et al. | ||
Conclusions
Glycopolymers have been investigated in several areas of therapeutic development and delivery. Despite considerable potential it must be noted that the majority of the studies discussed are preliminary—many have not been pursued beyond simple in vitro studies—and extensive research is still required if glycopolymer-based therapeutics are to reach the clinic.The most advanced example, that of PK2, was found to be unsatisfactory for its planned target but has demonstrated that it is possible to produce a relatively complex drug-conjugate to a standard that permits clinical evaluation, that is cGMP. PK2 was synthesised using methodologies that much of the synthetic polymer community would now consider archaic and ill-defined but their simplicity may be an advantage in seeking FDA, or equivalent, approval. Combined with the advances in polymerisation technologies that have occurred over recent decades the production of materials of consistent, defined quality may allow translation of some of aforementioned examples to clinical development.
Notes and references
- S. G. Spain, M. I. Gibson and N. R. Cameron, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2059–2072 CrossRef CAS.
- V. Ladmiral, E. Melia and D. M. Haddleton, Eur. Polym. J., 2004, 40, 431–449 CrossRef CAS.
- Y. Miura, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5031–5036 CrossRef CAS.
- M. Ambrosi, N. R. Cameron and B. G. Davis, Org. Biomol. Chem., 2005, 3, 1593–1608 RSC.
- S. R. S. Ting, G. Chen and M. H. Stenzel, Polym. Chem., 2010 10.1039/c0py00141d.
- J. J. Lundquist and E. J. Toone, Chem. Rev., 2002, 102, 555–578 CrossRef CAS.
- N. Yamazaki, S. Kojima, N. V. Bovin, S. Andre, S. Gabius and H. J. Gabius, Adv. Drug Delivery Rev., 2000, 43, 225–244 CrossRef CAS.
- P. Dhal, S. Holmes-Farley, C. Huval and T. Jozefiak, Adv. Polym. Sci., 2006, 192, 9–58 CAS.
- R. Duncan, Nat. Rev. Drug Discovery, 2003, 2, 347–360 CrossRef CAS.
- R. Satchi-Fainaro, R. Duncan and C. Barnes, Adv. Polym. Sci., 2006, 193, 1–65 CAS.
- J. Khandare and T. Minko, Prog. Polym. Sci., 2006, 31, 359–397 CrossRef CAS.
- W. W. Thompson, D. K. Shay, E. Weintraub, L. Brammer, N. Cox, L. J. Anderson and K. Fukuda, JAMA, J. Am. Med. Assoc., 2003, 289, 179–186 Search PubMed.
- P. Palese and M. L. Shaw, in Field's Virology, ed. D. M. Knipe and P. M. Howley, Lippincott, Williams & Wilkins, 5th edn, 2007, vol. 2, pp. 1653–1674 Search PubMed.
- N. K. Sauter, M. D. Bednarski, B. A. Wurzburg, J. E. Hanson, G. M. Whitesides, J. J. Skehel and D. C. Wiley, Biochemistry, 1989, 28, 8388–8396 CrossRef CAS.
- P. M. Colman, Protein Sci., 1994, 3, 1687–1696 CrossRef CAS.
- R. Wagner, M. Matrosovich and H. D. Klenk, Rev. Med. Virol., 2002, 12, 159–166 CrossRef CAS.
- M. N. Matrosovich, L. V. Mochalova, V. P. Marinina, N. E. Byramova and N. V. Bovin, FEBS Lett., 1990, 272, 209–212 CrossRef CAS.
- S. K. Choi, M. Mammen and G. M. Whitesides, J. Am. Chem. Soc., 1997, 119, 4103–4111 CrossRef CAS.
- W. J. Lees, A. Spaltenstein, J. E. Kingery-Wood and G. M. Whitesides, J. Med. Chem., 1994, 37, 3419–3433 CrossRef CAS.
- M. Mammen, G. Dahmann and G. M. Whitesides, J. Med. Chem., 1995, 38, 4179–4190 CrossRef CAS.
- M. A. Sparks, K. W. Williams and G. M. Whitesides, J. Med. Chem., 1993, 36, 778–783 CrossRef CAS.
- G. B. Sigal, M. Mammen, G. Dahmann and G. M. Whitesides, J. Am. Chem. Soc., 1996, 118, 3789–3800 CrossRef.
- A. Spaltenstein and G. M. Whitesides, J. Am. Chem. Soc., 1991, 113, 686–687 CrossRef CAS.
- J. E. Kingery-Wood, K. W. Williams, G. B. Sigal and G. M. Whitesides, J. Am. Chem. Soc., 1992, 114, 7303–7305 CrossRef CAS.
- T. J. Pritchett and J. C. Paulson, J. Biol. Chem., 1989, 264, 9850–9858 CAS.
- M. A. Gauthier, M. I. Gibson and H.-A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS.
- P. Theato, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6677–6687 CrossRef CAS.
- S. K. Choi, M. Mammen and G. M. Whitesides, Chem. Biol., 1996, 3, 97–104 CrossRef CAS.
- S. L. Johnston, Virus Res., 2002, 82, 147–152 CAS.
- O. Wang, J. S. Dordick and R. J. Linhardt, Org. Lett., 2003, 5, 1187–1189 CrossRef.
- K. Matsuoka, C. Takita, T. Koyama, D. Miyamoto, S. Yingsakmongkon, K. I. P. J. Hidari, W. Jampangern, T. Suzuki, Y. Suzuki, K. Hatano and D. Terunuma, Bioorg. Med. Chem. Lett., 2007, 17, 3826–3830 CrossRef CAS.
- J. Sakamoto, T. Koyama, D. Miyamoto, S. Yingsakmongkon, K. I. Hidari, W. Jampangern, T. Suzuki, Y. Suzuki, Y. Esumi, K. Hatano, D. Terunuma and K. Matsuoka, Bioorg. Med. Chem. Lett., 2007, 17, 717–721 CrossRef CAS.
- UNAIDS, Report on the Global AIDS Epidemic, 2008.
- K. D. McReynolds and J. Gervay-Hague, Chem. Rev., 2007, 107, 1533–1552 CrossRef CAS.
- T. Yoshida, T. Akasaka, Y. Choi, K. Hattori, B. Yu, T. Mimura, Y. Kaneko, H. Nakashima, E. Aragaki, M. Premanathan, N. Yamamoto and T. Uryu, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 789–800 CrossRef CAS.
- C. P. Ferri, M. Prince, C. Brayne, H. Brodaty, L. Fratiglioni, M. Ganguli, K. Hall, K. Hasegawa, H. Hendrie, Y. Huang, A. Jorm, C. Mathers, P. R. Menezes, E. Rimmer and M. Scazufca, Lancet, 2005, 366, 2112–2117.
- J. Hardy and D. J. Selkoe, Science, 2002, 297, 353–356 CrossRef CAS.
- S. T. Ferreira, M. N. Vieira and F. G. De Felice, IUBMB Life, 2007, 59, 332–345 Search PubMed.
- R. Kayed, E. Head, J. L. Thompson, T. M. McIntire, S. C. Milton, C. W. Cotman and C. G. Glabe, Science, 2003, 300, 486–489 CrossRef CAS.
- G. M. Shankar, S. Li, T. H. Mehta, A. Garcia-Munoz, N. E. Shepardson, I. Smith, F. M. Brett, M. A. Farrell, M. J. Rowan, C. A. Lemere, C. M. Regan, D. M. Walsh, B. L. Sabatini and D. J. Selkoe, Nat. Med., 2008, 14, 837–842 CrossRef CAS.
- J. McLaurin, T. Franklin, X. Zhang, J. Deng and P. E. Fraser, Eur. J. Biochem., 1999, 266, 1101–1110 CrossRef CAS.
- J. McLaurin, T. Franklin, P. E. Fraser and A. Chakrabartty, J. Biol. Chem., 1998, 273, 4506–4515 CrossRef CAS.
- A. Kakio, S. Nishimoto, K. Yanagisawa, Y. Kozutsumi and K. Matsuzaki, Biochemistry, 2002, 41, 7385–7390 CrossRef CAS.
- A. Kakio, S. I. Nishimoto, K. Yanagisawa, Y. Kozutsumi and K. Matsuzaki, J. Biol. Chem., 2001, 276, 24985–24990 CrossRef CAS.
- J. Diaz-Nido, F. Wandosell and J. Avila, Peptides, 2002, 23, 1323–1332 CrossRef CAS.
- Y. Miura, K. Yasuda, K. Yamamoto, M. Koike, Y. Nishida and K. Kobayashi, Biomacromolecules, 2007, 8, 2129–2134 CrossRef CAS.
- B. C. Widemann and P. C. Adamson, Oncologist, 2006, 11, 694–703 CrossRef CAS.
- W. M. Pardridge, NeuroRx, 2005, 2, 3–14 CrossRef.
- R. Polt, M. Dhanasekaran and C. M. Keyari, Med. Res. Rev., 2005, 25, 557–585 CrossRef CAS.
- J. F. Poduslo and G. L. Curran, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 2218–2222 CrossRef CAS.
- J. F. Poduslo and G. L. Curran, Mol. Brain Res., 1994, 23, 157–162 CrossRef CAS.
- L. Costantino, F. Gandolfi, L. Bossy-Nobs, G. Tosi, R. Gurny, F. Rivasi, M. A. Vandelli and F. Forni, Biomaterials, 2006, 27, 4635–4645 CrossRef CAS.
- H. Ringsdorf, J. Polym. Sci., Part C: Polym. Symp., 1975, 51, 135–153 Search PubMed.
- G. Pasut and F. M. Veronese, Prog. Polym. Sci., 2007, 32, 933–961 CrossRef CAS.
- K. G. Mann, Thromb. Haemostasis, 1999, 82, 165–174 Search PubMed.
- H. Dam, Biochem. J., 1935, 29, 1273–1285 CAS.
- M. A. Brousson and M. C. Klein, Can. Med. Assoc. J., 1996, 154, 307–315 Search PubMed.
- R. M. Puckett and M. Offringa, Cochrane Database Syst. Rev., 2000, CD002776 Search PubMed.
- G. Ashwell and J. Harford, Annu. Rev. Biochem., 1982, 51, 531–554 CrossRef CAS.
- M. Hashida, H. Hirabayashi, M. Nishikawa and Y. Takakura, J. Controlled Release, 1997, 46, 129–137 CrossRef CAS.
- C. Fleming, A. Maldjian, D. Da Costa, A. K. Rullay, D. M. Haddleton, J. S. John, P. Penny, R. C. Noble, N. R. Cameron and B. G. Davis, Nat. Chem. Biol., 2005, 1, 270–274 CrossRef CAS.
- L. W. Seymour, D. R. Ferry, D. J. Kerr, D. Rea, M. Whitlock, R. Poyner, C. Boivin, S. Hesslewood, C. Twelves, R. Blackie, A. Schatzlein, D. Jodrell, D. Bissett, H. Calvert, M. Lind, A. Robbins, S. Burtles, R. Duncan and J. Cassidy, Int. J. Oncol., 2009, 34, 1629–1636 CAS.
- R. Duncan, J. Kopeček, P. Rejmanova and J. B. Lloyd, Biochim. Biophys. Acta, 1983, 755, 518–521 CrossRef CAS.
- R. Duncan, L. C. W. Seymour, L. Scarlett, J. B. Lloyd, P. Rejmanova and J. Kopeček, Biochim. Biophys. Acta, 1986, 880, 62–71 CrossRef CAS.
- E. A. Lefrak, J. Pitha, S. Rosenheim and J. A. Gottlieb, Cancer, 1973, 32, 302–314 CrossRef CAS.
- R. Injac and B. Strukelj, Technol. Cancer Res. Treat., 2008, 7, 497–516 Search PubMed.
- J. W. Hopewell, R. Duncan, D. Wilding and K. Chakrabarti, Hum. Exp. Toxicol., 2001, 20, 461–470 CrossRef CAS.
- R. Duncan, I. C. Hume, P. Kopeckova, K. Ulbrich, J. Strohalm and J. Kopecek, J. Controlled Release, 1989, 10, 51–63 CrossRef CAS.
- J. Kopecek and R. Duncan, J. Controlled Release, 1987, 6, 315–327 CrossRef CAS.
- L. W. Seymour, D. R. Ferry, D. Anderson, S. Hesslewood, P. J. Julyan, R. Poyner, J. Doran, A. M. Young, S. Burtles and D. J. Kerr, J. Clin. Oncol., 2002, 20, 1668–1676 CrossRef CAS.
- P. J. Julyan, L. W. Seymour, D. R. Ferry, S. Daryani, C. M. Boivin, J. Doran, M. David, D. Anderson, C. Christodoulou, A. M. Young, S. Hesslewood and D. J. Kerr, J. Controlled Release, 1999, 57, 281–290 CrossRef CAS.
- K. Greish, J. Drug Targeting, 2007, 15, 457–464 Search PubMed.
- F. Suriano, R. Pratt, J. P. K. Tan, N. Wiradharma, A. Nelson, Y.-Y. Yang, P. Dubois and J. L. Hedrick, Biomaterials, 2010, 31, 2637–2645 CrossRef CAS.
- M. E. Davis, Curr. Opin. Biotechnol., 2002, 13, 128–131 CrossRef.
- D. Grande, S. Baskaran, C. Baskaran, Y. Gnanou and E. L. Chaikof, Macromolecules, 2000, 33, 1123–1125 CrossRef CAS.
- Y. Gnanou, D. Grande and R. Guerrero, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 1999, 40, 99–100 CAS.
- D. Grande, S. Baskaran and E. L. Chaikof, Macromolecules, 2001, 34, 1640–1646 CrossRef CAS.
- S. Baskaran, D. Grande, X. L. Sun, A. Yayon and E. L. Chaikof, Bioconjugate Chem., 2002, 13, 1309–1313 CrossRef CAS.
- X. L. Sun, D. Grande, S. Baskaran, S. R. Hanson and E. L. Chaikof, Biomacromolecules, 2002, 3, 1065–1070 CrossRef CAS.
- O. Saksela, D. Moscatelli, A. Sommer and D. B. Rifkin, J. Cell Biol., 1988, 107, 743–751 CrossRef CAS.
- R. Guan, X. L. Sun, S. J. Hou, P. Y. Wu and E. L. Chaikof, Bioconjugate Chem., 2004, 15, 145–151 CrossRef CAS.
- K. Nakamae, M. Takashi, A. Jikihara and A. S. Hoffman, J. Biomater. Sci., Polym. Ed., 1995, 6, 79–90 CrossRef.
- T. Miyata, A. Jikihara and K. Nakamae, Macromol. Chem. Phys., 1997, 197, 1135–1146.
- Y. L. Zhang, Q. J. Yao, C. F. Xia, X. Jiang and P. G. Wang, ChemMedChem, 2006, 1, 1361–1366 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |