Synthesis of glycopolymers and their multivalent recognitions with lectins
S. R. Simon
Ting
,
Gaojian
Chen†
and
Martina H.
Stenzel
*
Centre for Advanced Macromolecular Design, School of Chemical Engineering, University of New South Wales, Sydney, NSW 2052, Australia. E-mail: m.stenzel@unsw.edu.au; Fax: +61 2 93856250; Tel: +61 2 93854344
First published on 30th June 2010
Abstract
Synthetic carbohydrate ligands – also widely known as glycopolymers – are known to undergo numerous recognition events when interacting with their corresponding lectins. Interactions are greatly enhanced due to the multivalent character displayed by the large number of repeating carbohydrate units along the polymers (pendant glycopolymers); therefore, resulting what is called the “glycocluster effect”. Moreover, the strength and the availability of these multivalent recognitions can be tuned via the architecture of the glycopolymers. Hence, understanding the mechanistic interactions between the types of lectins (plant, animal, toxin and bacteria) with their synthetic ligands is crucial. This review focuses on the synthesis of pendant glycopolymers via various synthetic pathways (free radical polymerization, NMP, RAFT, ATRP, cyanoxyl mediated polymerization, ROP, ROMP and post-polymerization modification) and their interactions with their respectively lectins.
 S. R. Simon Ting | S. R. Simon Ting was born in Singapore in 1979. Before completing his Ph.D. in Polymer Chemistry, he graduated from the University of New South Wales in 2006 with a B.E. (Hons) in Chemical Engineering (Core-shell particles from suspension polymerization) under the supervision of Assoc. Prof. Martina H. Stenzel. He remained at the Centre for Advanced Macromolecular Design and completed his doctoral study in 2010 under the same supervisor, with the thesis entitled, “Synthesis of Glyco-Particles via Controlled/Living Free-Radical Polymerization”. He is currently a research associate in the same centre working with Assoc. Prof. Per B. Zetterlund on the synthesis of nano-particles using low energy polymerizations in dispersed systems. |
 Gaojian Chen | Gaojian Chen joined Prof. David Haddleton's group for PhD Study from 2004 to 2007 at the University of Warwick, being granted the Overseas Research Students Awards and Warwick Postgraduate Research Fellowship. He continued his postdoctoral research with Martina Stenzel at the Centre for Advanced Macromolecular Design, University of New South Wales until 2009. He is currently Associate Professor at the Centre for Soft condensed Matter Physics and Interdisciplinary Research at Soochow University. His research mainly focused on the preparation of well-defined polymers via living radical polymerization and click chemistry, with special interest on drug delivery applications. |
 Martina H. Stenzel | Martina Stenzel studied chemistry at the University of Bayreuth, Germany, before completing her PhD in 1999 at the University of Stuttgart. Since 2000, she has worked at the University of New South Wales in the Centre for Advanced Macromolecular Design (CAMD), where she is currently an Associate Professor and ARC Future Fellow. Her research interest is focused on the synthesis of functional polymers such as glycopolymers and other polymers for biomedical applications. Martina Stenzel has published more than 150 peer reviewed papers mainly on RAFT polymerization. She is currently the chair of the Polymer division of the Royal Australian Chemical Institute and editor of the Australian Journal of Chemistry. |
Introduction
Carbohydrates have become a hot topic for research within the scientific community. This is due to the myriad of biological communication events, including: cellular recognition, inflammation, signal transmission and infection of pathogens displayed by them.1–5 In the treatment of diseases, such as cancer, cytotoxic chemotherapy or radiotherapy can be life threatening as the therapeutics used are normally not site-specific. To improve the distribution of drugs in a biological system, the use of ligand (e.g. carbohydrate and peptide targeted therapeutics for the recognition of malignant cells), could be an important step towards the improved treatment of cancer and other diseases.6,7Moreover, many studies have shown that lectins on cell surfaces mediate cell-cell interactions by combining with complementary carbohydrates; in other words, the incorporation of ligands such as carbohydrates or other targeting moieties could result in increased cellular uptake via receptor-endocytosis.8–12 Sharon and Lee have long established that lectins on cell surfaces can act as receptors with an affinity towards their carbohydrate ligands.13,14
In order for the interactions between the drug-targeting ligands and the receptors on the cell surfaces to be strong, several parallel interactions take place.13 The interaction between one protein and one carbohydrate molecule is rather weak. However, when these single carbohydrate molecules are placed along a polymer backbone or any other entity, collective binding is much stronger, which is termed the “glycocluster effect”. Polymer scientists can contribute to the design of multivalent ligands, hence, allow for the enhanced interactions with the lectin receptors on cell surfaces.
Glycopolymers are synthetic polymers carrying carbohydrate functional groups. They include: glycodendrimers, linear glycopolymers and spherical glycopolymers in the form of micelles, vesicles and micro/nano particles.15 These advanced materials have the ability to produce multivalent interactions with the lectins on the cells, notably termed the “cluster glycoside effect”.
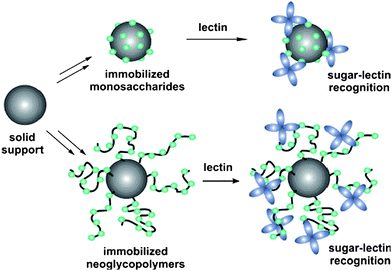
In this review article, attention is focused on polymers containing pendant carbohydrate moieties, which were studied regarding their interaction with lectins. Ironically, not all the synthetic glycopolymers documented have a strong binding ability towards lectins, as modification of the carbohydrate six-membered ring to allow the attachment of vinyl compounds, or other functionalities, may ultimately affect the bioactivity of the glycopolymers toward lectin interactions.16 Hence, essentially only glycopolymers synthesized and used for lectin binding assays are discussed in depth here. The first section will give a general discussion on the various polymerization methods used to synthesize glycopolymers from carbohydrate-containing monomers followed by the postfunctionalization of reactive polymer scaffolds with carbohydrates. We then discuss the interaction of different shaped glycopolymers with lectins i.e. (A) glycopolymers in their original linear forms, (B) spherical or surface assembly of glycopolymers and (C) glycopolymers adhered on to material surfaces (Fig. 1). Dendrimers are not part of this review despite representing a very important group of glycomaterials due to their very different synthetic strategies.17
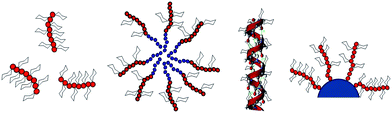 | ||
| Fig. 1 Glycopolymer architectures | ||
Lectins
Lectins (latin: legere (to select)) are sugar-binding proteins that bind with carbohydrates reversibly but with high specificity. This class of proteins can be found in all biological systems and they play a pivotal role in many biological events such as cell adhesion. The reaction between lectins and carbohydrates form the basis of cell agglutination such as hemagglutination.2,18 Sharon and Lis highlighted the importance of recognition events involving lectins in the biological system.14 Cell recognition as a concept of lock-and-key complementarities was first mentioned by Emil Fisher in 1897. More recently, Ambrosi et al. have described lectins as tools for the molecular understanding of the glycocode. In his review, particular interest into the mechanism of carbohydrate-binding with legumes was presented describing the energy of binding between saccharides and lectins.18 Another important review article in 2002 on cluster glycoside effect has also highlighted the importance of carbohydrate-lectin interactions. Lundquist and Toone underlined here the difficulties in understanding the nature of these interactions.3 An important step towards understanding the origin of the cluster effect was achieved by using remodelled glycoproteins. A “bind-and-slide” mechanism was proposed for the stronger interactions of multivalent glycoconjugates compared to monovalent ligands.19 This section of the review will provide the reader with a general idea and the availability of lectins used for carbohydrates binding studies, for further details about lectins please refer to the individual articles mentioned along the section.Hundreds of lectins have now been identified and isolated from organisms such as plants, animals and microorganisms. While all these lectins have certain biological properties in common such as the binding to carbohydrates, they are very diverse in terms of structure and size. Many of them can be grouped into families depending on their function or certain functional parameters. Carbohydrate binding proteins are also divided into two major subgroups according to the three-dimensional structures of the interaction between protein and carbohydrate. The group I carbohydrate-binding proteins completely entraps the carbohydrate ligands in deep binding pockets, while the group II carbohydrate-binding proteins bind their ligands in shallow pockets or grooves on the protein surface. This division not only applies to lectins but also to other proteins such as enzymes. The three-dimensional structure of the binding between carbohydrate and lectin has been the subject of intense investigations.20,21
Plant lectins
Legumes 22
The largest family among the simple lectins is the legume family with more than 70 lectins being isolated and many of them have been structurally characterized.23 They are simple lectins isolated from plants belonging to the Fabaceae family, mainly from seeds. Their molecular weights are usually below 40 kDa, occasionally with an additional domain on top of their carbohydrate binding sites. Interactions with carbohydrates require often the presence of Ca2+ and Mn2+ ions. They usually consist of 2 or 4 subunits with typically 1 binding site per subunit.The main representatives of the legume family are:
Concanavalin A (ConA), the lectin extracted from jack beans, is the most widely abundant lectin within the legume family. The abundance is due to the ease of isolation and the interactions with a wide range of saccharides which has led to many in-depth studies for this member of the legume family.15,18 ConA has a strong affinity to mannose, but also binds glucose.
Peanut agglutinin (PNA, Arachis hypogaea) is another legume, which binds specifically to galactose, preferably to galactosyl (β-1,3) N-acetylgalactosamine. PNA does not require any divalent cations for binding, but binding is enhanced in the presence of Ca2+ ions.
Other legumes: Griffonia simplicifolia agglutinin-I (GSA-I) and the isolectins GSA-I-AB3 and GSA-I–B4, isolated from the seeds of an African shrub, is a lectin that is specific for galactose/N-acetylgalactosamine and was found to adhere to endothelial cells and some epithelial cells.24 Dimerization is observed in Erythrina corallodendron (Coral Bean Tree) (EcorL), a legume which binds specifically to lactose [Gal β−(1 → 4)Glc], galactose and N-acetylgalactoseamine.25 The structurally almost identical Lathyrus ochrus lectin I (LOLI) in contrast, binds to glucose and mannose.23
Cereal lectins
Cereal lectins consist of two subunits with usually 2 binding sites per subunit. The presence of ions such as Ca2+ is not required. Cereal lectins are known to be rich in disulfide bonds.Wheat germ agglutinin (WGA) consists of two identical subunits whilst being rich in cysteine.26 It exists in three isoforms, WGA1, WGA2 and WGA3 with a high specifity to N-acetylglucosamine and N-acetylneuraminic acid (a sialic acid).
Other lectins
The lectins found in the bulbs of plants of the amaryllis, orchid, and garlic families (all belonging to the Amaryllidaceae family and related families) bind mannose (but not glucose).An example of the Moraceae family is Jacalin, extracted from the Jackfruit (Artocarpus integrifolia).27 Jacalin apparently does not bind to galactosyl-N-acetylglucosamine but recognizes galactose (β-1 → 3) N-acetylgalactosamine. Interestingly, it also binds to Immunoglobulin A (IgA).28
Famous examples from the Euphorbiaceae family are the lectins produced by the beans of the castor tree (Ricinus communis). The two lectins, ricin and Ricinus communis agglutinin (RCA) are closely related with ricin being one of the most toxic lectins.29
Function of plant lectins
The function of plant lectins is unknown, but it has been suggested that they act as defence system for the plant.Animal lectins
Animal lectins were originally divided into C-type lectins (need Ca2+-ions) and S-type lectins (sulfydryl-dependent). With increased knowledge in regards to the structure of lectins, more and more groups were identified and the list now includes: C-type, S-type (galectins), I-type (siglecs and others), P-type (phosphomannosyl receptors), pentraxins, egg lectins, calreticulin and calnexin, ERGIC-53 and VIP-36, discoidins, eel aggutinins (fucolectins), annexin lectins, fibrinogen-type lectins and a some single lectins that cannot yet be assigned to any of these groups.30 Only C-Type and Galectins are discussed here:C-Type
C-Type lectins are dependent on Ca2+ ions for their reactions with carbohydrates. They can have complex structures consisting of a carbohydrate recognition domain (CRD) of around 120 amino acids. C-Type lectins therefore, can have a variable number of subunits with 1–8 binding sites per subunit.23 C-type lectin subfamilies are divided into 7 subcategories : I Hyalectans, II Asialoglycoprotein receptors, III Collectins, IV Selectins, V NK group transmembrane receptors, VI Macrophage mannose receptor and VII Simple (single domain) lectins.30A C-type lectin is the endocytic lectin, which is more frequently described as the hepatic asialoglycoprotein (hepatic lectin), a lectin specific for galactose/N-acetylgalactosamine.2,12 In-depth studies have been carried out using copolymers with galactose moieties for the interactions with hepatic lectins and results suggested that high sugar concentrations facilitated binding.9
Macrophages express a range of receptors including several C-type lectins. The foremost C-type lectin on macrophages is the mannose receptor, which can also be found on hepatic and lymphatic endothelia, on mesangial cells in the kidneys, on tracheal smooth muscle cells, and on retinal pigment epithelium.31 It recognizes mannose, but also fucose and N-acetylglucosamine.
Selectins are cell adhesion molecules and share similar properties to C-type lectins. Selectins are divided into three subtypes: E-selectin (in endothelial cells), L-selectin (in leukocytes) and P-selectin (in platelets and endothelial cells). Selectins interact with the tetrasaccharide sialyl-Lewisx (sialyl-CD15), which consists of sialic acid, galactose, fucose, and N-acetyl-galactosamine.
Galectins (S-Type)32
Galectins, which were formerly called S-type lectins, have an incomprehensible array of activities ranging from inflammation response to a suggested role in cancer. A common trait in galectins is the affinity for β-galactosides, preferably as lactose and N-acetyl lactosamine, and a significant sequence similarity in the carbohydrate-binding site. They can be subdivided into a further three groups: (1) the dimeric form that consists of only the carbohydrate recognition domains (Galectins-1, -2, -5, -7, -10, -13, -14, GRIFIN, HSPC159, PP13, PPL13, OVGAL11); (2) galectins, where the CRD is attached to its N-terminal end which possess a highly sequence-repetitive domain (Galectin-3); (3) galectins, that comprise of two tandemly connected CRDs (Galectins-4, -6, -8, -9, -12).13Functions of animal lectins30
Animal lectins play a pivotal role in a variety of functions including:• Self/non-self recognition
• Intracellular routing of glycoconjugates
• Molecular chaperones during glycoprotein synthesis
• Mediation of endocytosis
• Cellular growth regulation
• Extracellular molecular bridging
• Cell –cell interactions for homing and trafficking
• Scavenging of cellular debris; anti-inflammatory action
• Recognition molecules within the immune system:
∘ direct defence
∘ recognition and trafficking within the immune system
∘ immune regulation (suppression or enhancement)
∘ prevention of autoimmunity
AB5 toxins
AB5 toxins contain the disease causing A unit and five B units, which attach to carbohydrates. Examples of AB5 toxins are the Cholera toxin (CT), which binds to galactose, the heat-labile enterotoxin of E. coli (LT-I) and the Shiga toxin.Bacterial adhesion lectins
The attachment of bacteria onto tissue is often mediated by carbohydrate protein interactions. FimH protein is a type 1 fimbriae, hair-like lectin that sits on bacteria, e.g. on Escherichia coli (E. coli) cell surfaces.2,33,34 The lectin only contains one binding site for mannose therefore, the multivalent binding is achieved by the arrangement of many fimbriae sitting alongside each other in the cell membrane. Pseudomonas aeruginosa produces two galactose and fucose binding lectins, called lecA and lecB (also named PA-IL and PA-IIL).Alternative grouping of lectins2
Alternative ways of grouping lectins have been envisaged with lectins being grouped according to their molecular structure. Lis and Sharon proposed a three-tiered division into simple lectins, mosaic and macromolecular assembly lectins.Glycopolymer-lectin interactions
The use of the lock-and-key metaphor is complementary to the described glycopolymer-lectin interactions, coined by Emil Fisher in the 1897. It has now evolved into more sophisticated descriptions such as the carbohydrate-encoded information/lectin as a decoder representation.14,18 The unique interactions between carbohydrates and lectins are highly specific, but normally weak. Hence, multiple interactions between carbohydrates and lectins are necessary to achieve strong binding. Multiple interactions provide extra specificity between carbohydrates and their binding proteins and also ultimately increase the strength of the interactions by many folds.18 Pieters recently described a way that leads to enhanced binding, or inhibition, as chelation, a process whereby two binding sites are bridged by a multivalent ligand.15 Enhancements displayed by proteins that exhibited two or more binding sites could be as high as 103–106, for both carbohydrate-based systems and non-carbohydrate-based systems.13,15,35–38Fig. 2 displays carbohydrate functionalized particles showing enhanced interactions with cell surface lectin receptors. | ||
| Fig. 2 Ligand-targeted therapeutics. The uptake by the cell via receptor-mediated endocytosis with multivalent interactions (left) and therapeutics without targeting ligands, the internalization by the cell via slow fluid-phase pinocytosis (right). | ||
The interactions between saccharides and their lectins are indeed profound when viewed in the perspective of glycobiology. The studies of carbohydrates influencing the properties of proteins to which they are attached and also the involvements of carbohydrates in recognition events are the two main areas thoroughly discussed in glycobiology.1 In the field of polymer science, the research into synthetic glycopolymers and the binding ability with various lectins has been carried out by many groups. Herein, we present a detailed discussion into the interactions of the different types of glycopolymers used for binding lectins, namely: (a) linear glycopolymers, (b) glycopolymer assemblies and (c) the attachments of glycopolymers onto the surface of materials (Table 1).
| Glycopolymer architectures | Synthesis method | Carbohydrate epitope | Lectin | Assay |
|---|---|---|---|---|
| Linear polymer143 | FRP | α-D-Glc(1 → 4)D-Glc, β-D-Gal(1 → 4)D-Glc, α-D-Glc(1 → 4)D-Glc(1 → 4)D-Glc | ConA | Turbidity |
| Linear polymer74 | ROMP | α-D-GlcNAc | ConA | HIA |
| Linear polymer53 | FRP | β-D-Gal(1 → 4)D-GlcNAc | ECorL | Fluorescence |
| Linear polymer69 | ROMP | α-Glc, α-man | ConA | HIA |
| Linear polymer73 | ROMP | α-GluNAc, α-manNAc | ConA | HIA |
| Linear polymer45 | FRP | β-Lac, β-chitobiose | WGA, PNA, RCA120, ECA, DSA | DDA, HIA |
| Linear polymer68 | ROMP | 3’ sulfated β-Gal | P-selectin | Cell binding |
| Linear polymer58 | FRP | β-Gal(1 → 4)GlcNAc, β-Gal(1 → 6)GlcNAc, β-Gal(1 → 4)Gal(1 → 4)β-Glc | RCA120 | HIA |
| Linear polymer114 | FRP, PF (Condensation) | LacNAc | ECA, PNA, RCA120, WGA | HIA, DDA |
| Linear polymer125 | ROMP, PF | α-man | ConA | HIA |
| Linear polymer72 | ROMP | SO3−β-Gal(1 → 4)α-Fuc(1 → 3)SO3−β-Glc | L-selectin | ELISA |
| Linear polymer144 | FRP, PF | α-Glc, β-Glc, β-Gal, β-Lac | ConA, RCA120 | HIA |
| Linear polymer148 | Oxidative P., PF | Sialic acid, Man | WGA, ConA | Turbidity |
| Linear polymer56 | FRP | Man | ConA, LCA | Turbidity |
| Linear polymer49 | FRP | β-D-Man | ConA | ELLA |
| Linear polymer44 | ROMP | α-Man | ConA | Turbidity |
| Linear polymer9 | FRP | D-Gal, β-D-Lac | HA | Fluorescence |
| Linear polymer126 | FRP, PF | β-Gal(1 → 3)α-GalNAc | PNA | Turbidity |
| Linear polymer127 | FRP, PF | β-D-Gal(1 → 4)D-Glc | RCA120 | ELLA |
| Linear polymer8 | FRP | P-Gal, P-Lac, P-TriGal | Galectin-3 | Fluorescence |
| Linear polymer62 | FRP | Maltitol, Lactitol | ConA, RCA120 | Immunodiffusion |
| Linear polymer93 | ATRP, PF (Click) | α-Man, β-Gal | ConA | Turbidity |
| Linear polymer51 | FRP | β-D-Gal | PNA | Turbidity/ITC |
| Linear polymer118 | ATRP, PF (Click) | α-D-Man, β-D-Gal | ConA | SPR, ELISA |
| Linear polymer52 | FRP | α-D-Glc, α-D-Gal | RCA120 | — |
| Linear polymer132 | ATRP, PF (Click) | α-Man, β-Gal, β-Lac | ConA, RCA I | Turbidity |
| Linear polymer146 | FRP, PF | D-Glc, D-Gal, D-Man | ConA, RCA120 | Turbidity |
| Linear polymer130 | FRP, PF | β-D-Gal(1 → 4)D-Glc, α-D-Glc(1 → 4)D-Glc | ConA, RCA120 | Turibidty |
| Linear polymer150 | PF | β-Gal(1 → 4)GlcNAc | SNA, HA | HIA |
| Linear polymer133 | CCTP, PF (Click) | Cellobiose, Man, Gal | RCA I, ConA | Turbidity |
| Linear polymer147 | Suzuki P., PF (Click) | Gal | PNA, ConA | Turbidity |
| Linear polymer164 | Suzuki P., PF (Click) | Glc | ConA, PNA | Turbidity |
| Linear polymer55 | FRP | α-D-Gal(1 → 1)α-D-Glc, α-D-Gal(1 → 1)β-D-Glc | BSI–B4, Shiga toxin-1 | Turbidity |
| Liposome157 | Chemical enzymatic | α-L-Fuc | E-selectin | ELISA |
| Core-shell nano spheres57 | FRP | Glc | ConA | ELLA |
| Micelles and other self-assembled structures119,120 | ATRP | β-D-Gal(1 → 4)β-D-Glc | RCA120 | Turbidity |
| nanofibres59 | FRP | α-Glc | ConA | Electrophoresis |
| Micelles and other self-assembled structures89 | ATRP | Glc | ConA | fluorescence |
| Micelles and other self-assembled structures90 | ATRP | D-Glc | ConA | Turbidity |
| Micelle135 | RAFT | Glc | ConA | Turbidity |
| Micelle121 | RAFT | α-D-Man | ConA | Turbidity |
| Micelle117 | NMP | β-Gal | PNA | Turbidity, fluorescence |
| Grafted to silver nanoparticle60 | FRP | D-Glc | ConA | Turbidity |
| Coated onto polystyrene dish54 | FRP | D-Glc, α-D-Glc(1 → 4)D-Glc, β-D-Gal(1 → 4)D-Glc | ConA | ELLA |
| Coated onto octadecyltrimethoxysilane and cationic aminopropyl trimethoxysilane monolayers64 | FRP | VLA, Heparin | RCA120 | FITC |
| Grafted to oligodeoxynucleotides165 | Telomerization | β-D-Gal, α-D-Man | ConA | Electrophoresis |
| Grafted to PET surface via biotin-avidin83 | Cyanoxyl-MP | β-Gal(1 → 4)β-Glc | Psohocarpus tetragonolobus | fluorescence |
| Polymer beads131 | Copper-catalyzed Huisgen [2 + 3] cycloaddn | α-D-Man | ConA | fluorescence |
| UV-induced graft polymerization from polymer membrane47 | FRP | α-D-Glc | ConA | Coomassie blue complex |
| Grafting onto gold surface via thiol functionality 61,115 | FRP | α-Man | ConA | QCM |
| Grafting onto gold surface via thiol functionality 92 | ATRP | Lactoselactone | RCA120 | SPR |
| Grafting onto gold nanoparticles via thiol functionality110 | RAFT | α-Man, β-GlcNAc | ConA, WGA | Turbidity |
| Grafting from gold surfaces91 | ATRP | β-Gal(1 → 4)Glc | RCA120 | SPR |
| Glyco nanoparticles coated to surface via biotin/avidin46 | RAFT | β-Gal(1 → 4)Glc | RCA120, ConA, Jacalin | Turbidity |
| Grafted from honeycomb structured films163 | RAFT | Glc | ConA | Fluorescence |
Binding assays
Carbohydrate-lectin binding assays can be conducted through a wide variety of methods, ranging from the earliest hemagglutination inhibition assay (HIA), evaluated by Landsteiner, to the sophisticated surface plasmon resonance (SPR), which uses materials absorbed onto metal.39,40 The basic principle behind lectin binding assays is the formation of isolated complexes between lectins and their ligands.41 However, care should be taken when comparing measurements among the analytical techniques, as the assays used operate at different concentrations and physical properties.3HIA is one of the earliest methods used for assessing the interactions between viruses/viral antigens and their corresponding ligands. Ligand solutions are initially placed at different concentrations into the microwells, and this is followed by the addition of soluble lectin to allow precipitation of aggregates. Once the precipitation is completed, the minimum concentration of carbohydrate that inhibits the hemagglutinination reaction is reported.3 Enzyme-linked immunosorbent assay (ELISA) was employed before its variant enzyme-linked lectin assay (ELLA) came about.42 More accurate measurements from reading the IC50 values on fitted curve were attained using ELLA. In order to determine physical parameters, carbohydrate-lectin binding constants are normally measured using isothermal titration microcalorimetry (ITC), quantifying the heat generated (enthalpy) from the binding.43 SPR utilizes the flow of lectin solution over a gold surfaced chip with immobilized ligands resulting in a change in the refractive index at the surface. The removal of the bound lectin during the flow of buffer solution provides the binding constant.39
The above paragraph listed the four most commonly used techniques for lectin binding assays, but there is a broader array of analytical techniques to choose from. Turbidimetric assays generated with the aid of UV-vis spectroscopy is now more frequently used as a method in determining the successful binding of glycopolymers with lectins.44 Two-dimensional immunodiffusion tests (double diffusion agar, DDA) can also be used for identifying specific binding between carbohydrates and lectins.45 There are other complementary techniques, such as using a quartz crystal microbalance (QCM), measuring the weight of the attached lectin, and electrophoresis to determine the molecular size of proteins adhered. In addition to the various binding assay used, the solution used for conducting the binding assay has to be carefully selected. In two separate articles, Narain and Xu found that in their system, the use of a certain concentration of Ca2+ and Mn2+ salts with the same anion (Cl−) greatly enhanced the aggregation of the glycopolymer when they interacted with their proteins.46,47
Glycopolymer
Glycopolymer interactions with lectins are greatly influenced by the rigidity of the polymer, the density of sugar molecules, the architecture, the molecular weight and other factors. Therefore, significant efforts have been dedicated to the synthesis of different glycopolymer architectures. This section is dedicated to the preparation of glycopolymers highlighting examples that involve glycopolymer with measured bioactivity. Glycopolymers carrying pendant sugar moieties are either synthesized by carbohydrate-containing monomers or by the post polymerization glycosylation of synthetic polymers.48Glycopolymers from the polymerization of glycomonomers
The polymerization of carbohydrate-containing monomers can be carried out by a range of polymerization techniques. This includes: free-radical polymerization (FRP),45,47,49–64 living anionic polymerization,65 ring-opening polymerization (ROP),66,67 ring-opening metathesis polymerization (ROMP),44,68–74 and controlled/living free-radical polymerization (CLRP) which, encompasses nitroxide mediated controlled free-radical polymerization (NMP),75–83 atom transfer radical polymerization (ATRP),84–93 and reversible addition-fragmentation chain transfer (RAFT) polymerisation.16,46,94–111 Significant efforts have been devoted to the synthesis of glycomonomers since they are usually not commercially available.112 Glycomonomers can be synthesized using established organic chemistry, for example, conducting an esterification of acryloyl chloride with 1,2:3,4-di-O-isopropylidene-α-D-galactopyranose (AIpGP) or performing a glycosylation reaction with 2-hydroxyethyl methacrylate (HEMA) using boron trifluoride diethyl etherate as a catalyst.51,107 The use of oxanorbornene-derived monomers was also widely performed using the esterification of 3,6-oxy-1,2,3,6-tetrahydrophthalic anhydride with sugar alcohols, as developed by Kiessling and co-workers.69,73 Transesterification in the presence of lipases was later used for the synthesis of vinyl saccharides, Miura et al. used six lipases in pyridine to synthesize vinyl esters of carbohydrates with yields reaching 90%.62 More complex methods to synthesize glycomonomers include ether bond formation at temperature as high as 130 °C.81,108In the subsequent subsections, emphasis has been placed on the synthesis of glycopolymers used specifically for lectin binding.
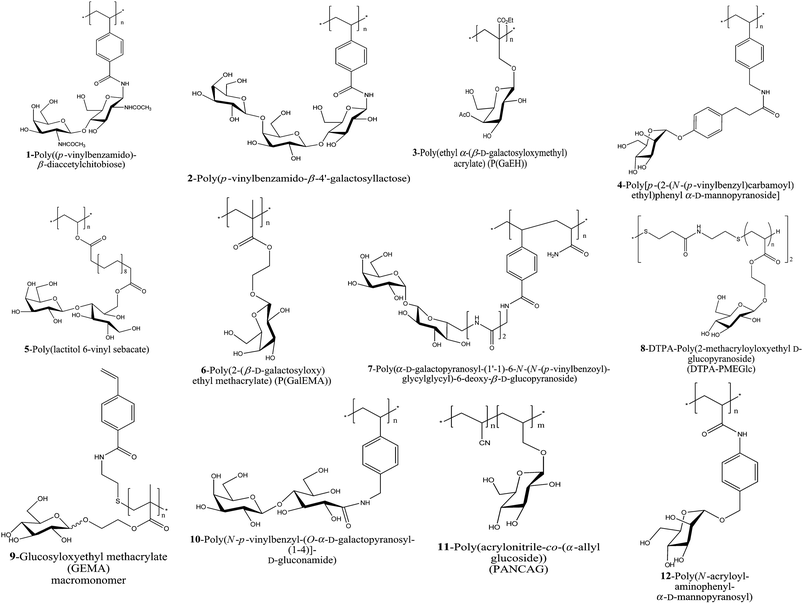 | ||
| Fig. 3 A schematic of the glycopolymers used for lectin conjugation synthesized via FRP. | ||
An examples for polymers made by free radical polymerization is the efficient preparation of poly(ω-(acrylamido)alkyl O-(β-D-galactopyranosyl)-(1→4)-2-acetamido-2-deoxy-β-D-glucopyranoside). The cluster glycopolymers were very soluble in water and had a high sugar density when compared to known n-pentenyl glycosides.53
Nishimura and Nagahori employed a template effect utilizing mannose/ConA and lens culinaris agglutinin (LCA) interactions to tailor glycopolymers. This tailored glycopolymer was aimed to exhibit high affinity and tight binding specificity against guest carbohydrate-binding proteins.56 Monomers bearing di- and tri-vinyl saccharides, 1 and 2 were homo or copolymerized with acrylamide in DMSO using 2,2′-azobisisobutyronitrile (AIBN) as a initiator. They were subsequently used for the binding with WGA and RCA120 respectively, and the inhibition ability was found to have increased by 103 when compared to their oligosaccharide.45,58
By employing protective chemistry, poly(N-p-vinylbenzyl-D-glucuronamide) (PV6Gna) was synthesized and tested regarding its bioactivity with ConA. Interestingly, a strong dependency between the positions where PV6Gma was substituted and its bioactivity was observed. Specific binding to hepatocytes was allowed only when C-6 of glucose was substituted and binding failed at position C-1 and C-3 of the substituted glucose.54 Cuervo-Rodriguez et al. have also employed acetylated protected glycomonomers to generate deacetylated glycopolymer 3 for the binding with RCA120.52
Other polymers of interest are mannose containing structures. Monomers with α-D-mannopyranose 4 with various substitutions at the C-2-position of the glycopolymer were studied concerning their bioactivity with ConA. The fluoride substituted derivative was found to be a highly potent inhibitors resistant to exo-glycosidase digestion.49
Chemoenzymatically modified maltitol and lactitol based glycomonomers were polymerized to give a α-glucose-containing polymer and β-galactose-containing polymer 5 and the polymers showed specific binding towards ConA and RCA120 selectively, moreover, the adhesion with hepatocyte was found to be positive.62
The polymerization of glycomonomer using conventional free-radical polymerization has also been documented by Cameron and co-workers. The investigation of 2-(2′,3′,4′,6′-tetra-O-acetyl-β-D-galactosyloxy)ethyl methacrylate (AcGalEMA) a protected carbohydrate-containing monomer was polymerized in chloroform and deacetylated in dichloromethane–methanol mixture. An alternative route was conducted by first deacetylating AcGalEMA to 2-(β-D-galactosyloxy)ethyl methacrylate (GalEMA) and subsequently polymerizing it in water–methanol mixture. The synthesis of P(GalEMA) 6 was successfully conducted via the latter method and the binding with peanut agglutinin (PNA) was investigated with their thermodynamic binding parameters calculated.50,51
A novel class of 1,1′-linked non-reducing disaccharides 7 with an α-galactoside epitope was developed by Nishida and co-workers. A “module effect” of the second sugar showed an integrated detoxifying activity to an E. coli toxin.55
Attractive from an application point of view is the surface modification with glycopolymers. Chemisorption was proven to be a quick method in attaching glycopolymers on desired substrates. Glycopolymer 8 was adhered to the surface by taking advantage of the disulfide group for the chemisorption onto silver colloids. A “surface-enhanced Raman (SER) effect” was observed as the emergence of a new peak at 647 cm−1 corresponded to a stretching vibration of C–S bond.60
Glycopolymeric nanospheres were synthesized viain situ FRP of styrene with 9 GEMA in an ethanol–water (Ratio 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) mixture with AIBN as initiator. Particle sizes ranging from 300 to 620 nm were produced by altering the monomer ratio. The polystyrene core-glycopolymer corona nanospheres can be employed as a versatile tool for the study of sugar-biomolecule recognition.57
:2) mixture with AIBN as initiator. Particle sizes ranging from 300 to 620 nm were produced by altering the monomer ratio. The polystyrene core-glycopolymer corona nanospheres can be employed as a versatile tool for the study of sugar-biomolecule recognition.57
Miura and co-workers self-assembled 10 onto hydrophobic silicon templates to construct a protein micropatterning substrate used for molecular recognition (Fig. 4).63,64
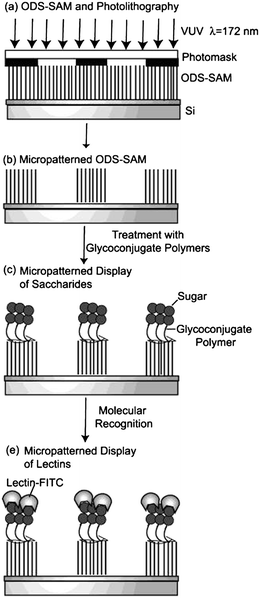 | ||
| Fig. 4 A schematic illustration of a micropatterned display: (a) photolithography on ODS-SAM, (b) micropatterned ODS-SAM, (c) micropatterned display of carbohydrate, and (d) micropatterned display of lectin.63 Reproduced with permission from American Chemical Society (ACS), copyright 2004. | ||
Glycopolymers synthesized by FRP were also successfully processed via electro-spinning. These nano sized fibrous sugar sticks spun by 11 contained glucose pendants that were used for ConA recognitions. The nanofibres can be envisaged as materials used for protein isolation and other lectin related applications.59
Crosslinked mannose-conjugated polymer 12 was immobilized onto the surfaces of gold as thin glycopolymer layer. Additional selectivity could be achieved by controlling the amount of crosslinking reagent used.114
A UV-induced graft polymerization of allyl glucoside was also employed using FRP. The polymerization of allyl glucoside resulted in the deposition of glucopyranoside onto polypropylene microporous membrane. The sugar density on the surface can be adjusted by changing the monomer concentration and the UV irradiation time.47
Alkanethiol containing glycopolymers were bound onto gold surfaces of a quartz crystal microbalance (QCM) and ConA was loaded onto the modified gold surfaces. Frequency changes corresponding to the mass of ConA attached to the surfaces were calculated according to the Sauerbrey equation,115 an equation typically used for converting frequency to mass, which was developed by G. Sauerbrey in 1959.
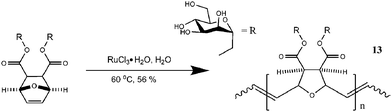 | ||
| Fig. 5 Mannose based glycopolymers generated by aqueous ruthenium-catalyzed ROMP.73 | ||
Mannose and glucose based glycopolymers 13 synthesized by ROMP were tested for their bioactivity with ConA. Detailed studies by Kiessling's group have shown that the glycopolymers synthesized via ROMP acted as cell and lectins agglutination inhibitors.44,69,73,74,83 More work has been carried out by Manning and Sanders from the group of Kiessling on sulfated saccharides (Fig. 6). Using a similar strategy, ROMP was used to synthesize sulfated glycopolymers 14 and 15 showing efficient binding towards L- or P-selectin.68,72
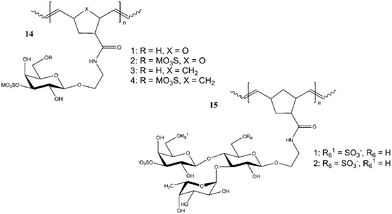 | ||
| Fig. 6 Sulfated glycopolymers synthesized using ROMP.68,72 | ||
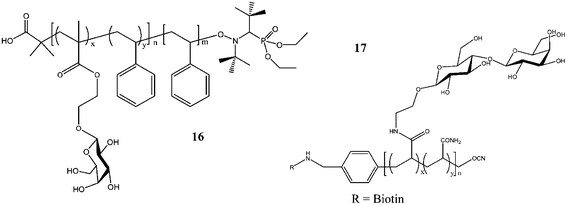 | ||
| Fig. 7 Glycopolymers synthesized via nitroxide-mediated controlled radical polymerization 16 and cyanoxyl-mediated polymerization 17.83,117 | ||
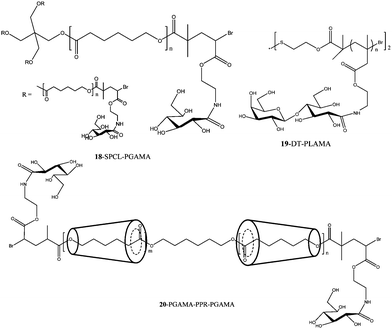 | ||
| Fig. 8 Star-shaped and Au–S bonded surface modified glycopolymers synthesized by ATRP.89–92 | ||
Extensive work was carried out by Haddleton and co-workers on the combination of click chemistry and ATRP to prepare glycopolymers.118 Co-clicking of mannose and galactose based azides allowed the alteration of the epitope density, which was shown to influence the interaction with rat mannose-binding lectin (MBL).
A disaccharide glycomonomer, 2-lactobionamidoethyl methacrylate (LAMA) 19 (Fig. 8) was synthesized using a disulfide-carrying ATRP initiator. The glycopolymer with its disulfide end-functionality was conjugated to a gold surface forming a polymer bush. The association and dissociation of PLAMA with RCA120 was investigated. Control studies with 2-lactobionamidoethyl disulfide (Cys-Lac) reveal that the multivalent effect displayed by the glycopolymer gave a reversible association whereas the cover glass coated with monovalent galactose did not possess this characteristic.92 LAMA glycomonomer was again used by Mateescu et al. for surface modifications onto gold substrates. The binding interactions were also proven to be highly efficient due to the glycopolymer's multivalent effect when interacting with RCA120. Control experiments with ConA did not result in the increase of the resonance angle from the surface plasmon spectroscopy analysis, which indicated a nonspecific adsorption of lectin.91
Glycopolymers with pendant disaccharide sugar moieties with poly(L-glutamate) blocks on both ends of the triblock copolymers were synthesized by ATRP followed by the ring-opening polymerization of β-benzyl-L-glutamate N-carboxyanhydride. The water soluble triblock copolymers self-assembled into lactose-containing polymeric aggregates, which interacted well with RCA120 lectins.119,120
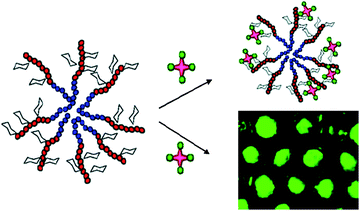
Random copolymers from p-acrylamidophenyl α-mannoside, p-acrylamidophenyl N-acetyl-β-glucosamine and acrylamide were obtained using (thiobenzoyl)thioglycolic acid as the RAFT agent. After the treatment with NaBH4, the thiol endfunctional glycopolymers were reacted with gold nanoparticles to form Au–S bonds to yield 21 (Fig. 9). Bimolecular recognition with ConA was conducted, revealing a multivalent effect caused by the pendant mannose and glucose moieties on the gold particles.110
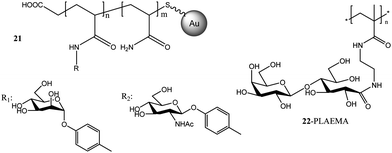 | ||
| Fig. 9 Glycopolymers conjugated with gold nanoparticles via RAFT polymerization.46,110 | ||
After investigation the RAFT polymerization of LAMA in detail, Narain and co-workers extended their work to a methacrylamide based 2-lactobioamidoethyl methacrylamide (LAEMA). Polymerizations resulted in well-defined glycopolymers 22 with disaccharides pendants (Fig. 9). The binding affinity with three different lectins, namely, RCA120, ConA and Jacalin (Artocarpus Integrifolia) were investigated.46
Our group has recently reported a new class of glycomonomer, synthesized by Cu(I) catalyzed click chemistry to from a stable 4-vinyl-1,2,3-triazole ring. This monomer was homopolymerized and chained extension with N-isopropyl acrylamide (NIPAAm) to give a thermo-responsive block copolymer. Subsequent protein binding with ConA resulted in efficient conjugation. The conjugation between the mannose containing polymer and ConA was dissolved by using 1-methyl-D-mannopyranoside, a competitive binding ligand.121
Post-functionalization of pre-formed polymers using sugar moieties (PF)
Although polymerization of sugar-containing monomers has been proven successful, modification of preformed polymers using saccharide-containing reagents offers an excellent alternative synthetic route. The post-functionalization approach is convenient to produce libraries of glycopolymers with the same macromolecular architecture by attaching different sugar moieties to pre-formed polymer scaffolds. It also generally provides a simpler procedure as some sugar-containing monomers have the tendency to self-polymerise during the purification procedures. Gauthier et al. recently published a review about the synthesis of functional polymers via post-polymerization approach.122Many attempts to modify preformed polymers with sugar moieties have been focused on amino saccharides resulting in amide linkages between the polymer backbone and sugar. Due to the good nucleophilicity of amines compared to other functional groups such as alcohols, selectivity is provided without the need for protecting groups. This is convenient for sugar related modifications due to the large amount of hydroxy groups in sugar molecules. Polymers with pending active carbonyl coumpunds such as carboxylic acid, N-hydroxysuccinimide (NHS) ester and anhydrides have been used to react with aminosaccharides (Fig. 10). Examples for amino sugars include N-acetyllactosamine (Fig. 10a)45 or 2-deoxy-D-glucose (Fig. 10b).123 The high reactivity of poly(acryloyl chloride) was used to prepare hydrophilic polyacrylamide compounds having glucose (PAAm-glucose) and galactose (PAAm-galactose) (Fig. 10c) as pendent groups in order to test that attachment of L929 and CHO-K1 cells.124
 | ||
| Fig. 10 Synthesis of glycopolymers via amide linkages. | ||
Strong and Kiessling synthesized a N-hydroxysuccinimide (NHS)-functionalized polymer via ring-opening metathesis polymerization. Subsequent treatment with a mannose derivative (Fig. 10d) resulted in polymers with high activity for ConA as tested using hemagglutination assays.125 The coupling reaction between NHS and amine groups was also employed by Baek and Roy. Poly(N-acryloxysuccinimide), synthesized via free radical polymerization, was reacted with the aminated carbohydrate ligand 3-(2-aminoethylthio)propyl β-D-Gal-(1 → 3)-α-D-GalNAc (Fig. 10e). Furthermore, a random glycopolymer bearing T-antigen was synthesized in a similar manner with both polymers demonstrating high activity with the peanut lectin from Arachis hypogaea.126
Auzely-Velty et al. prepared a copolymer of N-vinylpyrrolidone and maleic anhydride via radical polymerization method. Subsequent reaction with N-(4-aminobutyl)-O-β-D-galactopyranosyl-(1→4)-D-gluconamide (Fig. 10f) resulted in glycopolymers with good inhibitory properties against model RCA120 lectin.127
An attractive polymer for functionalization with carbohydrates is poly(vinyl alcohol) (PVA), albeit the low reactivity of the hydroxyl functionality requires the recourse to prior activation. Sachez-Chaves and coworkers synthesized partially functionalized polymers with monosuccinate groups by the reaction of PVAL with succinic anhydride. The polymers were then modified with 2-amino-2-deoxy-D-glucose to afford the desired glycopolymer, which selectively recognize ConA.128
A different way of activating PVA was reported by Arranz et al. who used 4-nitrophenyl chloroformate to convert PVA into a reactive ester for further reaction with 2-amino-2-deoxy-D-glucose (Fig. 10g).129 Similarly, Cerrada et al. used the commercially available random ethylene-vinyl alcohol copolymer (EVOH) for further modification with p-nitrophenyl chloroformate followed by reaction with different aminosaccharides. The glycopolymers showed specific interaction with ConA and RCA. 130
Alternate routes include click reactions, which are highly efficient while robust against most functionalities. Haddleton and co-workers investigated the construction of glycopolymers from alkyne backbone-functional polymers via Cu-catalyzed azide–alkyne click (CuAAC) chemistry in detail.93,118,131–133 Well-defined polymer backbones with alkyne functionalities were first synthesized via living radical polymerization or catalytic chain transfer polymerization followed by the subsequent reactions with different sugar azides (mannose, galactose, lactose) (Fig. 11) These polymers were tested regarding their activity towards a range of lectins.
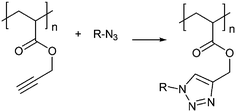 | ||
| Fig. 11 Synthesis of glycopolymers via Cu-catalyzed azide–alkyne click chemistry. | ||
Another reaction, the so-called thiol–ene coupling or thiol–ene click reaction, has been recently employed by researchers for the synthesis of functional polymers, either via a radical or base/nucleophilic approach.134 The thiol-ene click reaction is highly efficient and orthogonal to a wide range of functional groups, and is compatible with water and oxygen. In addition, the coupling reaction is simple and metal free.
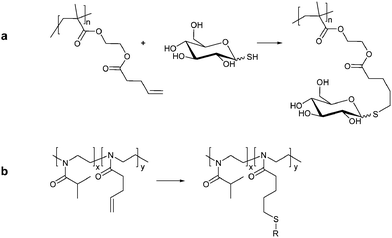
The radical thiol-ene click has been used for the synthesis of glycopolymers. A block copolymer based on poly(ethylene glycol)methacrylate and HEMA synthesized via RAFT polymerization were used by Chen et al. as a backbone for reaction with 4-pentenoic anhydride resulting in polymers bearing alkene side chains. Grafting of glucothiose onto the alkene functional scaffolds via a thiol–ene click was complete in less than 2 h. The resulting micelles showed high bioactivity with ConA (Fig. 12a).135 A similar approach was employed by Diehl and Schlaad using a series of poly[2-(isopropyl/3-butenyl)-2-oxazolines]. The copolymer was then modified by photoaddition of a thioglucose derivate, 2,3,4,6-tetra-O-acetyl-1-thio-β-D-glucopyranose (Fig. 12b).136
A range of other methods have been investigated to functionalize polymers via a post-polymerisation modification reaction with carbohydrates. Examples include the chemical modification of poly(vinyl alcohol) (PVA) by glycosidation with triacetylated sugar oxazoline,137 the reaction between poly[(9,9-bis-(6′-bromohexyl)-2,7-fluorenylene)-alt-1,4-phenylene] and thio-sugar,138 the use of carbohydrates bearing aldehyde groups which react efficiently with polymers with pendant amino residues139 and the functionalization of poly(ethylene terephthalate) (PET) fibres with glycosyl azides.140 None of these polymers were however tested for their bioactivity.
Interaction of lectins with different polymer architectures
Linear glycopolymer
Linear synthetic glycopolymers were the first and are the most widely tested glycopolymers regarding their ability to bind to lectins.141 While usually present as isolated macromolecules, high concentration can lead to clustering of polymers due to hydrogen bonding between hydroxyl groups from the sugar moieties or hydrophobic interactions from the polymer backbone.142The interactions between ConA and maltose/maltotriose-containing polymers monitored via the turbidity assay using a UV-vis spectrophotometer were used by Kobayashi et al. The accessibility of ConA's deep binding cavity was enhanced by the terminal glucose residue of the pendant trisaccharide chain. Non-specific binding from hydrophobic forces was also observed in one of the oligosaccharide synthesized.143 In subsequent studies, a double diffusion agar (DDA) was carried out using PNA and WGA for the identification of specific binding of a galactose-based polymer with PNA and a glucose-based glycopolymer with WGA. Further quantifications of their binding concentrations were measured using inhibition of the hemagglutination assay (HIA).45
A detailed investigations into RCA120, PNA, ECA (Erythrina cristagalli from seeds of coral tree), WGA and DSA (from Jimson weed) lectins were performed with several different glycopolymers bearing disaccharides groups with the aid of HIA and DDA assays.58
Rigid helical poly(glycosyl phenyl isocyanide)s were compared with flexible phenylacrylamide glycopolymers. The compatibility of orientation and spacing of clustering saccharide chains were found to be essential for specific molecular multivalent recognition by lectin.144
Linear glycopolymers were widely synthesized by Kiessling and co-workers via ROMP. Glucose-based glycopolymers polymerized from 7-oxanorbornene derivative were used for binding with ConA. Multivalent effects by the glycopolymer were documented via agglutination inhibition assay.69,74 Decreasing the saccharide density along the aliphatic backbone has been observed to increase the activity towards ConA due to the accessibility of ligand along the polymeric backbone (Fig. 13).73 The multivalent interactions between glycopolymers and ConA were eventually maximised by adjusting the binding epitope density of the polymeric carbohydrate ligands; turbidimetric assays and quantitative precipitation (QP) were conducted to investigate the interactions.44
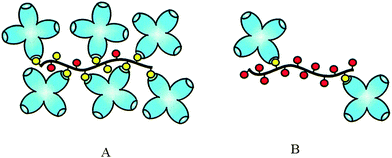 | ||
| Fig. 13 A schematic representation of ConA clustering by multivalent ligands. (A) High-density polymers can recruit many receptors to a single molecule; however, steric effects prevent binding of every residue. (B) Low-density polymers bind fewer total receptors per molecule. Increasing spacing between residues allows for more efficient binding.44 Reproduced with permission from ACS, copyright 2002. | ||
Mosaic lectins such as P-selectins and L-selectins were inhibited by sulfated neoglycopolymers analyzed using ELISA, static binding assay and cell rolling inhibition assay.68
Binding between glycopolymers and their lectins could normally be easily observed using turbidimetric assay or DDA, however, the determination of binding constants need to be quantified by other methods. Fluorescence spectroscopy was used to determine the binding constants when galactose/glucose disaccharides glycopolymers were interacted with Erythrina corallodendron (ECorL). The association constant was calculated using the Steck-Wallack equation.53
 | (1) |
Nagahori et al., aimed at controlling the carbohydrate-protein interaction by using the template effect of imprinted polymers. By controlling the size and the shape of the lectin-binding cavities using synthetic glycopolymer networks, high affinity and tight binding could be achieved.56
Linear glycopolymer of pendant sugar moieties varying in the ratio of α-mannoside and β-galactoside were synthesized via a combination of controlled/living radical polymerization (transition-metal-mediated, TMM-LRP also termed as ATRP) and a “co-clicking” chemistry.93,118,132 Haddleton and co-workers clicked azido-sugar (galactose/mannose) derivatives onto the polymer backbone bearing alkyne functional groups. QP experiments were carried out and found that the average number of ConA bound to the polymers increased with the mannose content in the polymers reaching a maximum number of bound ConA remain constant after a mannose content of 70%.93
Mammalian lectins are endocytic lectins and they are in the family of C-type lectin. They were used for the interactions with mannose/galactose-based glycopolymer prepared by the same TMM-LRP and “co-clicking” method. Successful multivalent binding were analyzed using ELISA and SPR binding studies.118
Geng et al. studied the rate of clustering between N-glycosyl 1,2,3-triazole glycopolymer and ConA. It was found that the rate of clustering was comparable with that of an analogous ligand prepared from the same polyalkyne precursor.132 It is also not surprising that glycopolymers synthesized via catalytic chain transfer polymerization (CCTP), Huisgens cycloaddition and thiol-ene double click reactions interacted well with ConA and RCA 1 with the binding being analyzed with turbidimetric assay and affinity chromatography (high performance liquid chromatography, HPLC), respectively. The galactose containing polymer was retained on a column with immobilized RCA I. By adding an increasing amount of free galactose to the mobile phase, more and more glycopolymers were eluted from the column confirming the specificity of the interactions between the glycopolymers and the lectin.133
Using an activated ester and aminosugars to generate glycopolymers in a postfunctionalization approach, Davis and Boyer also showed that glucosamine-based glycopolymers were successful in binding ConA, while galactosamine-based glycopolymers did not bind to ConA.145
Glycopolymers bearing β-D-glucopyranoside and β-D-galactopyranoside were synthesized and interacted with ConA and RCA120, respectively. Results show that the glucose-based polymer was not specific towards ConA due to the β-anomer of glucose and the β-galactose-based polymer interacted well with RCA120.52 Using a turbidimetric assay, reduction in specific interactions with ConA were observed with a mannose-based glycopolymer modified at the second position of the sugar ring.146
Hepatic lectin in the form of human hepatocarcinoma cells were selected to study the interaction with N-(2-hydroxypropyl)methacrylamide (HPMA) polymers with galactose. By employing flow cytometry and confocal microscopy, the authors found that glycopolymers with multiple sugar moieties such as tri-saccharides allow much better binding when compared to glycopolymer carrying monosaccharides.8,9 This result was also observed by Lee et al.13
Chen et al. employed Suzuki coupling polymerization of peracetylated galactopyranosyl-carrying monomer and deacetylation under Zemplén conditions. The reaction of the polymer with PNA resulted in significant fluorescence quenching with a Stern–Volmer quenching constant of 1.56 × 105 M−1.147
Glycopolythiophenes synthesized from the oxidative copolymerization of methyl (3-thienyl)acetate and thiophene-carbohydrate monomers were used for detecting influenza virus and E. coli.. Bioassays (Fig. 14) were performed to examine the biochromic capability of the glycopolymers. Upon binding with virus and bacterium, a lengthening of the effective conjugated length was observed as evidenced by the unusual red-shift in the visible absorption of the polymer backbone.148
 | ||
| Fig. 14 A schematic representation of the double sandwiched biochromic sensory device based on mannose-PT.148 Reproduced with permission from ACS, copyright 2000. | ||
The presence of type 1 fimbrae in E. coli, which contains proteins such as fimH, a lectin used for binding with mannose and glucose residue, has enticed scientist to study the interaction of E. coli with glycopolymers. Alexander and co-workers designed thermoresponsive glycopolymers which are capable of reversible aggregation with a specific bacterial strain. The glycopolymers represented a robust and reusable cell-sensing material. Green fluorescent protein (GFP) E. coli was used to study the interactions with thermoresponsive glycopolymer and results indicated that above the lower critical solution temperature (LCST), the glucose units were hidden within the poly(N-isopropyl acrylamide) (PNIPAAm) aggregates as there are no observable green fluorescent cluster by the E. coli cells. On the other hand, when the solution temperature is below the LCST, distinct green fluorescent cluster from the GFP E. coli were observed indicating interactions between the glycopolymers and E. coli.. Glycopolymers with longer spacers between glucose units and the polymer backbone were suggested to bind more efficiently when compared to short spacer units.149
Bark lectin from elderberry (SNA) was tested using HIA concerning their binding with spacer-N-linked glycopolymers. Findings showed that the spacer-N-linked glycopolymers bind better with avian and human virus HA than the spacer-O-linked glycopolymers. Furthermore, the synthetic pathway for synthesizing the former glycopolypetides proved feasible when produced in an industrial scale. Low immunogenicity was also discussed as some acrylamide monomers can be cytotoxic when used in vivo.150,151
Intensive studies into the modification of the C-2 position of β-D-mannopyranoside residue and the consequences regarding the interactions with ConA were conducted by Akai et al. 3-, 4- and 6-hydroxyl groups of D-mannopyranoside are essential binding sites for ConA. In addition, β-D-mannopyranoside interaction with ConA is four times weaker than the anomeric counterpart, α-D-mannopyranoside; due to the steric hindrance of the glycosyl bond in the binding site.152 The inhibition potency of C-2 fluoro substituted β-D-mannopyranoside-based glycopolymers was found to be similar with the α-D-mannopyranoside-based glycopolymers showing that by modifying the C-2 position the binding affinity with ConA could potentially be altered. It was suggested that the fluorine atom at C-2 forms a hydrogen bond with an amino acid at the entrance of the ConA binding site, resulting in enhanced binding affinity.49
Detailed studies using double-radial immunodiffusion assays were performed on random copolymers of N-acryloylated monomers and acrylamide or alternatively on glycopolymers obtained by grafting sugar of an end-group-aminated T-antigen (Galβ(1 → 3)-GalNAcα together with biocytin. Glycopolymer bearing different spacer arms were tested regarding their binding ability with PNA. Low affinity was observed with glycopolymer bearing shorter spacer arms from their polymer backbone due to lack of accessibility to PNA binding sites. This copolymers could ideally be used as an antibody-carbohydrate recognition antigen.126
Auzely-Velty et al. carried out studies with a focus on the influence of chemical modification of sugar in order to attach it to the backbone onto the conjugation with RCA120. With the aid of ELLA, it was found that the loss of glucosyl residue as a result of the chemical derivation of lactose for amidation reaction onto polymer backbone resulted in no significant multivalent effect compared to galactose alone. Nonetheless, binding was still achieved with similar binding affinity compared to the reference galactose monovalent ligand.127 In contrast, very promising results were reported when studying the interactions of P(GalEMA), a galactose-based glycopolymer, with PNA. A 50-fold affinity enhancement when compared to the monovalent saccharide D-galactose was measured. ITC was used to determine the thermodynamics of binding, in addition, UV-difference spectrum were also analysed indicating strong interactions between P(GalEMA) and PNA. Monovalent lectin such as galectin-3 was suggested for further examinations on the precipitation process for binding with P(GalEMA).51
Miyachi et al. made the discovery that by carefully designing the multivalent galacto-trehaloses (GT), GT glycopolymers could bind much stronger to BSI–B4 lectin (Bandeiraea implicifolia) and Shiga toxin-1. In addition, a detoxifying activity towards the E. coli toxin as the result of a “module effect” from the second sugar was also observed.55
What influences binding of linear glycopolymers?
It is evident from these results that glycopolymers are superior to single ligands when it comes to binding as long as the functionalization of the sugar does not interfere with the process. Unfortunately, the multivalency effect has not yet been fully understood. Polyvalent binding, which is covered by enthalpy and different entropy parameter, goes hand in hand with steric stabilization of the complex, which is the formation of a protective polymer layer around the target. The array of techniques employed in literature to test the binding between protein and glycopolymer as well as the different experimental parameters chosen hinder the direct comparison of the glycopolymers prepared. However, it seems that there are certain trends crystallizing when analyzing the published work.The observations in literatures confirm mostly what Whitesides and co-workers have elaborated on in a review article.4 A good match between two ligands and two binding sides are one of the determining parameters to achieve good binding. This means that optimum binding can be achieved if the distance between two sugar units is equivalent to the distance between two binding sides. This excludes very stiff polymers as the preferred choice for efficient binding. If a polymer is very stiff it is unlikely that the distance between two sugar units fits exactly to the distance between two binding sites unless the geometry of both, receptor and ligand, is an exact match. Indeed, a rigid helical poly(glycosyl phenyl isocyanate) was observed to have very little specific interactions with lectins while the equivalent polymer with a flexible phenylacrylamide showed good binding.144 However, this is not always a rule of thumb as demonstrated by the work of Kiick and co-workers who showed that a helical backbone can be superior to coiled structures.153
The importance of matching the geometry between ligand and receptor was also demonstrated in a range of studies where the amount of sugar molecules, the epitope density, was varied along the backbone. Comparing all these studies, it becomes clear that a high sugar density is not required. A significant amount of sugar molecular is simply not involved in the binding process, therefore have been “wasted”. A high epitope density nonetheless comes in useful when fast binding is required. The amount of mannose on a polymer backbone was found to be almost linearly correlated to the rate in which the polymer binds to ConA.44,154
The influence of the epitope density can be best understood when looking at experiments that calculate the amount of lectins attached per polymer chain. With increasing carbohydrate concentration on the backbone, the amount of lectin conjugated increases in an almost linear fashion until a point has been reached where the glycopolymer becomes too crowded with lectin and no further lectin can be added. Increasing the sugar concentration on the polymer further only leads to excess ligand with no purpose.44,93 The advantage of using copolymers instead of a glyco homopolymer has been demonstrated for the interaction between N-acetyl lactosamine containing polymers and ECorL,53 for the reaction between mannose containing polymers and ConA73,154 and for different galactose containing polymers and PNA or ECA.58 Moreover, with decreasing mol% of galactose in a polymer (from 52 down to 12 Mol%) the activity of the cholera toxin was more and more reduced.155 These results should not create the impression that the lower the amount of sugar the better. Tests are usually carried out with similar ligand concentrations, therefore the amount of actual polymer needs to increase with decreasing epitope density. If the polymer concentration is kept constant, but the amount of sugar on the polymer increases, an increase of activity is well observed. This was demonstrated in the case where a constant concentration of PHPMA with varying amounts of attached galactose were incubated with HepG2 cells, which carry ASGPR, the hepatic lectin, on the surface. With increasing amounts of galactose on the polymer, a significant increase of cellular uptake was recorded.9
Another effect that seems to promote binding is the molecular weight. Kiessling and co-worker found an increasing capability of the polymer to inhibit erythrocyte agglutination when the molecular weight increases.125 This initially linear relation though levels off at a certain value with no more changes being observed at higher molecular weights.156 Another distinct influence was noticed when introducing a spacer between sugar and polymer backbone. An increase in flexibility of the ligand by introduction a spacer enhances the binding of lectin considerably.53,155 The increased flexibility allows better adjustment to the lectin geometries albeit there does not seem to be a clear correlation between length or stiffness of spacer and the activity.148
While linear glycopolymers show very good bioactivity and they display the multivalent effect efficiently, their use for applications such as drug delivery or biosensing might be limited. More complex polymeric structures such as micelle and particles are sometimes necessary to be able to encapsulate drugs or to prepare nanomaterials for applications such as sensing. One way to achieve a more functional material when used in the biological system is by making use of self assembling of linear glycopolymers. Nonetheless, linear glycopolymers are still indispensable as their role in providing fundamental studies into lectin interactions would enhance the design of supramolecular structures. In the next section, the self-assembly of linear polymers are discussed and the importance of their bio-applications emphasized.
Nano-objects: from micelles to fibre
Amphiphilic diblock copolymers composed from a hydrophilic glycopolymer block and a hydrophobic block can self-assemble into various architectures such as micelles, vesicles/liposomes, α-helix or worm-like aggregates.89,90,117,119,120,135,157,158 Furthermore, in situ assembly during polymerizations could also result in nano-fibres and nano-spheres.57,59 These complex architectures derived from glycopolymers may display much higher affinity towards binding lectins, since spherical and three dimensioned structures gave greater surface area for lectins to access their binding ligands. Analytical techniques need to be carefully considered when monitoring the interaction between the synthetic ligands and lectins, as glycopolymers are no longer in their linear configuration but presented in the form of aggregated structures.The triblock copolymer poly(L-glutamate)-poly(2-acryloyloxyethyllactoside)-poly(L-glutamate) (PLG-PAEL-PLG), where the PLG blocks took on a α-helical structure, was able to self-assembled into lactose-installed polymeric aggregates with a high specificity to RCA120.120 Dong and Chaikof then extended this study and altered the initial copolymer concentration, which leads to different morphology changes from sphere to lamellae, then to worm-like micelle. Investigations into the interactions with RCA120via turbidity lead to the conclusion that the lactose moieties were positioned at the surfaces of these aggregates119
Poly(ε-caprolactone)-block-poly(gluconamidoethyl methacrylate) star shaped block copolymers self-assembled into micelles and vesicles were analyzed using UV-vis spectroscopy and dynamic light scattering (DLS) to prove their binding with ConA. The use of DLS was feasible as the glycopolymer were in the form of nano-sized spherical shape. Materials based on poly(ε-caprolactone) can degrade in the biological system, which makes these glycopolymers very appealing for application in the biological system.89,90
Thermo-responsive glycopolymer micelles were synthesized by Stenzel and co-workers by combining a glycopolymer block – obtained via thiol-ene click reaction – with PDEGMA, a polymer with a LCST of 29 °C in aqueous solution. The micelle formation and the bioactivity was tested with ConA via turbidimetric assay analyzed using UV-vis spectroscopy and DLS. Specific interactions were enhanced when the glycopolymers were aggregated into micelles above the lower critical solution temperature (LCST).135 Another class of thermo-responsive glycopolymer micelles based on poly(2′-(4-vinyl-[1,2,3]-triazol-1-yl)ethyl-O-α-D-mannopyranoside)-block-poly(N-isopropyl acrylamide) copolymer showed very similar results.121
Functional galactose-based glycopolymers were used for micellization and honeycomb structured porous films construction. These biomaterials were tested concerning their bioactivity with PNA. Control experiments using turbidimetric assay (for micelles) and confocal fluorescent microscopy (for honeycomb films) with ConA as binding lectin did not show specific binding with galactose moieties (Fig. 15).117
 | ||
| Fig. 15 A schematic representation of PNA lectins binding with galactosylated micelles and porous films.117 Reproduced with permission from ACS, copyright 2009. | ||
An interesting behaviour using micelles made from poly(ethylene oxide)-block-poly(2-glucosyl-oxyethyl acrylate) were reported. The turbity assay did not reveal any significant binding, but fluorescence measurements confirm the occurring interaction with lectins. The absence of turbity was explained by the stabilizing property of PEO preventing the precipitation of aggregates.142
Double hydrophilic block copolymer vesicles were found to have good bacteria interaction with E. coli. Type 1 fimbriae of E. coli containing fimH protein shows great affinity with glucose moieties from the poly(2-glucosyloxyethyl methacrylate) containing vesicle. By employing fluorescent microscopy, the aggregation of fluorescent E. coli glued together by the vesicles was evident.158
Another method of obtaining macromolecular structures from glycopolymers is by the in situ self assembly polymerization of sugar-containing monomer. Styrene was copolymerized with a macromonomer bearing terminal glucose functionalities. ELLA was used to determine the percentage inhibition of ConA binding to poly(N-p-vinylbenzyl-D-maltonamide) (PVMA) by the inhibitors using the following equation:57
 | (2) |
Nanofibrous sugar sticks electrospun from glycopolymers were produced by Xu and Smith. Using electrophoresis, comparison studies with the sugars on the nanofibres bearing pyranose ring configuration and sugars with linear configuration (pyranose formation prevented due to type of sugar modification) showed that only sugar with pyranose ring structure displayed specific binding towards ConA.59,159
What influences the binding with lectins?
Similar to linear molecules, a high density of sugar molecules is not required to achieve optimum binding as demonstrated on nanospheres with different sugar concentrations on the surface.57 Self-assembled structures introduce an additional parameter to lectin binding, the conformational change of the polymer chains below and above the critical micelle concentration. A more rigid chain conformation displayed by the more brush-like conformation of micelle has been proposed as potential factor that enhances binding when compare to their corresponding linear polymers. Turbidity assay was conducted to monitor the interaction rate between the poly(2′-(4-vinyl-[1,2,3]-triazol-1-yl)ethyl-O-α-D-mannopyranoside)-block-poly(N-isopropyl acrylamide) and ConA. The micelle displayed faster binding rate when compared with the equivalent unassembled block copolymer, the unimer.121 In addition, similar result were also observed when crosslinked glyco-particles synthesized via ab intio emulsion polymerization were used for binding with ConA.160 However, care should be exercised when interpreting result obtained via turbidimetric assay since the stabilizing effect of one block can potenatially prevent the formation of a cloudy solution.142Modified glyco-surfaces
Modifying the surface of materials with carbohydrates moieties is a widely studied approach to combine the properties of the underlying material with the bioactivity of the glycopolymer layer on the surface. Glycopolymers had been grafted onto noble metals such as gold and silver using chemisorptions,60 disulfide chemistry92 or crosslinking reaction.61 Alternatively, organic surfaces namely, polystyrene and Wang resin, were modified with carbohydrates54,131 Micro-patterning of proteins using grafted glycopolymers as ligands for recognizing specific proteins are widely investigated,63,64 in addition, glycopolymers were also grafted onto solid substrates such as polyethylene terephthalate (PET) membrane or polypropylene microporous membrane (PPMM) to be recognized by appropriate lectins.47,83 In the following sections, graft glycopolymers are discussed with respect to the different classes of materials used for grafting.Noble metal
The complexation of poly(2-methacryloyloxyethyl D-glucopyranoside) (PMEGlc)-silver (Ag) colloids with ConA were estimated by the Scatchard plot.60 A tool used for calculating affinity constants of a ligand with a protein.161 Conventional purification techniques by using the centrifuge to separate un-conjugated ConA with the sugar coated Ag colloids were performed by Kitano and co-workers.60 The concentration of ConA in the supernatant was determined from the absorbance at 280 nm by using UV-vis spectroscopy.Lectin binding on gold surface of QCM as potential detection tool was investigated using an alkanethiol containing glycopolymers. The use of Langmuir adsorption model, binding constant KA between ConA and mannose was estimated to be (1.4 ± 0.6) × 106 M−1. However, this value only provides an approximate values, as binding rate between the four binding site of ConA are different. It then depends on the flexibility of the glycoligand if the second and subsequent binding sites of ConA can be accessed.115
High affinity binding with lectin was also achieved using gold surface grafted crosslinked mannose-conjugated polymers. QCM, atomic force microscopy (AFM) and SPR were used to quantify the binding constant.61
Binding affinity between the different sizes of surface modified gold nanoparticles and lectins was investigated by Toyoshima and Miura. Studies found that by mixing gold particles of two size distributions, 40 and 100 nm amplifies binding between ConA and the ligands on the particles' surfaces. Further investigations into the interactions between the ligands with fimH protein in E. coli were visualized using TEM. Results revealed that binding only occurs when an E. coli with mannose binding ability was used. Coloration from the surface modified gold nanoparticles in aqueous medium changes from pink to purple indicating the formation of complexes.110
The association constant was again calculated using the Steck-Wallack equation (eqn 1) when SPR experiments were carried out with D-gluconamidoethyl methacrylate (GAMA) and 2-lactobionamidoethyl methacrylate (LAMA) coated gold sensor chips. The association constant gave a similar binding constant to that reported in the literature suggesting high accessibility of the sugar moieties on the sensor chip.91
Heterobifunctional crosslinker with high efficiency in biomedical applications were developed by Deng et al. Biotin and galactose coated gold nanoparticles were tested towards their bioactivity with their respective combining receptors, avidin and RCA120, respectively.46
Solid organic substrates
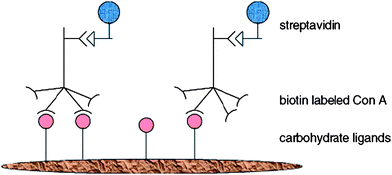
Glycopolymer-coated surfaces on PET membranes were created by using the interactions of biotin and streptavidin to anchor glycopolymers onto the membrane's surface. Fluorescein isocyanate (FITC)-Psophocarpus tetragonolobus, a lectin from winged bean was used for the interactions with galactose moieties on the membrane's surfaces. Fluorescent images of the lectin bound membranes reveal specific binding in the regions of glycopolymer immobilization.83Microfabrication by photolithography has also allowed lectins to be patterned onto silicone substrates. Strong hydrophobic forces were the key for attaching glycopolymers onto the polymer backbones of polystyrene and poly(vinyl alcohol). From the results obtained from SPR, tight and specific binding between the glycopolymers and corresponding lectins were evident. With the aid of the Scatchard plot, association constants were obtained. Due the highly hydrophobic nature of polystyrene, glycopolymers result in better glycocluster when compared to the glycocluster formed from poly(vinyl alcohol), hence resulting in higher association constants. X-Ray photoelectron spectroscopy (XPS) analysis together with fluorescent images allowed the detailed location of regions where ConA and RCA120 combination with glycopolymer occurred.63
When the grafting density of α-D-allyl glucoside grafted onto polypropylene porous membrane exceeded a critical value of 90 μg cm−2, glycoside cluster effect occurred and gave high absorbance of ConA on to the films. A protein-Coomassie brilliant blue complex was used to assist the measurement taken from the spectroscopic analytical method according to Bradford's method. Mn+2 and Ca+2 were ions found to be essential for the saccharide-binding activity of ConA, in addition, NaCl in buffer medium stabilizes ConA in solution.46,47
In order to marry carbohydrate-lectin interactions with biology, four-tiered hierarchical structures composed of lipids, carbohydrates, proteins and cells via orthogonal self-assembly strategy were designed for the interaction with RCA120 and basic fibroblasts growth factors (bFGF). The specific interaction between the glycopolymer (lactose-substituted styrene homopolymer, PVLA) with FTIC-RCA120 was observed using fluorescence microscopy. Green fluorescence in the octadecyltrimethoxysilane (ODS) region, which is enriched with PVLA, and the red fluorescence of tetramethylrhodamine isothiocyanate (TRITC)-bFGF in the region composed of heparin-aminopropyltrimethoxysilane (APS) confirm the suborder (Fig. 16). In vitro studies was extended by using hepatocytes (contains C-type, hepatic lectin) and fibroblasts for adhesion with PVLA and heparin and strikingly uniform arrangements of cells were observed under the optical microscope.64
 | ||
| Fig. 16 Fluorescence images of (RCA120-PVLA-ODS)/(bFGF-heparin-APS) substrate: viewed with (a) a green filter and (b) a red filter and (c) their overlay.64 Reproduced with permission from ACS, 2007. | ||
Galactose and N-acetyl-β-galactosamine are ligands known to recognized hepatic lectins mediated by asialoglycoprotein receptor (ASGPRS). Studies by Kim et al. revealed that poly-[N-p-vinylbenzyl-D-glucuronamide] (PV6Gna), a glucose based glycopolymer with its glucose unit modified at C-6 position also display hepatocytes adhesion. A direct lectin-enzyme assay shows that the glycopolymers are coated on the surface of the polystyrene microplate. The mechanism of hepatocytes adhesion onto PV6Gna surface is proposed to be integrin-independent mediated only by glucose moiety, however, further investigation needs to be carried out to confirm the proposed mechanism.54
Haddleton and co-workers optimized surface modified Wang resin with mannose in order to prepare column material for protein purification chromatography. Mannose was attached onto Wang resin using click chemistry (Fig. 17). HPLC equipped with fluorescence detector and in addition confocal microscopy investigations confirmed that ConA was specific when interacted with mannose units on the particles surfaces. ConA substrates on the mannose functionalized particles were removed by using large excess of α-methyl-D-mannopyranoside.131 A similar study was carried out by Gu et al.162 The Cu(I) catalyzed click conjugation chemistry was replaced by thiol-ene reaction leading to microspheres with high specificity for ConA.
 | ||
| Fig. 17 Immobilized sugar supports using the Wang resin via click chemistry.131 Reproduced with permission from ACS, copyright 2007. | ||
Min et al. grafted thermo-responsive glycopolymers onto honeycomb structured porous films. Depending on the temperature, binding of ConA was either permitted or prevented.163
Conclusions
In this review, we have provided an in depth discussions into the various synthetic pendant glycopolymer architectures used for binding lectins. The syntheses of these glycopolymers were generally documented into two main sections (a) polymerization from sugar-containing monomers and by (b) post polymerization glycosylation reactions. Lectins ranging from plants mainly legume, animal, toxins and bacteria were highlighted to provide readers with an overview of these lectins. Interactions between the various glycopolymer architectures and their corresponding lectins were thoroughly discussed. With the aid of the various type of lectin binding assays, many discoveries into what is called the “glycocluster effect” was made in relations to the implications of the different pendant sugar unit, i.e. mono-, di- or tri-saccharides and, the different functionalized carbon positions in the carbohydrate six-membered ring. Rigidity of the synthetic glycopolymers needs to be carefully considered when designing glycopolymer based materials. Finally, epitope density, molecular weight and architecture of glycopolymers were also debated to provide a general overview on the interactions between glycopolymer and their lectins. It seems, however, that it is difficult to compare different polymer systems since different authors employed different analytical techniques and different concentrations.In summary, polymer chemistry provides now a versatile toolbox to allow the creation of different bioactive glycopolymer structures, which enables the careful fine-tuning of the interaction between lectins and glycopolymers.
Acknowledgements
The authors thank the Australian Research Council (ARC, DP0771155) and the Centre for Advanced Macromolecular Design (CAMD) for support. The authors thank Dr Andrew Gregory for help with the manuscript. M. H. Stenzel acknowledges an ARC Future Fellowship.Notes and references
- R. A. Dwek, Chem. Rev., 1996, 96, 683–720 CrossRef CAS.
- H. Lis and N. Sharon, Chem. Rev., 1998, 98, 637–674 CrossRef CAS.
- J. J. Lundquist and E. J. Toone, Chem. Rev., 2002, 102, 555–578 CrossRef CAS.
- M. Mammen, S.-K. Chio and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754–2794 CrossRef.
- Y. Miura, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5031–5036 CrossRef CAS.
- T. M. Allen, Nat. Rev. Cancer, 2002, 2, 750–763 CrossRef CAS.
- R. Duncan, Nat. Rev. Cancer, 2006, 6, 688–701 CrossRef CAS.
- A. David, P. Kopeckova, J. Kopecek and A. Rubinstein, Pharm. Res., 2002, 19, 1114–1122 CrossRef CAS.
- A. David, P. Kopeckova, A. Rubinstein and J. Kopecek, Bioconjugate Chem., 2001, 12, 890–899 CrossRef CAS.
- X. Montet, M. Funovics, K. Montet-Abou, R. Weissleder and L. Josephson, J. Med. Chem., 2006, 49, 6087–6093 CrossRef CAS.
- Y. Shamay, D. Paulin, G. Ashkenasy and A. David, J. Med. Chem., 2009, 52, 5906–5915 CrossRef CAS.
- L. A. J. M. Sliedregt, P. C. N. Rensen, E. T. Rump, P. J. Van Santbrink, M. K. Bijsterbosch, A. R. P. M. Valentijn, G. A. Van der Marel, J. H. Van Boom, T. J. C. Van Berkel and E. A. L. Biessen, J. Med. Chem., 1999, 42, 609–618 CrossRef CAS.
- R. T. Lee and Y. C. Lee, Glycoconjugate J., 2000, 17, 543–551 CrossRef CAS.
- N. Sharon and H. Lis, Science, 1989, 246, 227–234 CAS.
- R. J. Pieters, Org. Biomol. Chem., 2009, 7, 2013–2025 RSC.
- A. M. Granville, D. Quemener, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromol. Symp., 2007, 255, 81–89 CrossRef CAS.
- Y. M. Chabre and R. Roy, Curr. Top. Med. Chem., 2008, 8, 1237–1285 CrossRef CAS.
- M. Ambrosi, N. R. Cameron and B. G. Davis, Org. Biomol. Chem., 2005, 3, 1593–1608 RSC.
- T. Iskratsch, A. Braun, K. Paschinger and I. B. H. Wilson, Anal. Biochem., 2009, 386, 133–146 CrossRef CAS.
- J. M. Rini, Annu. Rev. Biophys. Biomol. Struct., 1995, 24, 551–577 CrossRef CAS.
- W. I. Weis and K. Drickamer, Annu. Rev. Biochem., 1996, 65, 441–473 CrossRef CAS.
- R. Loris, T. Hamelryck, J. Bouckaert and L. Wyns, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1998, 1383, 9–36 Search PubMed.
- N. Sharon, Trends Biochem. Sci., 1993, 18, 221–226 CrossRef CAS.
- L. Laitinen, Histochem. J., 1987, 19, 225–234 CrossRef CAS.
- B. Shaanan, H. Lis and N. Sharon, Science, 1991, 254, 862–866 CrossRef CAS.
- C. S. Wright, J. Mol. Biol., 1987, 194, 501–529 CrossRef CAS.
- R. Sankaranarayanan, K. Sekar, R. Banerjee, V. Sharma, A. Surolia and M. Vijayan, Nat. Struct. Biol., 1996, 3, 596–603 CrossRef.
- K. Tachibana, S. Nakamura, H. Wang, H. Iwasaki, K. Tachibana, K. Maebara, L. Cheng, J. Hirabayashi and H. Narimatsu, Glycobiology, 2006, 16, 46–53 CAS.
- The Lectins. Properties, Functions, and Applications in Biology and Medicine, ed. I. E. Liener, N. Sharon and I. J. Goldstein, Academic Press Inc., 1986 Search PubMed.
- D. C. Kilpatrick, Biochim. Biophys. Acta, Gen. Subj., 2002, 1572, 187–197 Search PubMed.
- P. R. Taylor, L. Martinez-Pomares, M. Stacey, H. H. Lin, G. D. Brown and S. Gordon, Annu. Rev. Immunol., 2005, 23, 901–944 CrossRef CAS.
- H. Leffler, S. Carlsson, M. Hedlund, Y. N. Qian and F. Poirier, Glycoconjugate J., 2002, 19, 433–440 CrossRef CAS.
- J. Bouckaert, J. Berglund, M. Schembri, E. De Genst, L. Cools, M. Wuhrer, C.-S. Hung, J. Pinkner, R. Slaettegard, A. Zavialov, D. Choudhury, S. Langermann, S. J. Hultgren, L. Wyns, P. Klemm, S. Oscarson, S. D. Knight and H. De Greve, Mol. Microbiol., 2005, 55, 441–455 CrossRef CAS.
- G. E. Soto and S. J. Hultgren, J. Bacteriol., 1999, 181, 1059–1071 CAS.
- C. Maierhofer, K. Rohmer and V. Wittmann, Bioorg. Med. Chem., 2007, 15, 7661–7676 CrossRef CAS.
- A. A. Profit, T. R. Lee and D. S. Lawrence, J. Am. Chem. Soc., 1999, 121, 280–283 CrossRef CAS.
- J. Rao, J. Lahiri, L. Isaacs, R. M. Weis and G. M. Whitesides, Science, 1998, 280, 708–711 CrossRef CAS.
- N. Schaschke, G. Matschiner, F. Zettl, U. Marquardt, A. Bergner, W. Bode, C. P. Sommerhoff and L. Moroder, Chem. Biol., 2001, 8, 313–327 CrossRef CAS.
- W. Jager, Carbohydr. Chem. Biol., 2000, 1045–1057 Search PubMed.
- K. Landsteiner, The Specificity of Serological Reactions, Dover,New York, 1962 Search PubMed.
- L. R. Olsen, A. Dessen, D. Gupta, S. Sabesan, J. C. Sacchettini and C. F. Brewer, Biochemistry, 1997, 36, 15073–15080 CrossRef CAS.
- J. P. McCoy, Jr., J. Varani and I. J. Goldstein, Exp. Cell Res., 1984, 151, 96–103 CrossRef CAS.
- E. Freire, O. L. Mayorga and M. Straume, Anal. Chem., 1990, 62, 950A–959A.
- C. W. Cairo, J. E. Gestwicki, M. Kanai and L. L. Kiessling, J. Am. Chem. Soc., 2002, 124, 1615–1619 CrossRef.
- K. Kobayashi, A. Tsuchida, T. Usui and T. Akaike, Macromolecules, 1997, 30, 2016–2020 CrossRef CAS.
- Z. Deng, S. Li, X. Jiang and R. Narain, Macromolecules, 2009, 42, 6393–6405 CrossRef CAS.
- Q. Yang, M.-X. Hu, Z.-W. Dai, J. Tian and Z.-K. Xu, Langmuir, 2006, 22, 9345–9349 CrossRef CAS.
- S. G. Spain, M. I. Gibson and N. R. Cameron, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2059–2072 CrossRef CAS.
- S. Akai, Y. Kajihara, Y. Nagashima, M. Kamei, J. Arai, M. Bito and K.-I. Sato, J. Carbohydr. Chem., 2001, 20, 121–143 CrossRef CAS.
- M. Ambrosi, A. S. Batsanov, N. R. Cameron, B. G. Davis, J. A. K. Howard and R. Hunter, J. Chem. Soc., Perkin Trans. 1, 2002, 45–52 RSC.
- M. Ambrosi, N. R. Cameron, B. G. Davis and S. Stolnik, Org. Biomol. Chem., 2005, 3, 1476–1480 RSC.
- R. Cuervo-Rodriguez, V. Bordege and M. Fernandez-Garcia, Carbohydr. Polym., 2007, 68, 89–94 CrossRef CAS.
- T. Furuike, N. Nishi, S. Tokura and S.-I. Nishimura, Macromolecules, 1995, 28, 7241–7247 CrossRef CAS.
- S.-H. Kim, M. Goto, C.-S. Cho and T. Akaike, Biotechnol. Lett., 2000, 22, 1049–1057 CrossRef CAS.
- A. Miyachi, H. Dohi, P. Neri, H. Mori, H. Uzawa, Y. Seto and Y. Nishida, Biomacromolecules, 2009, 10, 1846–1853 CrossRef CAS.
- N. Nagahori and S.-I. Nishimura, Biomacromolecules, 2001, 2, 22–24 CrossRef CAS.
- T. Serizawa, S. Yasunaga and M. Akashi, Biomacromolecules, 2001, 2, 469–475 CrossRef CAS.
- A. Tsuchida, S. Akimoto, T. Usui and K. Kobayashi, J. Biochem., 1998, 123, 715–721 CAS.
- Q. Yang, J. Wu, J.-J. Li, M.-X. Hu and Z.-K. Xu, Macromol. Rapid Commun., 2006, 27, 1942–1948 CrossRef CAS.
- A. Yoshizumi, N. Kanayama, Y. Maehara, M. Ide and H. Kitano, Langmuir, 1999, 15, 482–488 CrossRef CAS.
- L. Yu, M. Huang, P. G. Wang and X. Zeng, Anal. Chem., 2007, 79, 8979–8986 CrossRef CAS.
- Y. Miura, T. Ikeda and K. Kobayashi, Biomacromolecules, 2003, 4, 410–415 CrossRef CAS.
- Y. Miura, H. Sato, T. Ikeda, H. Sugimura, O. Takai and K. Kobayashi, Biomacromolecules, 2004, 5, 1708–1713 CrossRef CAS.
- H. Sato, Y. Miura, N. Saito, K. Kobayashi and O. Takai, Biomacromolecules, 2007, 8, 753–756 CrossRef CAS.
- S. Loykulnant and A. Hirao, Macromolecules, 2000, 33, 4757 CrossRef CAS.
- K. Aoi, K. Tsutsumiuchi, E. Aoki and M. Okada, Macromolecules, 1996, 29, 4456–4458 CrossRef CAS.
- K. Tsutsumiuchi, K. Aoi and M. Okada, Macromolecules, 1997, 30, 4013–4017 CrossRef CAS.
- D. D. Manning, X. Hu, P. Beck and L. L. Kiessling, J. Am. Chem. Soc., 1997, 119, 3161–3162 CrossRef CAS.
- K. H. Mortell, R. V. Weatherman and L. L. Kiessling, J. Am. Chem. Soc., 1996, 118, 2297–2298 CrossRef CAS.
- J. J. Murphy, H. Furusho, R. M. Paton and K. Nomura, Chem.–Eur. J., 2007, 13, 8985 CrossRef CAS.
- N. L. Pohl and L. L. Kiessling, Synthesis, 1999, 1515–1519 CrossRef CAS.
- W. J. Sanders, E. J. Gordon, O. Dwir, P. J. Beck, R. Alon and L. L. Kiessling, J. Biol. Chem., 1999, 274, 5271–5278 CrossRef CAS.
- M. C. Schuster, K. H. Mortell, A. D. Hegeman and L. L. Kiessling, J. Mol. Catal. A: Chem., 1997, 116, 209–216 CrossRef CAS.
- K. H. Mortell, M. Gingras and L. L. Kiessling, J. Am. Chem. Soc., 1994, 116, 12053–12054 CrossRef CAS.
- Y. Chen and G. Wulff, Macromol. Chem. Phys., 2001, 202, 3273–3278 CrossRef CAS.
- Y. Chen and G. Wulff, Macromol. Chem. Phys., 2001, 202, 3426–3431 CrossRef CAS.
- H. Götz, E. Harth, S. M. Schiller, C. W. Frank, W. Knoll and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3379–3391 CrossRef CAS.
- A. Narumi, T. Matsuda, H. Kaga, T. Satoh and T. Kakuchi, Polymer, 2002, 43, 4835–4840 CrossRef CAS.
- A. Narumi, T. Satoh, H. Kaga and T. Kakuchi, Macromolecules, 2002, 35, 699–705 CrossRef CAS.
- K. Ohno, T. Fukuda and H. Kitano, Macromol. Chem. Phys., 1998, 199, 2193–2197 CrossRef CAS.
- K. Ohno, Y. Izu, S. Yamamoto, T. Miyamoto and T. Fukuda, Macromol. Chem. Phys., 1999, 200, 1619–1625 CrossRef CAS.
- K. Ohno, Y. Tsujii, T. Miyamoto, T. Fukuda, M. Goto, K. Kobayashi and T. Akaike, Macromolecules, 1998, 31, 1064–1069 CrossRef CAS.
- X.-L. Sun, K. M. Faucher, M. Houston, D. Grande and E. L. Chaikof, J. Am. Chem. Soc., 2002, 124, 7258–7259 CrossRef CAS.
- J. Q. Meng, F. S. Du, Y. S. Liu and Z. C. Li, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 752 CrossRef CAS.
- S. Muthukrishnan, D. P. Erhard, H. Mori and A. H. E. Muller, Macromolecules, 2006, 39, 2743 CrossRef CAS.
- S. Muthukrishnan, G. Jutz, A. Andre, H. Mori and A. H. E. Muller, Macromolecules, 2005, 38, 9 CrossRef CAS.
- K. Ohno, Y. Tsujii and T. Fukuda, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 2473–2481 CrossRef CAS.
- V. Vazquez-Dorbatt and H. D. Maynard, Biomacromolecules, 2006, 7, 2297 CrossRef CAS.
- X.-H. Dai and C.-M. Dong, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 817–829 CrossRef CAS.
- X.-H. Dai, C.-M. Dong and D. Yan, J. Phys. Chem. B, 2008, 112, 3644–3652 CrossRef CAS.
- A. Mateescu, J. Ye, R. Narain and M. Vamvakaki, Soft Matter, 2009, 5, 1621–1629 RSC.
- K. Mizukami, H. Takakura, T. Matsunaga and H. Kitano, Colloids Surf., B, 2008, 66, 110–118 CrossRef CAS.
- V. Ladmiral, G. Mantovani, G. J. Clarkson, S. Cauet, J. L. Irwin and D. M. Haddleton, J. Am. Chem. Soc., 2006, 128, 4823–4830 CrossRef CAS.
- M. Al-Bagoury, K. Buchholz and E. J. Yaacoub, Polym. Adv. Technol., 2007, 18, 313 CrossRef CAS.
- L. Albertin and N. R. Cameron, Macromolecules, 2007, 40, 6082 CrossRef CAS.
- L. Albertin, C. Kohlert, M. Stenzel, L. J. R. Foster and T. P. Davis, Biomacromolecules, 2004, 5, 255 CrossRef CAS.
- L. Albertin, M. Stenzel, C. Barner-Kowollik, L. J. R. Foster and T. P. Davis, Macromolecules, 2004, 37, 7530 CrossRef CAS.
- L. Albertin, M. H. Stenzel, C. Barner-Kowollik and T. P. Davis, Polymer, 2006, 47, 1011 CrossRef CAS.
- L. Albertin, M. H. Stenzel, C. Barner-Kowollik, L. J. R. Foster and T. P. Davis, Macromolecules, 2005, 38, 9075 CrossRef CAS.
- L. Albertin, M. H. Stenzel, C. Barner-Kowollik, L. J. R. Foster and T. P. Davis, Polymer, 2005, 46, 2831 CrossRef CAS.
- J. Bernard, X. J. Hao, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Biomacromolecules, 2006, 7, 232 CrossRef CAS.
- A. B. Lowe, B. S. Sumerlin and C. L. McCormick, Polymer, 2003, 44, 6761 CrossRef CAS.
- Z. Ozyurek, H. Komber, S. Gramm, D. Schmaljohann, A. H. E. Muller and B. Voit, Macromol. Chem. Phys., 2007, 208, 1035 CrossRef.
- S. Pearson, N. Allen and M. H. Stenzel, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1706–1723 CrossRef CAS.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Chem. Commun., 2008, 2477 RSC.
- S. R. S. Ting, A. M. Granville, D. Quemener, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Aust. J. Chem., 2007, 60, 405–409 CrossRef CAS.
- S. R. S. Ting, A. M. Gregory and M. H. Stenzel, Biomacromolecules, 2009, 10, 342–352 CrossRef CAS.
- N.-Y. Xiao, A.-L. Li, H. Liang and J. Lu, Macromolecules, 2008, 41, 2374–2380 CrossRef CAS.
- L. Zhang, J. Bernard, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromol. Rapid Commun., 2008, 29, 123 CrossRef CAS.
- M. Toyoshima and Y. Miura, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1412–1421 CrossRef CAS.
- S. G. Spain, L. Albertin and N. R. Cameron, Chem. Commun., 2006, 4198–4200 RSC.
- G. Wulff, J. Schmid and T. Venhoff, Macromol. Chem. Phys., 1996, 197, 259–274 CrossRef CAS.
- V. Ladmiral, E. Melia and D. M. Haddleton, Eur. Polym. J., 2004, 40, 431–449 CrossRef CAS.
- X. Zeng, T. Murata, H. Kawagishi, T. Usui and K. Kobayashi, Biosci., Biotechnol., Biochem., 1998, 62, 1171–1178 CrossRef CAS.
- M. Huang, Z. Shen, Y. Zhang, X. Zeng and P. G. Wang, Bioorg. Med. Chem. Lett., 2007, 17, 5379–5383 CrossRef CAS.
- M. Okada, Prog. Polym. Sci., 2001, 26, 67–104 CrossRef CAS.
- S. R. S. Ting, E.-H. Min, P. Escale, M. Save, L. Billon and M. H. Stenzel, Macromolecules, 2009, 42, 9422–9434 CrossRef CAS.
- J. Geng, G. Mantovani, L. Tao, J. Nicolas, G. Chen, R. Wallis, D. A. Mitchell, B. R. G. Johnson, S. D. Evans and D. M. Haddleton, J. Am. Chem. Soc., 2007, 129, 15156–15163 CrossRef CAS.
- C.-M. Dong and E. L. Chaikof, Colloid Polym. Sci., 2005, 283, 1366–1370 CrossRef CAS.
- C.-M. Dong, K. M. Faucher and E. L. Chaikof, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5754–5765 CrossRef CAS.
- M. Hetzer, G. Chen, C. Barner-Kowollik and M. H. Stenzel, Macromol. Biosci., 2010, 10, 119–126 CrossRef CAS.
- M. A. Gauthier, M. I. Gibson and H.-A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS.
- G. Martinez, M. Fernandez-Garcia and M. Sanchez-Chaves, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 18–27 CrossRef CAS.
- R. Bahulekar, T. Tokiwa, J. Kano, T. Matsumura, I. Kojima and M. Kodama, Carbohydr. Polym., 1998, 37, 71–78 CrossRef CAS.
- L. E. Strong and L. L. Kiessling, J. Am. Chem. Soc., 1999, 121, 6193–6196 CrossRef CAS.
- M.-G. Baek and R. Roy, Macromol. Biosci., 2001, 1, 305–311 CrossRef CAS.
- R. Auzely-Velty, M. Cristea and M. Rinaudo, Biomacromolecules, 2002, 3, 998–1005 CrossRef CAS.
- M. Sanchez-Chaves, F. Arranz and M. Cortazar, Polymer, 1998, 39, 2751–2757 CrossRef CAS.
- M. C. Garcia-Oteiza, M. Sanchez-Chaves and F. Arranz, Macromol. Chem. Phys., 1997, 198, 2237–2247 CrossRef CAS.
- M. L. Cerrada, M. Sanchez-Chaves, C. Ruiz and M. Fernandez-Garcia, Biomacromolecules, 2009, 10, 1828–1837 CrossRef CAS.
- G. Chen, L. Tao, G. Mantovani, J. Geng, D. Nystroem and D. M. Haddleton, Macromolecules, 2007, 40, 7513–7520 CrossRef CAS.
- J. Geng, J. Lindqvist, G. Mantovani, G. Chen, C. T. Sayers, G. J. Clarkson and D. M. Haddleton, QSAR Comb. Sci., 2007, 26, 1220–1228 Search PubMed.
- L. Nurmi, J. Lindqvist, R. Randev, J. Syrett and D. M. Haddleton, Chem. Commun., 2009, 2727–2729 RSC.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS.
- G. Chen, S. Amajjahe and M. H. Stenzel, Chem. Commun., 2009, 1198–1200 RSC.
- C. Diehl and H. Schlaad, Macromol. Biosci., 2009, 9, 157–161 CrossRef CAS.
- A. Takasu, T. Niwa, H. Itou, Y. Inai and T. Hirabayashi, Macromol. Rapid Commun., 2000, 21, 764–769 CrossRef CAS.
- C. Xue, V. R. R. Donuru and H. Liu, Macromolecules, 2006, 39, 5747–5752 CrossRef CAS.
- J. Zhu, C. Gosen and R. E. Marchant, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 192–199 CrossRef CAS.
- L. Renaudie, C. Le Narvor, E. Lepleux and P. Roger, Biomacromolecules, 2007, 8, 679–685 CrossRef CAS.
- A. Gress, B. Smarsly and H. Schlaad, Macromol. Rapid Commun., 2008, 29, 304–308 CrossRef CAS.
- L.-C. You, F.-Z. Lu, Z.-C. Li, W. Zhang and F.-M. Li, Macromolecules, 2003, 36, 1–4 CrossRef CAS.
- K. Kobayashi, H. Sumitomo and Y. Ina, Polym. J., 1985, 17, 567–575 CrossRef CAS.
- T. Hasegawa, S. Kondoh, K. Matsuura and K. Kobayashi, Macromolecules, 1999, 32, 6595–6603 CrossRef CAS.
- C. Boyer and T. P. Davis, Chem. Commun., 2009, 6029–6031 RSC.
- C. Ruiz, M. Sanchez-Chaves, M. L. Cerrada and M. Fernandez-Garcia, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7238–7248 CrossRef CAS.
- Q. Chen, Y. Xu, Y. Du and B.-H. Han, Polymer, 2009, 50, 2830–2835 CrossRef CAS.
- M.-G. Baek, R. C. Stevens and D. H. Charych, Bioconjugate Chem., 2000, 11, 777–788 CrossRef CAS.
- G. Pasparakis, A. Cockayne and C. Alexander, J. Am. Chem. Soc., 2007, 129, 11014–11015 CrossRef CAS.
- M. Ogata, K. I. P. J. Hidari, W. Kozaki, T. Murata, J. Hiratake, E. Y. Park, T. Suzuki and T. Usui, Biomacromolecules, 2009, 10, 1894–1903 CrossRef CAS.
- K. Totani, T. Kubota, T. Kuroda, T. Murata, K. I. P. J. Hidari, T. Suzuki, Y. Suzuki, K. Kobayashi, H. Ashida, K. Yamamoto and T. Usui, Glycobiology, 2003, 13, 315–326 CrossRef CAS.
- R. D. Poretz and I. J. Goldstein, Biochemistry, 1970, 9, 2890–2896 CrossRef CAS.
- S. Liu and K. L. Kiick, Macromolecules, 2008, 41, 764–772 CrossRef CAS.
- J. E. Gestwicki, C. W. Cairo, L. E. Strong, K. A. Oetjen and L. L. Kiessling, J. Am. Chem. Soc., 2002, 124, 14922–14933 CrossRef CAS.
- B. D. Polizzotti and K. L. Kiick, Biomacromolecules, 2006, 7, 483–490 CrossRef CAS.
- M. Kanai, K. H. Mortell and L. L. Kiessling, J. Am. Chem. Soc., 1997, 119, 9931–9932 CrossRef CAS.
- S. A. DeFrees, L. Phillips, S. Zalipsky and L. Guo, J. Am. Chem. Soc., 1996, 118, 6101–6104 CrossRef CAS.
- G. Pasparakis and C. Alexander, Angew. Chem., Int. Ed., 2008, 47, 4847–4850 CrossRef CAS.
- I. J. Goldstein, C. E. Hollerman and E. E. Smith, Biochemistry, 1965, 4, 876–883 CrossRef CAS.
- S. R. S. Ting, E. H. Min, P. B. Zetterlund and M. H. Stenzel, Macromolecules, 2010, 43, 5211–5221 CrossRef CAS.
- G. Scatchard, J. S. Coleman and A. L. Shen, J. Am. Chem. Soc., 1957, 79, 12–20 CrossRef CAS.
- W. Gu, G. Chen and M. H. Stenzel, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5550–5556 CrossRef CAS.
- E. H. Min, S. R. S. Ting, L. Billon and M. H. Stenzel, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3440–3455.
- Q. Chen and B.-H. Han, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2948–2957 CrossRef CAS.
- T. Akasaka, K. Matsuura and K. Kobayashi, Bioconjugate Chem., 2001, 12, 776–785 CrossRef CAS.
Footnote |
| † Current Address: Center for Soft Condensed Matter Physics and Interdisciplinary Research, Soochow University, Suzhou 215006, China |
| This journal is © The Royal Society of Chemistry 2010 |