Synthetic polymers for biopharmaceutical delivery
Johannes Pall
Magnusson
,
Aram Omer
Saeed
,
Francisco
Fernández-Trillo
and
Cameron
Alexander
*
School of Pharmacy, University of Nottingham, University Park, Nottingham, NG7 RD, UK. E-mail: cameron.alexander@nottingham.ac.uk
First published on 4th October 2010
Abstract
The increasing complexity of biological molecules being developed for medical applications requires more sophisticated carrier materials to take these potent but delicate therapeutics to in vivo targets. Recent advances in polymer synthesis chemistries are enabling carrier vehicles of greater sophistication in architecture and function to be prepared. In this outline review, the application of polymer chemistries to protein and nucleic acid therapeutics are considered.
Introduction
The basic functions of natural polymers such as proteins and nucleic acids are well understood in biology, but increasingly, these sophisticated macromolecules are being considered for therapies. Proteins play an essential role in regulating processes at a cellular level and provide the foundations which are necessary to maintain the homeostasis of an organism. The increased understanding of the roles of proteins in healthy and diseased states has led to the identification of new cellular targets and subsequently to new treatment options. In addition, recent breakthroughs in recombinant DNA techniques have made possible the production of therapeutic proteins in large quantities.1,2 Protein therapeutics have advantages over small molecule drugs in certain cases as some protein–protein interactions are more specific than the interactions of a protein with a small drug molecule.For nucleic acids, the potential opportunities for therapies are perhaps even greater, since the gene, gene regulators and expressed gene products, can all be used to correct imbalances in the body. Advances in molecular biology and genetics have provided important information on the control of cellular processes and disease pathogenesis.3 Identification of target genes is generating novel therapeutic options for the treatment of many inherited and acquired diseases.4,5 Gene therapy aims to manipulate cellular processes by altering gene expression in specific cell populations, via delivery of DNA, RNA, or antisense sequences. Originally, gene therapy approaches were focused on treating inherited genetic disorders such as cystic fibrosis and haemophilia, but recently acquired disease conditions such as HIV, cancer and wound healing have gained increased attention.6–8
Biomedical applications are not just linked to therapy: proteins and peptides have found numerous other uses in applications such as diagnostics,9–11 due to their high specificity and catalytic activity. In this regard, DNA and RNA based diagnostic systems are pushing the limits of detection for disease-specific markers.12–16
Despite these successes and the many potential advantages of biopolymer therapeutics over small molecule agents, the successful applications of these moieties as therapeutic agents in vivo have been somewhat limited. This stems primarily from their rapid elimination, degradation and poor cellular uptake when formulated via conventional routes. The inherent instability of these delicate biopolymers outside their normal biological environment means that chemical modifications or carrier agents are required to package, protect and deliver them in vivo, and it is these agents that form the subject of this outline review.
Proteins and nucleic acids—the need for delivery systems
The biggest challenge for new biotherapeutics is to enable delivery to the desired target site at the right time and in the right dose. Simple administration of native proteins via the oral route results in degradation due to stomach acidity, enzymes in the gastro-intestinal tract and poor absorption. Intravenous injection offers the possibility of systemic action, but many proteins are rapidly eliminated through recognition by the reticuloendothelial system and by renal clearance. Multiple injections are required in these cases, which result in poor patient compliance. Protein delivery systems must thus stabilise and protect, as well as optimise sustained release. Similar issues arise for nucleic acids in terms of formulating the delicate biopolymer and its inherent instability in vivo due to degradation. For DNA-based therapies the target is the cell nucleus, whereas for RNA, delivery to the cytoplasm is required—in both cases significant barriers to delivery exist (Fig. 1).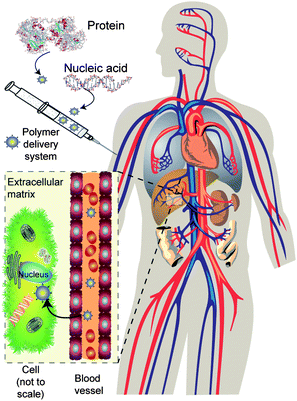 | ||
| Fig. 1 Barriers to biopharmaceutical delivery. A typical injectable formulation is shown: for delivery to the target the polymer carrier must protect the biopharmaceutical during injection, in transit through the bloodstream, and release it in the correct cell or intracellular compartment. | ||
To formulate DNA, the nucleic acid (which can be several million Daltons in molar mass) is normally condensed to a small size, to protect it from serum and intracellular nucleases. The DNA must be delivered to the target cell, cross the external cell membranes passively or actively, leave the endosomal compartments (avoiding degradative enzymes and escaping traffic to the lysosomes), then it must translocate into the nuclear compartment and be made available for transcription.
These requirements have led to polymeric delivery systems being designed with analogy to viruses, since the latter are natural DNA or RNA carriers able to deliver their payloads to cells with high efficiency. In addition, for an injectable DNA delivery formulation, polymers with encapsulated or complexed DNA must be capable of extended circulation in the bloodstream in order to have a chance to reach their cellular target. They must also be small enough to gain access to tissues and cells—typically this will put a size limit of <250 nm for the hydrodynamic diameter of the carrier (“vector”) system in the circulation. Not surprisingly, therefore, delivery systems of some complexity are being developed.
Sustained release of biotherapeutics—encapsulation in degradable polymers
A common, and perhaps the most simple, method of improving the delivery and pharmacokinetics of biopolymers involves creating a sustained release formulation of the therapeutic in a carrier matrix. The native biopolymer retains the same intrinsic pharmacokinetic properties as normal but while encapsulated in the formulation it cannot be cleared from the body. Thus the time of action is enhanced by the slow release of the biotherapeutic from the formulation. Polymeric matrices enable control over release rate, and are advantageous as long as the polymer itself or its degradation products are non-toxic and/or can be easily eliminated from the host. A number of polymer matrices do at least partially meet these requirements as encapsulants, the most important of which are outlined below.Polymers composed of lactide and glycolide units such as poly(lactic acid) (PLA), poly(glycolic acid) (PGA) and the copolymers of lactic and glycolic acid, i.e., poly(lactide-co-glycolide) (PLGA) have been the most widely used in biomedical applications (Fig. 2).17 PLGA-based polymers are favourable for sustained release applications due to their extended release rates which can span from days to months.18–21 Macromolecules such as proteins, peptides, plasmid DNA, human growth hormone factor, etc., have been successfully incorporated into PLGA based micro/nanoparticles.22–24 PLGA polymers produce lactic acid and glycolic acid upon hydrolytic degradation; these two substances are naturally occurring in metabolic pathways of the body, therefore, their applications as drug and gene delivery systems are generally considered to be safe and are approved for human use. Furthermore, PLGA polymers are commercially available in a range of molar masses, and lactide![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :glycolide ratio. PLGA polymers can be synthesised by polycondensation or by ring opening polymerisation of lactide and glycolide.25 The polycondensation method is an equilibrium process, and it can be difficult to remove the liberated water, thus limiting the molecular weight of the final polymers. On the other hand, the ring-opening polymerisation (ROP) of lactide and glycolide allows for a much higher control of the polymerisation than polycondensation. As a result, ROP is the most widely used method for the synthesis of well-defined PLGA materials. In practice, optimisation of polymerisation conditions allows for the controlled ring opening polymerisation of lactide and glycolide, the mean degree of polymerisation (DP) of the resulting polymers is usually equal, or at least proportional, to the monomer conversion multiplied by the monomer to initiator molar ratio. Polymerisation of lactide and glycolides can occur either in bulk or solution; however, bulk polymerisation is desired when the end product is intended for medical applications. Bulk polymerisation eliminates the use and potential residual presence of organic solvents such as toluene, benzene, or chlorobenzene in the polymer.26
:glycolide ratio. PLGA polymers can be synthesised by polycondensation or by ring opening polymerisation of lactide and glycolide.25 The polycondensation method is an equilibrium process, and it can be difficult to remove the liberated water, thus limiting the molecular weight of the final polymers. On the other hand, the ring-opening polymerisation (ROP) of lactide and glycolide allows for a much higher control of the polymerisation than polycondensation. As a result, ROP is the most widely used method for the synthesis of well-defined PLGA materials. In practice, optimisation of polymerisation conditions allows for the controlled ring opening polymerisation of lactide and glycolide, the mean degree of polymerisation (DP) of the resulting polymers is usually equal, or at least proportional, to the monomer conversion multiplied by the monomer to initiator molar ratio. Polymerisation of lactide and glycolides can occur either in bulk or solution; however, bulk polymerisation is desired when the end product is intended for medical applications. Bulk polymerisation eliminates the use and potential residual presence of organic solvents such as toluene, benzene, or chlorobenzene in the polymer.26
 | ||
| Fig. 2 Structures of common polymeric materials used for biopolymer delivery. | ||
The degradation rate of PLGA materials can be controlled via the molecular weight of the polymer and also through lactide:glycolide ratio. Usually low molecular weight and high content of glycolide accelerate the degradation rate. In vitro PLGA composed of a 50:50 ratio of lactide to glycolide exhibits the fastest degradation,27,28 whereas other combinations of lactide:glycolide ratios e.g. 75:25, or 80:20 exhibit significantly longer degradation times.
To date, PLGA-type materials for biomedical applications have not been a strong focus for very highly controlled polymerisation chemistries as these methods have not been necessary for their clinical use, however, recent studies have shown that molar mass, tacticity and polymer stereochemistry can be controlled by using selective metal Lewis acid catalysts.29–31 The applications of these newer PLGAs are likely to be an important area of biomaterials research in future years.32,33 Examples of other PLGA-type materials include functionalised polyesters prepared via ROP of derivatised lactones. Caprolactones with substitution at the 3-, 4- or 5- positions have been the most explored to date. Homopolymerisation of 5-keto-ε-caprolactone monomers via ROP and further derivatisation has enabled tailoring of the physical and chemical properties of the polymer. The Mayes group have outlined a facile and useful strategy for grafting aminoxy terminated PEG to co-poly(5-keto-ε-caprolactone-ε-caprolactone).34 The resultant co-polymers were soluble in water at 50 weight% PEG or above, and the degree of substitution could be easily controlled by choice of PEG graft. Other monomers such as 6-chloro-ε-caprolactone have also been investigated, for example for copolymerisation with ε-caprolactone to generate polyesters with pendant chloro groups. The chloro groups were successfully transformed into azides for subsequent ‘click’ reactions to install biological molecules of therapeutic interest.35 Other vinyl functionalised ε-caprolactone polyesters have also been prepared,36 thus allowing for further derivatisation or incorporation into cross-linked matrices.
Polyesters of these types as well as various natural and synthetic polymers can be formulated as matrices and microspheres for biomedical applications,37,38 and in these cases the physical properties of the formulated particles such as size, mass and porosity can also affect the release of the therapeutic payload.39 While the formulation of small drug molecules within polymers for sustained release is well established40 the fragile nature of many proteins and nucleic acids creates several compatibility issues for the encapsulation process in polymer matrices. It is important during the formulation, the storage and the administration of the biopolymer that the activity is not lost due to aggregation, irreversible precipitation or denaturation.
There are a number of approaches described for encapsulating biopolymers within PLGA based nanoparticles, these include spray drying, phase separation methods and double emulsion techniques.41–44 Initially, these methods were developed to encapsulate peptides and proteins within PLGA based micro- and nanoparticles. The encapsulation of proteins is typically carried out via emulsification or direct incorporation of the solid protein (Fig. 3). For the emulsification procedure the protein or other biopolymer is dissolved in an aqueous solution and dispersed in a non-miscible polymer organic solution to give a suspension of aqueous droplets in a polymer solution.45–47 Water is added to form a secondary emulsion and subsequently the organic solvent is evaporated. The evaporation of the organic solvent causes the PLGA to precipitate around the biopolymer to form microspheres. The most challenging aspect in the double emulsion method for proteins is the unfolding of the protein at the organic solvent water interface,48 though this tendency can be suppressed by careful choice of surfactants which stabilise the emulsion. Another drawback of the double emulsion technique, of special relevance for biopolymers such as proteins and nucleic acids, is the shear force used to disperse the aqueous solution. In the case of DNA this can result in strand breakage and loss of biological activity. There are some methods that can be used to minimise biopolymer damage during the dispersion process. Condensation of DNA can be carried out with cationic polymers such as poly(L-lysine) to reduce the effects of applied shear. Poly(L-lysine) is capable of condensing the DNA into sub-micron polyelectrolyte complexes while preserving the supercoiling of DNA and retaining its biological activity.49,50
 | ||
| Fig. 3 Encapsulation of therapeutic protein into polymeric particles by (a) emulsification and (b) solid incorporation method. | ||
Alternatively for solid incorporation, the protein powder is suspended directly in the organic polymer solution, the suspension is frozen and the organic solvent extracted leaving microspheres.51,52 As the hydration of the protein is limited the unfolding of the protein does not occur and therefore the activity of the protein is better maintained. The lyophilisation itself of the protein can, however, cause a loss in the activity. Generally speaking the solid incorporation route gives better encapsulation efficiency than the emulsification method.
In addition to formulation issues the protein can also be non-compatible with the polymer matrix or its degradation products, which in turn can decrease the efficiency of the formulation.
PLGA based particles are sensitive to moisture and undergo hydrolysis over time as mentioned above to generate lactic and glycolic acid units.53 This subsequently may lead to a substantial reduction in pH in the PLGA matrix in non-buffered environments, which in turn may cause degradation of encapsulated biopolymers, compromising their therapeutic effects.41,54 Therefore, it is important to keep formulated PLGA particles free of moisture during storage. Drying of the particles is usually carried out by lyophilisation for long-term storage. Sometimes, cryoprotectants such as sucrose or glucose can be added to the formulation to preserve the biopolymer structure (e.g. DNA supercoiling) during the drying process.55
Some of these limitations associated with encapsulation into nanoparticles can be addressed by employment of vesicles made from amphiphilic polymers. Biomolecules of interest can be kept in aqueous media within the vesicle membrane, thus improving their stability and viability. The fact that these vesicles (sometimes also known as polymersomes) can be prepared using a broad range of chemistries and monomers, including ‘smart’ materials that can respond to different stimuli such as pH, temperature, oxidation or light, highlights their potential for the delivery of therapeutic biopolymers.56,57 Proteins58 and nucleic acids59 have been successfully encapsulated within biodegradable polymersomes. In both instances, the kinetics of the release of biopolymers could be controlled either by changing the size and nature of the polymer, or by blending these biodegradable polymers within a matrix of inert poly(ethylene glycol)–polybutadiene (PEG–PDB) polymer.
In comparison to conventional liposomes prepared from lipids, polymersomes offer several advantages. Due to the higher molecular weights of polymers, polymersomes tend to have thicker membranes that render them more stable. In addition their surface properties can be easily tailored by the proper choice of monomers and chemistries.56,60
To their disadvantage, polymersomes tend to have lower encapsulation efficiencies than liposomes, resulting in low drug loading. Methods are being developed to improve vesicle loading, although most of them rely on the same principles applied for the preparation of nanoparticles, that is, use of cosolvents and freeze-drying cycles.56,60
Polymer bioconjugation chemistries
It should be noted that, regardless of the formulation employed, encapsulation by itself does not improve the native properties of a biopolymer. For example, if a protein is prone to cause immunogenic reactions or is rapidly cleared, on release from a polymer carrier these issues will persist. Other approaches are then required to improve the pharmaceutical properties. For instance, the native properties of proteins and nucleic acids can be improved via chemical modifications or conjugations.61For proteins the most successful approach has been the conjugation of PEG polymer chains to the protein, a process termed “PEGylation”. The popularity of PEG stems from its hydrophilic nature, inert character and FDA approval. The attachment of PEG increases the hydrodynamic volume of the protein and creates a steric barrier on the surface of the protein (Fig. 4).62 The effect of this conjugation is that the protein is less rapidly cleared via the kidneys and protease accessibility is decreased, the residence time of the protein in vivo therefore increases.
The synthesis of PEG is conventionally carried out by anionic ring opening polymerisation of ethylene oxide, and is initiated by the nucleophilic attack of a hydroxide ion on the ethylene oxide. Various derivatives of PEG have been synthesised using this method. The most common derivative used for the PEGylation of proteins has been methoxy-PEG (mPEG) that contains a methoxy and hydroxyl end groups.63 The hydroxyl group can be further activated so it can react with the protein. The polymerisation of the ethylene oxide has to be carried out under extremely dry conditions to control the properties of the PEG formed and to suppress the formation of dihydroxy PEG as much as possible. If a diol PEG is formed it can cause a number of problems during subsequent PEGylation such as crosslinking and precipitation.
The chemistry used for conjugation to a protein is quite varied and depends on the properties of the protein being conjugated. Generally the nucleophilicity and reactivity of the amino acid residues can be manipulated to control the sites of conjugation (Table 1).
 | ||
| Fig. 4 Effect of polymer conjugation on protein properties. | ||
| Amino acid | Functionality | pKa |
|---|---|---|
| Terminal amine |

|
7.6–8 |
| Lysine |

|
9.3–9.5 |
| Cysteine |

|
8.8–9.1 |
| Tyrosine |
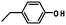
|
9.7–10.1 |
| Histidine |
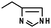
|
6.7–7.1 |
| Aspartic acid |

|
3.7–4 |
| Glutamic acid |

|
4.2–4.5 |
| Arginine |
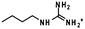
|
>12 |
The first reported PEGylating reagents contained a dichlorotriazine moiety (Scheme 1a) and were used to conjugate to the amine residues of the protein. Lysine residues are abundant and easily accessible in most proteins and therefore popular targets for modification. Acidic residues on proteins, although abundant in number, are normally not targeted for conjugation since their modification usually leads to extensive crosslinking of the protein.
 | ||
| Scheme 1 Functional PEGs for the modification of lysine (a–c) and cysteine (d–f) residues of proteins. (a) Dichlorotriazine, (b) succinimidyl succinate, (c) succinimidyl carbonate, (d) maleimide, (e) iodoacetamide, and (f) pyridyl disulfide. R denotes protein. | ||
Today the use of dichlorotriazine has been phased out due to the fact that it reacts with other amino acid residues such as histidine, cysteine, serine and tyrosine, leading to non-specific functionalization.64 In addition, the formed monochlorotriazine can further react with other nucleophilic residues within the protein, leading to undesired crosslinked materials. The latter can be avoided by reacting the residual chlorocarbon site prior to protein conjugation, and this way branched PEG reagents have been implemented.65 Branched PEG polymers offer significantly better steric protection against proteolysis than their linear PEG counterparts. Similar protection is achieved with multiple linear PEG attachment and fewer branched PEG, while the activity of the protein is better maintained with fewer polymer attachments.66
Activated esters such as PEG succinimidyl succinate (PEG-SS) (Scheme 1b) are quite popular reagents for protein PEGylation. Despite instability under basic conditions and a half life of around 20 minutes at pH 8.0, PEG-SS will react preferentially with primary amines, even in the presence of other nucleophiles such as water, leading to the desired conjugate. In general, the unwanted hydrolysed PEG-SS can be easily removed by standard means such as Size Exclusion Chromatography (SEC) or dialysis.
A way to decrease this detrimental side reaction is the employment of PEG succinimidyl carbonate (PEG-SC) (Scheme 1c) which is less prone to hydrolyse in water, thus the conjugation can be carried out at higher pH.67 PEG-SC reacts preferentially with lysine of the protein. In addition it has been shown that at acidic pH it predominantly reacts with histidine residues, although the bond formed between the PEG-SC and histidine is unstable at neutral pH in the presence of primary amines. This can be advantageous in vivo since breaking of the polymer linkage reverts the protein back into its native conformation, so that the PEGylated protein is, in effect, behaving as a prodrug.68
As pointed out above, lysine residues are usually abundant in protein and accessible for modification, making them the preferred candidates for PEGylation. Unfortunately, this abundance means that the reaction at the lysine residues leads inherently to a mixture of different isomers, depending on the number and position of the reacted residues. The extent of this can be controlled by varying the ratio of polymer to protein and by changing the pH, since the nucleophilicity of each amine is affected by its local environment.68–70 In particular, the N-terminal amine, due to its lower pKa, can be specifically modified by carrying out the conjugation at slightly acidic pH, where the otherwise competing lysine side chains amines are protonated and, therefore, non-nucleophilic.71 This process leads to monofunctional derivatives and has recently gained increased attention due to the fact that terminal amines are normally easily accessible. Different chemistries have been proposed for their specific functionalisation, with the reaction with aldehydes at pH around 5 being the most extensively studied. This conjugation creates a Schiff base which is normally reduced to form a stable amine linkage.72,73
Other popular targets for conjugation are free cysteines since reactions at these groups normally yield site specific monofunctionalised derivatives of the protein. Various thiol reactive PEG derivatives have been developed for the conjugation of free cysteine residues of proteins, including maleimide (Scheme 1d),74 iodoacetamide (Scheme 1e),75 and pyridyl disulfide (Scheme 1f).76 It is known that maleimide and iodoacetamide moieties have a slight tendency to react with amine residues, an issue that increases at higher pH, making them less selective. On the other hand, pyridyl disulfide functionality does not react with amines since the reaction occurs via thiol exchange. The reaction is therefore more specific while the bond formed is reducible, thus the native structure of the protein can be obtained again under reducing conditions. These approaches are, however, limited by the fact that free cysteine residues are not that common and not always accessible on the protein. In some cases, their functionalization can have a detrimental effect on the activity of the protein. Elegant work in the field of protein functionalisation which tackles this problem was reported by Brocchini and co-workers77,78 who demonstrated that, in the case of interferon α-2b, native disulfide bonds could be cleaved and PEGylated via a PEG monosulfone which bridged the disulfide back together in high yield (Scheme2). The bulkiness of the PEG reagents ensured that only one PEG chain could be conjugated to each disulfide. As an example the modified PEGylated interferon maintained the tertiary structure and activity of the native after the conjugation. This strategy is particularly significant as it does not require a free cysteine for the conjugation and is therefore applicable as a generic method for the site specific conjugation of proteins, as long as the disulfide bond is solvent accessible and not located in the active site of the protein. The methodology has recently been expanded to asparaginase and somatostatin with similar results.79

In addition to ‘chemistry’ based protocols, proteins have also been conjugated using enzymatic methods. Enzymes are very selective so that the site and numbers of conjugation can be predicted beforehand and controlled.80 One of the most popular enzymes used for conjugation is transglutaminase,81 as it can couple amine-functionalised PEG specifically to glutamine residues on a protein.
In addition to the residue targeted for conjugation, other parameters have to be considered, such as the linker chemistry or the final charge of the protein–polymer conjugate as they can have a marked effect on the protein stability and activity. In this context, the employment of cleavable linkers can have a very important impact. The attachment of the polymer chains normally decreases the activity of the protein, therefore it can be beneficial in vivo if the protein assumes its native conformation. The ideal situation is that the polymer protects the protein against degradation and clearance in circulation, but, when it reaches its site of action, the polymer is cleaved off and the activity of the protein is restored. In this way the polymer formulation behaves like a prodrug (Scheme 3).82–84
 | ||
| Scheme 3 A cleavable PEGylation linker (benzyl elimination). The linker reacts specifically to primary amines and is cleaved in vivo to release the native protein. R denotes protein. | ||
As is the case for therapeutic proteins, the use of polymers to aid delivery of nucleic acids to target cells and tissues is of interest. In comparison to proteins, the conjugation of polymers to DNA/oligonucleotides in the context of drug delivery has not been as extensively studied, although there have been elegant recent examples of DNA block co-polymers.85 Since oligonucleotides do not contain the same functional reactive groups as proteins, other conjugation chemistries have to be implemented. Nucleic acids can be modified in a number of ways via electrophilic or nucleophilic substitution reactions and many chemistries are accessible for this.86 However, modifications of the bases can require conditions that are difficult to scale up and addition of a bulky group such as a polymer can in addition affect the ability of the oligonucleotide to form complementary base pairs. The modification at the 5′ or 3′ end is normally preferred since those reactions lead to single site modification which have less effect on base pairing.87 The modification of the 5′ end has been carried out on resins using phosphoramidite solid phase chemistry or alternatively in solution.88,89
Controlled radical polymerisation chemistries—recent advances in bioconjugates and polymer–biopolymer complexes
Conventionally, cationic polymers are used in nucleic acid delivery because of their intrinsic ability to condense with polyanionic DNA and siRNA to form polymer–NA (nucleic acid) complexes, usually termed polyplexes. Examples of the most commonly used cationic polymers to complex with DNA are poly(ethyleneimine) (PEI), poly(L-lysine) (PLL) and chitosan (CS). Details of these cationic polymers for nucleic acid delivery, their transfection efficacy and cytotoxicity have been well-reviewed elsewhere.90 However, for systemic administration, these polyplexes require surface modification primarily to enhance the circulation time and to reduce unwanted specific (antibody mediated) and non-specific (reticuloendothelial) uptake. Generally, PEGylation techniques have been used to extend the circulation time mainly by providing steric barriers on the gene carriers that minimise opsonisation and which increase the hydrodynamic volume of the polymer–nucleic acid complex to reduce renal excretion. However, postfunctionalisation of the PEG chain is cumbersome in terms of reactivity, purification and limited modifications can be carried out on the polymer backbone itself, in terms of tailoring new properties into the final material. Recent developments in Living Radical Polymerisation techniques (LRPs) offer remedies to these shortcomings. LRP offer unprecedented control over the polymer topology and functionalities. Various new polymer structures have been engineered from these processes (Fig. 5).91
Though various LRP techniques exist they all operate through the same “caged” radical effect in which the radical formed at the end of the polymer can propagate or be reformed. Each chain is only active for a brief time in comparison to the polymer chains formed with conventional free radical polymerisations (FRPs), therefore the material produced by LRP is better defined than material produced by FRP.92 This is specially important for biomedical applications since a better defined material is likely to result in less variations in functional behavior in the body. The polymer morphology is also better controlled using LRP and complex structures such as blocks, branched, dendritic, stars and gradient copolymers can be obtained. The polymers synthesised with LRP are also easier to post-modify than PEG based polymers as higher control over the end group functionality is achieved.93–95
Of the recent LRP techniques, the most extensively studied are Atom Transfer Radical Polymerisation (ATRP) and Reversible Addition Fragmentation Transfer (RAFT). Both methodologies are compatible with many functional groups so a wide range of protein reactive polymers can be created with functionalised initiators. A wide variety of functional monomers can be polymerised by both techniques although the RAFT process is more generic with respect to types of monomers. These LRP techniques have been used for the synthesis of novel protein polymer conjugates. Normally the polymer is “grafted on” the protein, that is the polymer is grown from a protein reactive initiator, which is purified and subsequently conjugated to the protein. Among others, cysteine and amine reactive polymers have been synthesised and conjugated to proteins using this method. Polymers synthesised with LRP as alternatives to PEG based therapeutics include polymers such as poly(Hydroxyethyl methacrylate) (pHEMA) and poly(Polyethylene glycol methacrylate) (pPEGMA). Both these polymers exhibit a promising biocompatibility profile although further examination of their pharmacokinetic profile is required.96,97 Of the two, significant efforts have been made into creating PEGMA based protein polymer conjugates in the past few years, as potential alternatives to PEG based polymers, and data on pPEGMA conjugates in vivo is being generated.98
A number of pPEGMA protein conjugates have been reported in the literature.99,100 Most recently the Maynard group described the conjugation of pPEGMA made by RAFT to a lysozyme protein. The polymer was synthesised via a trithiocarbonate RAFT agent containing a protected maleimide moiety. After synthesis and purification the maleimide protecting group was removed and the polymer conjugated to a lysozyme mutant which contained a free cysteine residue (Scheme 4).101
 | ||
| Scheme 4 Synthesis of maleimide-functionalised polymer via RAFT polymerisation and the subsequent conjugation of the polymer to lysozyme T4L V131C mutant.101 | ||
Polymers made by LRP can, in addition to improving the existing properties of proteins, be used to confer new properties onto the protein. Multifunctional polymers synthesised via LRP include copolymers made in the presence of monomeric dyes, making the polymer suitable for diagnostic purposes, i.e. for biological tracing studies.102
As has been described above, polymers have in the past been mainly attached to proteins via the “grafting on” approach, where a preformed functionalised polymer is conjugated to the protein. This approach has certain limitations, in terms of purification when the protein and polymer are of similar sizes, as steric bulkiness of the polymer can also decrease the efficiency of the reaction thus limiting the extent of conjugation.
As an alternative, polymers can be “grafted from” proteins by the generation of free radicals in situ. Previous techniques adopted to do this were not able to control the number and sites of polymerisation, as free radicals were essentially generated in a random fashion and conditions used were not compatible with many proteins.103,104 With the introduction of LRP it has become possible to create biopolymer–polymer conjugates using the “grafting from” approach in a controlled manner and under conditions which are compatible with proteins. The low numbers of radicals active at any one time limits exposure to radicals and prevents protein damage. With the “grafting from” approach a functionalised LRP initiator is conjugated to the protein, the protein is then used as a macroinitiator to initiate the polymerisation.
In comparison to conventional conjugation the benefits of the LRP for the ‘grafting from’ approach include better control around the sites of conjugation (i.e. less local radical-induced damage), faster reaction times and easier purification of the conjugate since only monomers have to be removed after the polymerisation.105 The LRP approach has certain disadvantages in terms of analysis of the conjugate, since prior to polymerisation from the protein the properties of the polymer are not known. Therefore ideally the protein has to be functionalised by a cleavable bond which can be broken selectively after polymerisation to aid in the analysis of the polymer.
The first polymerisations from proteins using ATRP were reported separately in 2005 by Bontempo and Maynard106 and Lele et al.107 In the work of Maynard's group,106 NIPAm (N-isopropylacrylamide) was polymerised from a functionalised streptavidin to form a pNIPAm–protein conjugate. The protein was activated via a biotin functionalised ATRP initiator. The final product was characterised via SDS PAGE and GPC. The polymer was analysed separately by GPC after disruption of the non-covalent bond between streptavidin and biotin.
Lele and co-workers functionalised chymotrypsin at the amine residues with 2-bromoisobutyryl bromide, creating an amide ATRP initiator (Scheme 5).107 By varying the feed of 2-bromoisobutyryl bromide to protein, different degrees of functionalisation were achieved. The extent of functionalisation was confirmed by MALDI-TOF. The functionalised protein was initiated in the presence of a PEGMA monomer. The protein–polymer conjugate was analysed by SDS PAGE and the molecular weight of the conjugated material was estimated with SEC. The protease activity of chymotrypsin was partially retained after the polymerisation, though higher number of initiating sites decreased the activity in comparison to native chymotrypsin. The work demonstrated that growing polymers from amide-based initiators in the presence of protein functionalities was possible, however, the use of non-cleavable initiators meant that the polymer chains could not be analysed completely.
 | ||
| Scheme 5 Functionalisation of chymotrypsin by an ATRP initiator and subsequent PEGMA polymerisation. | ||
RAFT polymerisations have been implemented to grow polymers from proteins. In the work by Boyer and others,108 a pyridyl disulfide based RAFT initiator was conjugated to the free thiol of BSA. The trithiocarbonate RAFT used was linked to the dithiopyridyl reactive group via a 16 monomer unit PEG spacer, which conferred water solubility onto the molecule. A pNIPAm polymer was grown from the functionalised BSA RAFT protein using a room temperature activated azo initiator to initiate the polymerisation. In previous work by the same authors,109 γ radiation was used to initiate the polymerisation but this was found to cause some degradation of the protein: as a consequence low-temperature thermal initiation with an azo compound was found to be preferable.110 The protein–polymer conjugate was analysed via SDS PAGE, 1H-NMR and GPC, in which the polymer portion was analysed individually. Good polymerisation control (PDI < 1.2) was achieved at conversions below 50%.
Magnusson et al. have recently described ATRP routes to prepare polyPEGMA conjugates of recombinant human growth hormone (rhGH).111 These conjugates were prepared under solely aqueous conditions using a derivative of the AGET ATRP route, and good control over the polymer grown from the protein was obtained. Cleavage of the polymer from the rhGH indicated low polydispersity (Mw/Mn = 1.2) of the polymer chains formed under the ATRP conditions. The resulting conjugate exhibited enhanced stability to stress and proteases compared to native rhGH and, importantly, displayed useful biological activity. Rats injected with the poly(PEGMA)–rhGH conjugates exhibited a sustained response in terms of weight gain for a few days following the injections. These responses were better than control groups that received the same dosing frequency of the unconjugated rhGH and untreated animals.111
Despite the numerous advantages of LRP over conventional PEG reagents LRP techniques have certain limitations. ATRP requires a transition metal complex to catalyse the polymerisation, and this complex has to be removed efficiently and quantitatively for biomedical applications. This can be especially problematic when the polymer or protein shows a high affinity for the metal.
In the case of RAFT, initial in vitro studies have revealed that certain RAFT agents can exhibit cellular toxicity,112 thus polymers made by these routes require removal of the RAFT moiety prior to being used in a biomedical context. The method of polymerisation chosen to create the bioconjugate thus depends on the application requirement, length of treatment and amount to be administered.
Controlled polymer synthesis for nucleic acid delivery systems
The optimisation of nucleic acid carrier systems based on PEG and cationic polymers, to fine tune the pharmaceutical carriers, has proven to be difficult. It has also been limited by the availability of PEGs with appropriate functional chain ends and ranges of molar mass. In addition, functionalisation of cationic gene carriers with monofunctional PEG or heterobifunctional PEG is difficult and costly on a pharmaceutical scale. As a consequence, the synthesis of better gene carriers, with easy manipulation and control over their properties (architecture, composition, molecular weight as well as size and functionalities) remains a challenge and a major opportunity for polymer scientists. As for protein functionalisation the progress in LRP techniques has opened up a host of new chemistries available for the formulation of nucleic acids. Currently, numerous well-defined cationic based polymers and copolymers, synthesised via LRP routes, are being developed as nonviral gene delivery vector. Poly(dimethylamino)propyl methacrylamide (pDMAPMAm) and poly(N,N-dimethylaminoethyl methacrylate) (pDMAEMA) polymers are currently receiving much attention as gene delivery systems. Recently, Zhu et al.113 reported the preparation of cationic micelles as drug and gene dual delivery systems. Using RAFT, well-defined amphiphilic triblock copolymers were synthesized composed of poly(ε-caprolactone) (PCL) as the hydrophobic block, with pDMAPMAm and PEG as the hydrophilic blocks. The amphiphilic block copolymers self-assembled in aqueous solution to form cationic core–shell micelles which were subsequently loaded with the anti-cancer drug Doxorubicin (DOX), while pDMAPMAm was used to condense DNA (Scheme 6). The advantage of this synthesis was reflected in control over molar mass, in particular, that of the cationic block pDMAPMAm, which enabled good DNA-condensing properties. Improvements in the toxicological profile were obtained by changing the feed ratio of the monomers during the polymerisation process to control the overall level of cation content.![MPEG-b-p[pDMAPMAm-co-(HEMA–PCL)] polymer and the self-assembled cationic micelle consisting of a hydrophobic PCL core and hydrophilic pDMAPMAm and PEG.](/image/article/2011/PY/c0py00210k/c0py00210k-s6.gif) | ||
| Scheme 6 MPEG-b-p[pDMAPMAm-co-(HEMA–PCL)] polymer and the self-assembled cationic micelle consisting of a hydrophobic PCL core and hydrophilic pDMAPMAm and PEG. | ||
More recently, Üzgun and co-workers,114 copolymerised oligo(ethylene glycol) methyl ether methacrylate (OEGMA) and N,N-dimethylaminoethyl methacrylate (DMAEMA) by ATRP. This generated a series of well-defined p(DMAEMA-co-OEGMA) copolymers with different molecular structures, which were capable of condensing pDNA into nanoparticles of controlled particle size. This example indicates the power of ATRP: versatile routes to design gene transfer agents with excellent colloidal stability and low cytotoxicity have now been shown possible by structural tailoring of multiblock copolymers.
Other controlled polymerisations have been used to synthesize well-defined hydrophilic polymers that can be covalently linked to biopolymer therapeutics, for instance siRNA. Heredia et al.115 described the synthesis of a thiol-reactive poly(PEG acrylate) (pPEGA) via a RAFT polymerisation. The resultant polymer was efficiently conjugated to siRNA in high yield and in a reversible manner. This synthetic strategy might prove to be a useful alternative to conventional PEGylation of siRNA for various therapeutic applications.
LRP has also been used by Armes, Battaglia, and coworkers for the preparation of ‘smart’ amphiphiles able to encapsulate plasmid DNA after their assembly into vesicles.116,117 A pH responsive poly(2-(diisopropylamino)ethyl methacrylate) (pDPA) was polymerized from the chain-end of biocompatible poly(2-(methacryloyloxy)ethyl-phosphorylcholine) (pMPC) to generate the responsive block materials. Solutions of the block-copolymer with DNA at pH 6 were adjusted to pH 7.5 in order to promote block-copolymer self-assembly and, therefore, plasmid encapsulation. The authors reported encapsulation efficiencies of up to 20% without the need for a cosolvent. In addition, plasmid DNA was protectively shielded, either as a polyplex at pH below 7.5, or inside the polymeric vesicle at physiological pH. The authors were able to show both good cell viability and transfection efficiency for green fluorescent protein.
The combinations of these recent advances in synthetic polymer chemistries have provided opportunities for the development of truly well-defined synthetic materials across a wide range of different sizes, shapes, compositions and functionalities. Adopting the concepts from biological molecules combined with the latest chemistry tools is beginning to enable the synthesis of biomaterials that resemble strongly biological molecules in their structural components and functions. For instance, it has been possible to construct polymer based nano-materials that mimic lipoproteins, histones and viral capsids. Each of these biological structures forms as a result of complex assembly of proteins to afford nanostructure, core–shell morphology and specific surface chemistry. In a similar approach, synthetic materials such as well-defined amphiphilic block copolymers have been prepared to trigger self-assembly into micellar structures in aqueous medium. Moreover, surface functionalisations of the polymers with molecules/ligands of interest have been used to impart site recognition properties analogous to active binding site on protein structures.
The research group of Wooley has reported the design and synthesis of several nucleic acid delivery systems originated from biomimicry concepts, using histones as the model to prepare cationic shell crosslinked knedel-like (cSCK) nanoparticles.118 In nucleosomes, the octameric histones cores self-assemble into a disk like shape of approximately 10 nm diameter, around which DNA is condensed into a superhelix structure via electrostatic interactions with cationic arginine groups. For the synthetic polymer analogues, cSCKs were formed by the self-assembly of well-defined amphiphilic block copolymers in water, followed by crosslinking of the shell layer to enhance structural stabilisation. The amphiphilic block copolymer, poly(acrylamidoethylamine)-b-polystyrene (PAEA-b-PS), prepared by LRP, was used in these studies. This amphiphilic block copolymer displayed large numbers of primary amines, used partially for crosslinking the shell after micellar assembly, and to impart cationic character to the final cSCK nanostructure for electrostatic interaction with DNA to allow packaging into complexes for protection, transport and delivery (Scheme 7). The cSCKs exhibited ζ potentials of +21.3 mV and the diameters of 9 ± 1 nm; indicating their nanoscale similarity with self-assembled histone cores.
 | ||
| Scheme 7 Synthetic route for the preparation of well-defined cationic shell crosslinked nanoparticles (cSCKs). Reprinted with permission from ref. 118. | ||
Finally it should be noted that polymeric materials that respond to external stimuli (pH, temperature, and light) have been a focus of research for many applications, including sensing, diagnostics, computation, actuation, as well as controlled release.119 Within the biomedical sector, responsive polymers have been considered as ‘smart’ therapeutic agents, as carriers for drugs and gene delivery systems. The fact that pH gradients exist among various cell types, tissues and intracellular organelles, and that these are related to many physiological and pathological conditions such as endosomal processing, tumour tissue and inflammation, means that local disease states can be exploited to accumulate therapeutic agents via pH responsive carriers.
Recently, Beaudette and colleagues,120 have exploited this using acid degradable polymeric particles in the creation of protein-based vaccines and cancer immunotherapeutics. The developed system was based on polyacrylamide hydrogel microparticles cross-linked with acid-labile moieties which enabled fine-tuning of the degradation rate of the particles based on the pH of the biological environment. A chemical conjugation was used to attach immunostimulatory CpG DNA to the polymer backbone (Scheme 8). Following phagocytosis by antigen presenting cells (APCs), the particle underwent degradation in the endosomal compartments and released their payload.
 | ||
| Scheme 8 Acid-labile particles (a) degrade to release their protein payload and a polymer–CpG conjugate, which can interact with TLR9 (b). Synthesis of macromonomer 8 from 3′ amine-functionalized oligonucleotide 6 (c). Reprinted with permission from ref. 115. | ||
Another bioinspired approach is that developed by Ranquin et al. in which an activating enzyme such as Trypanosoma ViVax (TvNH) was encapsulated inside a biocompatible poly(2-methyloxazoline)-b-poly(dimethylsiloxane)-b-(2-methyloxazoline) (PMOXA–PDMS–PMOXA) vesicle decorated with the channel forming bacterial proteins OmpF and Tsx. Efficient cleavage of one prodrug, 2-fluoroadenosine, by these polymersomes was demonstrated.121,122
Discussion
As it is apparent from the above examples, sophisticated polymer chemistries have been developed to modify the properties of biopolymers, through encapsulation, complexation and/or conjugation. The use of controlled radical techniques such as RAFT and ATRP has to date been confined to making well-defined biomimics and polymer–protein conjugates, but there are clearly opportunities to develop precisely structured nucleic acid conjugates by controlled radical polymerisations. These nucleic acid derivatives should possess similarly useful combinations of properties compared to their parent biopolymers as do polymer–protein conjugates. A number of synthetic polymer–DNA hybrids have been prepared by the Herrmann group and these indeed show behaviours that are enhanced through the combination i.e. nucleic acid complementary sequence pairing and synthetic polymer self-assembly.85,123The increasing desire to use RNA, especially siRNA in therapeutic applications, e.g. to knock down a protein associated with a disease, suggests that polymer–RNA hybrids could be an important synthetic target in the future. The derivatisation of nucleic acids, and particularly RNA, is more problematic than protein conjugation, but for therapeutically unique siRNA sequences, the enhancement to properties or end application might be significant enough to make a hybrid clinically viable.
In this regard it is worth noting the increasing use of ‘click’ and related highly efficient reaction strategies in polymer synthesis and modification. Very well-defined polymers can be prepared by e.g. RAFT, to contain reactive groups such as azide, alkene, alkyne, which can subsequently be derivatised in high yield and site specificity. The ‘click’ philosophy and the development of other high yield derivatisation chemistries (such as active ester routes) have been extensively reviewed elsewhere but it is perhaps only relatively recently that their power has been realised for functional polymer synthesis.124–129 Polymer–protein and polymer–nucleic acid hybrids either prepared or modified via click routes are likely to be an important class of materials in the future.
One important issue for all polymers used in therapy is in vivo fate. Any material introduced into the body must either degrade to harmless fragments or be excreted unchanged once it has fulfilled its therapeutic use. This issue has meant that, to date, only a relatively small number of monomers and polymers have been used in the clinic. This in turn is because of the difficulty, time and expense of conducting full toxicological studies of new materials and determining the ultimate fate of all polymer, and polymer-derived, fragments in the body. One example of this problem can be seen for the field of polymer–protein conjugates. An enormous choice of chemistries exists for the covalent functionalisation of proteins but it is important to choose correctly depending on the specific protein being modified and its intended application. Many conjugation chemistries can in practice lead to a mixture of polymer–protein isomers. These isomers can potentially have very different pharmacodynamic properties in vivo dependent on the region of modification and its proximity to a recognition site. Similarly, different polydispersities of polymer modification will lead to rather different pharmacodynamic profiles in vivo, as blood circulation time and clearance are strongly molar mass dependent. Both of these potential issues have been improved in recent years130 through more specific linker chemistries and higher purity polymers. The issue of protein and polymer isomers can be tackled by ensuring reproducibility in the synthesis and the proper analysis of the conjugate. If each individual isomer can be analysed and the mixture produced each time is reproducible it is safe to consider the mixture as a single drug moiety. The product has to be properly analysed (each species) and the synthesis has to be reproducible each time. There are several such products already in the market, e.g. the PEGylated asparaginase “Oncaspar”. However, there is an increasing regulatory focus on being able to control every aspect of the process, not just the specification of the final product. In this respect, monodisperse polymer-bioconjugates would be much easier to analyse and would face considerably reduced regulatory hurdles towards the clinic than is currently the case. It is clear then, that the stimulus to produce the highest ‘quality’ of polymer conjugates, in terms of monodispersity, controllable functionality, and ease of analysis, remains very strong for biomedical applications.
Conclusions
In this outline article, we have attempted to demonstrate that there is a wide diversity in synthetic polymers for biopharmaceutical delivery. There also remain many challenges for the polymer chemist in developing better polymers for biomedical applications. True precision in the generation of synthetic architectures, placement of functionality, and ease of conjugation or complexation with a biotherapeutic are all necessary for optimum biomaterial formulations. In addition, ‘intelligent’ or at least active, behaviour is required in these materials for the most demanding applications, in order that the polymer can carry the biotherapeutic to a disease site, or repair a defective gene. There are clearly a host of difficulties in generating such materials synthetically, in characterising their structures and in evaluating their overall biological role—but this means there is plenty of room for creative polymer chemists to work in an exciting and rewarding research field.Acknowledgements
We thank the Engineering and Physical Sciences and Biotechnology and Biological Sciences Research Councils (Grants EP/H005625/1, EP/G042462/1, EP/D022347/1, D021847/1 and BB/F01855X/1) and the University of Nottingham, UK for funding.References
- T. Moks, L. Abrahmsen, B. Osterlof, S. Josephson, M. Ostling, S. O. Enfors, I. Persson, B. Nilsson and M. Uhlen, Bio/Technology, 1987, 5, 379–382 CrossRef CAS.
- F. M. Wurm, Nat. Biotechnol., 2004, 22, 1393–1398 CrossRef CAS.
- M. R. Fannon, Trends Biotechnol., 1996, 14, 294–298 CrossRef CAS.
- M. Schena, R. A. Heller, T. P. Theriault, K. Konrad, E. Lachenmeier and R. W. Davis, Trends Biotechnol., 1998, 16, 301–306 CrossRef CAS.
- L. Silverman, R. Campbell and J. R. Broach, Curr. Opin. Chem. Biol., 1998, 2, 397–403 CrossRef CAS.
- W. F. Anderson, Nature, 1998, 392, 25–30 CrossRef CAS.
- C. Andree, W. F. Swain, C. P. Page, M. D. Macklin, J. Slama, D. Hatzis and E. Eriksson, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 12188–12192 CrossRef CAS.
- J. Bonadio, E. Smiley, P. Patil and S. Goldstein, Nat. Med. (Tokyo, Jpn.), 1999, 5, 753–759 Search PubMed.
- G. H. Patterson and J. Lippincott-Schwartz, Science, 2002, 297, 1873–1877 CrossRef CAS.
- D. A. De Angelis, G. Miesenback, B. V. Zemelman and J. E. Rothman, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 12312–12316 CrossRef CAS.
- J. Lippincott-Schwartz and G. H. Patterson, Science, 2003, 300, 87–91 CrossRef.
- E. D. Goluch, J. M. Nam, D. G. Georganopoulou, T. N. Chiesl, K. A. Shaikh, K. S. Ryu, A. E. Barron, C. A. Mirkin and C. Liu, Lab Chip, 2006, 6, 1293–1299 RSC.
- D. Patra, C. Pagliuca, C. Subramani, B. Samanta, S. S. Agasti, N. Zainalabdeen, S. T. Caldwell, G. Cooke and V. M. Rotello, Chem. Commun., 2009, 4248–4250 RSC.
- M. De, S. Rana, H. Akpinar, O. R. Miranda, R. R. Arvizo, U. H. F. Bunz and V. M. Rotello, Nat. Chem., 2009, 1, 461–465 Search PubMed.
- F. McKenzie and D. Graham, Chem. Commun., 2009, 5757–5759 RSC.
- K. Faulds, R. Jarvis, W. E. Smith, D. Graham and R. Goodacre, Analyst, 2008, 133, 1505–1512 RSC.
- W. H. Wong and D. J. Mooney, Synthesis and properties of biodegradable polymers used as synthetic matrices for tissue engineering, in Synthetic Biodegradable Polymer Scaffolds, ed. A. Atala and D. J. Mooney, Birkhauser, Boston, 1997, pp. 51–82 Search PubMed.
- T. Govender, S. Stolnik, M. C. Garnett, L. Illum and S. S. Davis, J. Controlled Release, 1999, 57, 171–185 CrossRef CAS.
- R. A. Jain, Biomaterials, 2000, 21, 2475–2490 CrossRef CAS.
- D. H. Kim and D. C. Martin, Biomaterials, 2006, 27, 3031–3037 CrossRef CAS.
- U. Bilati, E. Allemann and E. Doelker, J. Microencapsulation, 2005, 22, 205–214 CrossRef CAS.
- M. Kumar, U. Bakowsky and C. M. Lehr, Biomaterials, 2004, 25, 1771–1777 CrossRef CAS.
- B. Bittner, B. Ronneberger, R. Zange, C. Volland, J. M. Anderson and T. Kissel, J. Microencapsulation, 1998, 15, 495–514 CrossRef CAS.
- I. Bala, S. Hariharan and M. Kumar, Crit. Rev. Ther. Drug Carrier Syst., 2004, 21, 387–422 CrossRef CAS.
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC.
- F. E. Kohn, J. G. Vanommen and J. Feijen, Eur. Polym. J., 1983, 19, 1081–1088 CrossRef CAS.
- T. G. Park, W. Q. Lu and G. Crotts, J. Controlled Release, 1995, 33, 211–222 CrossRef CAS.
- R. A. Miller, J. M. Brady and D. E. Cutright, J. Biomed. Mater. Res., 1977, 11, 711–719 CrossRef CAS.
- M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093–2107 CrossRef CAS.
- M. D. Jones, M. G. Davidson and G. Kociok-Kohn, Polyhedron, 2010, 29, 697–700 CrossRef CAS.
- B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229–3238 CrossRef CAS.
- E. S. Place, J. H. George, C. K. Williams and M. M. Stevens, Chem. Soc. Rev., 2009, 38, 1139–1151 RSC.
- C. K. Williams, Chem. Soc. Rev., 2007, 36, 1573–1580 RSC.
- I. Taniguchi, A. M. Mayes, E. W. L. Chan and L. G. Griffith, Macromolecules, 2005, 38, 216–219 CrossRef CAS.
- R. Riva, S. Schmeits, C. Jerome, R. Jerome and P. Lecomte, Macromolecules, 2007, 40, 796–803 CrossRef CAS.
- X. Lou, C. Detrembleur, P. Lecomte and R. Jerome, Macromolecules, 2001, 34, 5806–5811 CrossRef CAS.
- Y. S. Jong, J. S. Jacob, K. P. Yip, G. Gardner, E. Seitelman, M. Whitney, S. Montgomery and E. Mathiowitz, J. Controlled Release, 1997, 47, 123–134 CrossRef CAS.
- L. D. Shea, E. Smiley, J. Bonadio and D. J. Mooney, Nat. Biotechnol., 1999, 17, 551–554 CrossRef CAS.
- R. Langer, Nature, 1998, 392, 5–10 CAS.
- R. Langer, Science, 1990, 249, 1527–1533 CrossRef CAS.
- E. Walter, K. Moelling, J. Pavlovic and H. P. Merkle, J. Controlled Release, 1999, 61, 361–374 CrossRef CAS.
- Y. Y. Hsu, T. Hao and M. L. Hedley, J. Drug Targeting, 1999, 7, 313–323 Search PubMed.
- S. Freitas, H. P. Merkle and B. Gander, J. Controlled Release, 2005, 102, 313–332 CrossRef CAS.
- H. Sah and Y. Bahl, J. Controlled Release, 2005, 106, 51–61 CrossRef CAS.
- L. M. Sanders, B. A. Kell, G. I. McRae and G. W. Whitehead, J. Pharm. Sci., 1986, 75, 356–360 CrossRef CAS.
- Y. Ogawa, M. Yamamoto, H. Okada, T. Yashiki and T. Shimamoto, Chem. Pharm. Bull., 1988, 36, 1095–1103 CAS.
- J. L. Cleland and A. J. S. Jones, Pharm. Res., 1996, 13, 1464–1475 CrossRef CAS.
- L. C. Ter Beek, M. Ketelaars, D. C. McCain, P. E. Smulders, P. Walstra and M. A. Hemminga, Biophys. J., 1996, 70, 2396–2402 CAS.
- Y. Capan, B. H. Woo, S. Gebrekidan, S. Ahmed and P. P. DeLuca, Pharm. Dev. Technol., 1999, 4, 491–498 CrossRef CAS.
- Y. Capan, B. H. Woo, S. Gebrekidan, S. Ahmed and P. P. DeLuca, J. Controlled Release, 1999, 60, 279–286 CrossRef CAS.
- O. L. Johnson, W. Jaworowicz, J. L. Cleland, L. Bailey, M. Charnis, E. Duenas, C. Wu, D. Shepard, S. Magil, T. Last, A. J. S. Jones and S. D. Putney, Pharm. Res., 1997, 14, 730–735 CrossRef CAS.
- J. L. Cleland, E. T. Duenas, A. Park, A. Daugherty, J. Kahn, J. Kowalski and A. Cuthbertson, J. Controlled Release, 2001, 72, 13–24 CrossRef CAS.
- H. Okada and H. Toguchi, Crit. Rev. Ther. Drug Carrier Syst., 1995, 12, 1–99 CAS.
- D. Q. Wang, D. R. Robinson, G. S. Kwon and J. Samuel, J. Controlled Release, 1999, 57, 9–18 CrossRef CAS.
- S. Ando, D. Putnam, D. W. Pack and R. Langer, J. Pharm. Sci., 1999, 88, 126–130 CrossRef CAS.
- F. Meng, Z. Zhong and J. Feijen, Biomacromolecules, 2009, 10, 197–209 CrossRef CAS.
- J. Du and R. K. O'Reilly, Soft Matter, 2009, 5, 3544–3561 RSC.
- S. Rameez, H. Alosta and A. F. Palmer, Bioconjugate Chem., 2008, 19, 1025–1032 CrossRef CAS.
- Y. Kim, M. Tewari, J. D. Pajerowski, S. Cai, S. Sen, J. Williams, S. Sirsi, G. Lutz and D. E. Discher, J. Controlled Release, 2009, 134, 132–140 CrossRef CAS.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS.
- L. A. Canalle, D. Lowik and J. C. M. van Hest, Chem. Soc. Rev., 2010, 39, 329–353 RSC.
- F. M. Veronese and G. Pasut, Drug Discovery Today, 2005, 10, 1451–1458 CrossRef CAS.
- S. M. Chamow, T. P. Kogan, M. Venuti, T. Gadek, R. J. Harris, D. H. Peers, J. Mordenti, S. Shak and A. Ashkenazi, Bioconjugate Chem., 1994, 5, 133–140 CrossRef CAS.
- A. Abuchowski, T. Vanes, N. C. Palczuk and F. F. Davis, J. Biol. Chem., 1977, 252, 3578–3581 CAS.
- Q. Guoqiang, M. Jianbiao, Z. Juyan, H. Binglin and W. Daobin, Polym. Adv. Technol., 1997, 8, 581–586 CrossRef CAS.
- C. Monfardini, O. Schiavon, P. Caliceti, M. Morpurgo, J. M. Harris and F. M. Veronese, Bioconjugate Chem., 2002, 6, 62–69.
- R. B. Greenwald, A. Pendri, A. Martinez, C. Gilbert and P. Bradley, Bioconjugate Chem., 1996, 7, 638–641 CrossRef CAS.
- D. C. Wylie, M. Voloch, S. Lee, Y. H. Liu, S. Cannon-Carlson, C. Cutler and B. Pramanik, Pharm. Res., 2001, 18, 1354–1360 CrossRef CAS.
- P. Bailon, A. Palleroni, C. A. Schaffer, C. L. Spence, W. J. Fung, J. E. Porter, G. K. Ehrlich, W. Pan, Z. X. Xu, M. W. Modi, A. Farid and W. Berthold, Bioconjugate Chem., 2001, 12, 195–202 CrossRef CAS.
- S. Zalipsky, R. Seltzer and S. Menonrudolph, Biotechnol. Appl. Biochem., 1992, 15, 100–114 CAS.
- O. Kinstler, G. Molineux, M. Treuheit, D. Ladd and C. Gegg, Adv. Drug Delivery Rev., 2002, 54, 477–485 CrossRef CAS.
- H. Lee, I. H. Jang, S. H. Ryu and T. G. Park, Pharm. Res., 2003, 20, 818–825 CrossRef CAS.
- R. B. Pepinsky, D. J. Lepage, A. Gill, A. Chakraborty, S. Vaidyanathan, M. Green, D. P. Baker, E. Whalley, P. S. Hochman and P. Martin, J. Pharmacol. Exp. Ther., 2001, 297, 1059–1066 CAS.
- M. S. Rosendahl, D. H. Doherty, D. J. Smith, S. J. Carlson, E. A. Chlipala and G. N. Cox, Bioconjugate Chem., 2005, 16, 200–207 CrossRef CAS.
- V. N. Lapko, D. L. Smith and J. B. Smith, J. Mass Spectrom., 2000, 35, 572–575 CrossRef CAS.
- C. Woghiren, B. Sharma and S. Stein, Bioconjugate Chem., 1993, 4, 314–318 CrossRef CAS.
- S. Shaunak, A. Godwin, J. W. Choi, S. Balan, E. Pedone, D. Vijayarangam, S. Heidelberger, I. Teo, M. Zloh and S. Brocchini, Nat. Chem. Biol., 2006, 2, 312–313 CrossRef CAS.
- S. Balan, J. W. Choi, A. Godwin, I. Teo, C. M. Laborde, S. Heidelberger, M. Zloh, S. Shaunak and S. Brocchini, Bioconjugate Chem., 2007, 18, 61–76 CrossRef CAS.
- M. Zloh, S. Shaunak, S. Balan and S. Brocchini, Nat. Protoc., 2007, 2, 1070–1083 Search PubMed.
- A. Mero, B. Spolaore, F. M. Veronese and A. Fontana, Bioconjugate Chem., 2009, 20, 384–389 CrossRef CAS.
- G. Pasut and F. M. Veronese, Adv. Drug Delivery Rev., 2009, 61, 1177–1188 CrossRef CAS.
- R. B. Greenwald, H. Zhao, K. Yang, P. Reddy and A. Martinez, J. Med. Chem., 2004, 47, 726–734 CrossRef CAS.
- H. Zhao, K. Yang, A. Martinez, A. Basu, R. Chintala, H. C. Liu, A. Janjua, M. L. Wang and D. Filpula, Bioconjugate Chem., 2006, 17, 341–351 CrossRef CAS.
- R. B. Greenwald, A. Pendri, C. D. Conover, H. Zhao, Y. H. Choe, A. Martinez, K. Shum and S. Y. Guan, J. Med. Chem., 1999, 42, 3657–3667 CrossRef.
- F. E. Alemdaroglu, M. Safak, J. Wang, R. Berger and A. Herrmann, Chem. Commun., 2007, 1358–1359 RSC.
- Y. Singh, P. Murat and E. Defrancq, Chem. Soc. Rev., 2010, 39, 2054–2070 RSC.
- G. T. Hermanson, Bioconjugates Techniques, Academic Press, 2008 Search PubMed.
- G. M. Bonora, G. Tocco, S. Zaramella, F. M. Veronese, O. Pliasunova, A. Pokrovsky, E. Ivanova and V. Zarytova, Farmaco, 1998, 53, 634–637 CrossRef CAS.
- J. H. Jeong, S. W. Kim and T. G. Park, J. Controlled Release, 2003, 93, 183–191 CrossRef CAS.
- D. K. Kim and J. Dobson, J. Mater. Chem., 2009, 19, 6294–6307 RSC.
- T. E. Patten and K. Matyjaszewski, Adv. Mater., 1998, 10, 901–915 CrossRef CAS.
- K. Matyjaszewski, Macromolecules, 1999, 32, 9051–9053 CrossRef CAS.
- K. Matyjaszewski, S. Coca, Y. Nakagawa and J. H. Xia, Abstr. Pap., Jt. Conf. - Chem. Inst. Can. Am. Chem. Soc., 1997, 213, 83-PMSE Search PubMed.
- A. B. Lowe, B. S. Sumerlin, M. S. Donovan and C. L. McCormick, J. Am. Chem. Soc., 2002, 124, 11562–11563 CrossRef CAS.
- A. J. T. Dirks, S. S. van Berkel, N. S. Hatzakis, J. A. Opsteen, F. L. van Delft, J. Cornelissen, A. E. Rowan, J. C. M. van Hest, F. Rutjes and R. J. M. Nolte, Chem. Commun., 2005, 4172–4174 RSC.
- J. P. Montheard, M. Chatzopoulos and D. Chappard, J. Macromol. Sci., Rev. Macromol. Chem. Phys., 1992, C32, 1–34 Search PubMed.
- S. Abraham, S. Brahim, A. Guiseppi-Elie and IEEE, in 26th Annual International Conference of the IEEE-Engineering-in-Medicine-and-Biology-Society, IEEE, San Francisco, CA, 2004, pp. 5036–5039 Search PubMed.
- S. M. Ryan, X. Wang, G. Mantovani, C. T. Sayers, D. M. Haddleton and D. J. Brayden, J. Controlled Release, 2009, 135, 51–59 CrossRef CAS.
- G. Mantovani, F. Lecolley, L. Tao, D. M. Haddleton, J. Clerx, J. Cornelissen and K. Velonia, J. Am. Chem. Soc., 2005, 127, 2966–2973 CrossRef CAS.
- F. Lecolley, L. Tao, G. Mantovani, I. Durkin, S. Lautru and D. M. Haddleton, Chim. Oggi, 2005, 23, 45 Search PubMed.
- E. Bays, L. Tao, C. W. Chang and H. D. Maynard, Biomacromolecules, 2009, 10, 1777–1781 CrossRef CAS.
- B. Le Droumaguet, G. Mantovani, D. M. Haddleton and K. Velonia, J. Mater. Chem., 2007, 17, 1916–1922 RSC.
- Q. Z. Dong and Y. L. Hsieh, J. Appl. Polym. Sci., 2000, 77, 2543–2551 CrossRef CAS.
- J. M. Zhu and P. Li, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3346–3353 CrossRef CAS.
- J. Nicolas, G. Mantovani and D. M. Haddleton, Macromol. Rapid Commun., 2007, 28, 1083–1111 CrossRef CAS.
- D. Bontempo and H. D. Maynard, J. Am. Chem. Soc., 2005, 127, 6508–6509 CrossRef CAS.
- B. S. Lele, H. Murata, K. Matyjaszewski and A. J. Russell, Biomacromolecules, 2005, 6, 3380–3387 CrossRef CAS.
- C. Boyer, V. Bulmus, J. Q. Liu, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, J. Am. Chem. Soc., 2007, 129, 7145–7154 CrossRef CAS.
- J. Q. Liu, V. Bulmus, D. L. Herlambang, C. Barner-Kowollik, M. H. Stenzel and T. P. Davis, Angew. Chem., Int. Ed., 2007, 46, 3099–3103 CrossRef CAS.
- W. M. Garrison, Chem. Rev., 1987, 87, 381–398 CrossRef CAS.
- J. P. Magnusson, S. Bersani, S. Salmaso, C. Alexander and P. Caliceti, Bioconjugate Chem., 2010, 21, 671–678 CrossRef CAS.
- C. W. Chang, E. Bays, L. Tao, S. N. S. Alconcel and H. D. Maynard, Chem. Commun., 2009, 3580–3582 RSC.
- J. L. Zhu, H. Cheng, Y. Jin, S. X. Cheng, X. Z. Zhang and R. X. Zhuo, J. Mater. Chem., 2008, 18, 4433–4441 RSC.
- S. Uzgun, O. Akdemir, G. Hasenpusch, C. Maucksch, M. M. Golas, B. Sander, H. Stark, R. Imker, J. F. Lutz and C. Rudolph, Biomacromolecules, 2010, 11, 39–50 CrossRef CAS.
- K. L. Heredia, T. H. Nguyen, C. W. Chang, V. Bulmus, T. P. Davis and H. D. Maynard, Chem. Commun., 2008, 3245–3247 RSC.
- H. Lomas, I. Canton, S. MacNeil, J. Du, S. P. Armes, A. J. Ryan, A. L. Lewis and G. Battaglia, Adv. Mater., 2007, 19, 4238–4243 CrossRef CAS.
- H. Lomas, M. Massignani, K. A. Abdullah, I. Canton, C. L. Presti, S. MacNeil, J. Du, A. Blanazs, J. Madsen, S. P. Armes, A. L. Lewis and G. Battaglia, Faraday Discuss., 2008, 139, 143–159 RSC.
- K. Zhang, H. Fang, G. Shen, J.-S. A. Taylor and K. L. Wooley, Proc. Am. Thorac. Soc., 2009, 6, 450–457 CrossRef CAS.
- D. Cunliffe, S. Pennadam and C. Alexander, Eur. Polym. J., 2004, 40, 5–25 CrossRef CAS.
- T. T. Beaudette, E. M. Bachelder, J. A. Cohen, A. C. Obermeyer, K. E. Broaders, J. M. J. Frechet, E. S. Kang, I. Mende, W. W. Tseng, M. G. Davidson and E. G. Engleman, Mol. Pharmaceutics, 2009, 6, 1160–1169 CrossRef CAS.
- G. Huysmans, A. Ranquin, L. Wyns, J. Steyaert and P. Van Gelder, J. Controlled Release, 2005, 102, 171–179 CrossRef CAS.
- A. Ranquin, W. Versees, W. Meier, J. Steyaert and P. Van Gelder, Nano Lett., 2005, 5, 2220–2224 CrossRef CAS.
- F. E. Alemdaroglu, N. C. Alemdaroglu, P. Langguth and A. Herrmann, Macromol. Rapid Commun., 2008, 29, 326–329 CrossRef CAS.
- J. Geng, J. Lindqvist, G. Mantovani and D. M. Haddleton, Angew. Chem., Int. Ed., 2008, 47, 4180–4183 CrossRef CAS.
- V. Ladmiral, G. Mantovani, G. J. Clarkson, S. Cauet, J. L. Irwin and D. M. Haddleton, J. Am. Chem. Soc., 2006, 128, 4823–4830 CrossRef CAS.
- K. T. Wiss, O. D. Krishna, P. J. Roth, K. L. Kiick and P. Theato, Macromolecules, 2009, 42, 3860–3863 CrossRef CAS.
- C. R. Becer, K. Kokado, C. Weber, A. Can, Y. Chujo and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1278–1286 CrossRef CAS.
- A. Bousquet, C. Barner-Kowollik and M. H. Stenzel, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1773–1781 CrossRef CAS.
- M. Hetzer, G. J. Chen, C. Barner-Kowollik and M. H. Stenzel, Macromol. Biosci., 2010, 10, 119–126 CrossRef CAS.
- G. G. Kochendoerfer, S. Y. Chen, F. Mao, S. Cressman, S. Traviglia, H. Y. Shao, C. L. Hunter, D. W. Low, E. N. Cagle, M. Carnevali, V. Gueriguian, P. J. Keogh, H. Porter, S. M. Stratton, M. C. Wiedeke, J. Wilken, J. Tang, J. J. Levy, L. P. Miranda, M. M. Crnogorac, S. Kalbag, P. Botti, J. Schindler-Horvat, L. Savatski, J. W. Adamson, A. Kung, S. B. H. Kent and J. A. Bradburne, Science, 2003, 299, 884–887 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |