Flame retardancy of polylactide: an overview
Serge
Bourbigot
*abcd and
Gaëlle
Fontaine
abcd
aUniv Lille Nord de France, F-5900, Lille, France. E-mail: serge.bourbigot@ensc-lille.fr
bENSCL, ISP-UMET, F-59652, Villeneuve d'Ascq, France
cUSTL, ISP-UMET, F-59655, Villeneuve d'Ascq, France
dCNRS, UMR 8207, F-59652, Villeneuve d'Ascq, France
First published on 1st July 2010
Abstract
The paper reviews the different methods for flame retarding polylactide (PLA). They are critically discussed based on the open and patent literature. No real specific methods have been developed for PLA but some approaches provide high performance and especially intumescence. The mechanisms of action remain similar to those observed in other polymers.
 Serge Bourbigot | Prof. Bourbigot got his PhD in 1993 and he joined ENSCL as lecturer in 1993. He passed his French “Habilitation à Diriger des Recherches” in chemical engineering at the University of Lille in 1998. He took the position of Professor in chemical engineering at ENSAIT in 1999. In 2002, Prof. Bourbigot left his laboratory to go to NIST (Gaithersburg, MD – USA) on sabbatical leave for a year. In 2003, ENSCL offered him the position of Full Professor in flame retardancy. His research interests include reaction and resistance to fire of polymeric materials including thermoplastics, thermosets, textiles and coating. |
 Gaëlle Fontaine | Dr Fontaine got her PhD in 1998 in organic chemistry at the University of Caen. She did a postdoc at the University of Cambridge at the Institute of Biotechnology (1999–2000). She joined ENSCL as lecturer in 2001 as an organic chemist. She is in charge of the 'sustainable development' unit of ENSCL. Her research interests include the synthesis of new flame retardants and of biopolymers and the development of new flame retardant formulations for polymeric materials including intumescent and nanocomposite based materials. |
Introduction
The worldwide capacity of bio-based plastics, according to company announcements, will increase from 0.36 Mt in 2007 to 2.33 Mt in 2013 and to 3.45 Mt in 2020.1 This is equivalent to average annual growth rates of 37% between 2007 and 2013 and 6% between 2013 and 2020. In 2007, the most important products in terms of production volumes were starch plastics (0.15 Mt) and poly(lactic acid) (PLA) or polylactide (0.15 Mt). Based on the company announcements it is projected that the most important representatives by 2020 will be starch plastics (1.3 Mt), PLA (0.8 Mt), bio-based polyethylene (PE) (0.6 Mt) and polyhydroxyalkanoates (PHA) (0.4 Mt).1Of the above mentioned bio-based plastics, one of the outstanding achievements in the realm of polymers from renewable resources is undoubtedly the rapid progress related to the research and development activities for the production of PLA.2 This polymer is now familiar and well documented. PLA has been an industrial commodity with important applications, particularly in packaging3 and fiber technology.4,5 It is an aliphatic polyester produced via polymerization of the renewable fermentation product lactic acid. With the setup of NatureWorks' (formally Cargill Dow) production plant for PLA in 2002, PLA became the third type of bio-based polymer that was commercialized and is now produced on a large scale.6
The physical and mechanical properties of PLA make it a good candidate as replacement for petrochemical thermoplastics in several application areas. The properties profile of commercial PLA is in some aspects similar to synthetic thermoplastics (mechanical strength, elastic recovery and heat sealability), while it shares other properties with bio-based polymers (biodegradability, dyeability, barrier characteristics). According to two major companies producing PLA, namely Nature Works and PURAC, about the substitution potential for PLA, they agree on the potential for PLA or PLA components to partially replace low density polyethylene (LDPE), high density polyethylene (HDPE), polypropylene (PP), polyamide (PA) and polyethylene terephtalate (PET), as well as seeing possibilities for PLA to substitute for polymethylmethacrylate (PMMA). Even if PLA is primarily used in packaging and the textile sector today, its expected market should be rapidly extended to transportation and electrical and electronic equipment (E&E) sectors. For example in the electronic sector, Fujitsu is making injection molded computer keys and computer housing made from polycarbonate (PC)/PLA blends.7 One recent development which should enable wider application of PLA in electronics products is NEC's process for imparting flame resistance to PLA without the use of halogen or phosphorous compounds. The product is reported to have heat resistance, moldability and strength comparable to fiber-reinforced polycarbonate used in desktop-type electronic products. According to this brief survey of PLA's applications, it can be clearly seen that flame retarding PLA is (or will be) becoming an issue for this polymer. Acceptable solutions should then be found, as has been done for polymers which are supposed to be substituted by PLA. It is the main goal of this paper to review the flame retardancy of PLA.
In a general way, the flammability behavior of polymers is defined on the basis of several processes and/or parameters, such as burning rates (solid degradation rate and heat release rate), spread rates (flame, pyrolysis, burn-out, smolder), ignition characteristics (delay time, ignition temperature, critical heat flux for ignition), product distribution (in particular, toxic species emissions), smoke production, etc…8 Our goal is then to inhibit or even suppress the combustion process acting chemically and/or physically in the solid, liquid or gas phase. We can interfere with combustion during a particular stage of this process, e.g. during heating, decomposition, ignition or flame spread. Based on this and on our expertise in the field of flame retardancy,9 three main approaches can then be considered to reduce the flammability of PLA: (i) to chemically modify PLA; (ii) to incorporate flame retardants into polymers via usual procedures and (iii) to add a surface treatment to the polymer. This will be further commented on in the first section of this review to provide the reader with a basic understanding of flame retardancy. The second section will be devoted to the state of the art of the flame retardancy for PLA. The third part of the review will discuss solutions developed for PLA based on scientific approach used for other thermoplastics.
A basic explanation of flame retardancy
Various methods can be used to protect materials more effectively against attack by fire. Inherently flame retardant (FR) polymers or high performance polymers can be used but this implies the use of specific materials which might not have the required properties (e.g. aging, cost…). Nevertheless, they exhibit very low flammability and are very efficient.10 The second method is to chemically modify the existing polymer (or its monomer) to synthesize FR polymer. In this case, it requires a specific process of production and very often the new FR polymer can only be only synthesized at the laboratory scale.11,12 The last method is to use flame retardants and/or micro- or nano-particles directly incorporated in the materials (e.g. thermoplastics or thermosets) or in a coating covering their surface (e.g. structural steel or textiles).9,13 This approach is often preferred to provide low flammability to polymeric materials because it is an acceptable compromise between cost and properties and because it brings great flexibility to design materials with multifunctional properties. The first two approaches will be not commented because it is out of the scope of this review and no papers report such approaches for PLA. The last approach will be fully commented on in terms of mechanism of action in order to introduce the other sections of this article.(a) Polymer containing flame retardant
Depending on their nature, flame retardants can act chemically and/or physically in the solid, liquid or gas phase. Our intention here is to provide the reader with the general principles of the modes of action of flame retardants.Flame retardants interfere with combustion during a particular stage of this process, e.g. during heating, decomposition, ignition or flame spread. The various ways in which a flame retardant can act are described in the following. They do not occur singly but should be considered as a complex process in which many individual stages occur simultaneously with one dominating (e.g., using hydroxides exhibits an endothermic decomposition cooling down the substrate and diluting the ignitable gas mixture due to the formation of inert gases associated with the formation of oxide protective barrier). There are several ways in which the combustion process can be retarded by physical action:
(i) By formation of a protective layer. The additives can form under an external heat flux a shield with a low thermal conductivity which can reduce the heat transfer from the heat source to the material. It then reduces the degradation rate of the polymer and decreases the “fuel flow” (pyrolysis gases issued from the degradation of the material) able to feed the flame in flammable molecules. A typical example is the principle of the intumescence phenomenon (formation of an expanded carbonaceous protective coating).13 Phosphorus additives may act in similar manner. Their pyrolysis leads to pyro- or polyphosphoric species thermally stable which form a protective vitreous barrier. The same mechanism can be observed using boric acid based additives, inorganic borates, silicon compounds or low melting glasses.14
(ii) By cooling. The degradation reactions of the additive can play a part in the energy balance of combustion. The additive can degrade endothermally which cools down the substrate to a temperature below that requires for sustaining the combustion process. Aluminium trihydroxide (ATH) or magnesium hydroxide (MDH) act partially under this principle (endothermic decomposition with water release and formation of an oxide protective coating) and its efficiency depends on the amount incorporated in the polymer (generally 60 wt% in thermoplastics).15
(iii) By dilution. The incorporation of inert substances (e.g. fillers such as talc or chalk) and additives which evolve inert gases on decomposition, dilutes the fuel in the condensed and gaseous phases so that the lower ignition limit of the gas mixture is not reached.
The most significant chemical reactions interfering with the combustion process take place in the condensed and gaseous phases:
(i) Reaction in the condensed phase. Here two types of reaction can take place. Firstly, breakdown of the polymer can be accelerated by the flame retardant causing pronounced flow of the polymer and, hence, its withdrawal from the sphere of influence of the flame which breaks away. Secondly, the flame retardant can cause a layer of carbon (charring), a ceramic-like structure and/or a glass to be formed at the polymer surface.9
(ii) Reaction in the gaseous phase. The radical mechanism of the combustion process which takes place in the gas phase is interrupted by the flame retardant or its degradation products. The exothermic processes which occur in the flame are thus stopped, the system cools down, and the supply of flammable gases is reduced and eventually completely suppressed. In particular, halogens can act as flame inhibitors.16
Note that fire retardant additives may be used alone or in association with other systems (sometimes using low amounts of the last system) in polymeric materials to obtain a synergistic effect, i.e. the protective effect is higher that this assumed from addition of the separate effects of each system (this will be further commented on in the next section in the particular case of PLA).17–19
(b) Surface treatment for making flame retarded polymer
The surface treatments are easy and efficient ways to protect materials against fire.9 Indeed, it presents several advantages: it does not modify the intrinsic properties of the materials such as for example the mechanical properties, it is easily processed and it can be used onto several materials such as metallic materials,20 polymers,21 textile22 and wood.23 Moreover, while inflammation occurs usually on the surface of a material it is important to concentrate the protective action at this location.Cold plasma technologies are surface modification processes. Hence the inherent advantages of the substrates can be kept and materials exhibiting various properties can be made. Cold plasma reactive species can promote surface functionalization reactions or generate organic or inorganic thin layers as a result of recombination of radicals or molecular fragments species on the surfaces. This is this second approach that has been used to develop fire retardant protective coatings for polymeric materials.21,24–26 Silicon-containing organic compounds (organosilicones) are widely used as monomers in low-pressure plasma deposition due to their availability, liquid state, volatility at room temperature, safe handling and low cost. The structure of the formed deposits is very dependent on the energetic character of the plasma and can be turned from SiO2-like inorganic coatings to silicon-like polymers. The mode of action is the formation of a carbonaceous and silica like layer during combustion acting as a barrier limiting mass and heat transfers between the flame and the polymer and slows down the toxic gases emission released by polymer combustion.
Other plasma techniques have been developed like the plasma induced graft-polymerization technique for flame retardancy purposes. It consists of the simultaneous grafting and polymerization of functionalized monomers, such as acrylate monomers, on the surface of a material. This approach has been used advantageously to flame retard polymeric substrate using fluorinated acrylate27 and textiles using phosphorus-based acrylates.28,29
Intumescent coating is well known to provide efficient protection to metallic structure against fire.30,31 This approach has been recently used on polymeric substrate and it has been found large improvement at various flammability test.32 Nevertheless, specific surface treatment by cold plasma must apply prior to making the deposit (intumescent paint). It permits the good adhesion of the intumescent coating upon heating and it avoids its delamination. The mode of action is similar as that of intumescent paint on metallic part, namely the formation of heat barrier limiting mass and heat transfer from the heat source and/or the flame to the substrate.
Flame retardancy of PLA: the state of the art
A rapid survey of the literature (open literature and patent) using scientific database such as Web of Science, Scifinder or Scopus with the appropriate keywords show there are only few papers devoted to the flame retardancy of PLA. The reason is that the first applications of PLA were designed for disposable materials (e.g. packaging) or semi-durable materials (e.g. textile) and there was no really need for flame retardancy. Now the market turns to more durable materials and as it was mentioned in the introduction, it is expected to substitute thermoplastics like PP, PA or PET by PLA in sectors such as transportation and E&E. In those sectors, fire hazard is an issue and flame retardancy is required. In this section we will then review solution and approach used for flame retarding PLA.(a) Blending approach
The blending of high thermally stable polymer with low thermally stable polymer increases the temperature of degradation of the low thermally stable polymer. Based on this consideration, some authors have tried to extrapolate these results to flame retardancy. In the case of PLA, it has been blended with polycarbonate (PC) which is a char forming polymer leading to an intumescent behavior upon heating. Kimura and Horikoshi,7 Nodera and Hayata33 and Iga et al.34 showed limited improvement of the fire performance with virgin PC. To get acceptable performance, they used PC containing silicone and V-2 rating at the UL-94 test was achieved (vertical flammability test where the ranking is V-0, V-1, V-2 and non classified; V-0 is the best classification – see ASTM D3801-06 Standard Test Method for Measuring the Comparative Burning Characteristics of Solid Plastics in a Vertical Position).7 The mechanism of action was not examined in the papers and they report charring acting as a protective barrier. The purpose of this approach was to make phosphorus-free and halogen-free materials for use in notebook computer housings.(b) Use of conventional flame retardant
The market of the flame retardant (FR) is international and is mainly constituted by ATH, MDH, phosphorus-containing compounds, halogenated compounds, antimony trioxide and nitrogenated compounds which act in gas and/or condensed phase.9 Those conventional FRs were also evaluated in PLA. Nishida et al.35 used ATH to make it flame resistant and also to permit its depolymerization at high temperature (300 °C). In this particular case, they used the depolymerization for recycling purposes. In this work, they considered both the application of PLA for durable use and its recycling. No comment was given about the mechanism of action and the performance of the flame retarded PLA but we can reasonably assume that the fire behavior of the material is similar to typical thermoplastics flame retarded with ATH. Flame retardancy is due to physical effects requiring relatively large amounts of ATH (50–65 wt%). The activity of ATH consists of:15 (a) dilution of the polymer in the condensed phase; (b) decreasing the amount of available fuel; (c) increasing the amount of thermal energy needed to raise the temperature of the composition to the pyrolysis level, due to the high heat capacity of the fillers; (d) enthalpy of decomposition – emission of water vapor; (e) dilution of gaseous phase by water vapor –decrease of amount of fuel and oxygen in the flame; (f) possible endothermic interactions between the water and decomposition products in the flame; (g) decrease of feedback energy to the pyrolysing polymer; (h) insulative effect of the oxides (Al2O3) remaining in the char; (i) charring of the materials. More recently, Yanagisawa et al.36 investigated the flame retardancy of PLA using ATH at high loading (50 wt%) combined with phenolic resins. The application was housings of electronic products where the required level of flame retardancy is high. It is reported that the mechanism of action was mainly due to the formation of homogeneous char layers produced by the phenol resins on the surface of the composite upon heating. It permitted to reduce the loading of ATH. The mechanism was not completely described by the authors but we suspect that phenolic resin should provide high char yield which could be reinforced by the formation of alumina from ATH. This ceramified char becomes then protective enough to achieve good FR performance.Textile is a major application for PLA and it is also required to achieve flame retardancy. The flame retardancy of PLA fabrics were investigated by Kubokawa et al.37–39 using conventional FR treatments measuring the limiting oxygen index (LOI measured according to the ASTM Standard Method D2863-97). Untreated PLA fabric was measured at 24 vol% and when treated with several bromine-containing flame retardants and a phosphorus-containing flame retardant, triphenylphosphate (TPP), LOI increases slightly to 28 vol% with TPP and to 26 vol% with the bromine-containing FR (tetrabromobisphenol A or TBP-A which was revealed to be the most efficient). It was found that the thermal stability of the FR fabrics was not improved by the FR treatment at lower temperatures but it became much higher above 375 °C. They also noticed the exothermal reactions were retarded upon heating in air associated to the formation of a higher char yield. According to the explanations of the authors, we suspect that FR treatment promotes charring suggesting a mechanism of action in the condensed phase. Note that in the case of the brominated treatment action in the gaseous phase should also occur.
In our group, we have investigated phosphinate-based FRs designed for polyamides and polyesters (Exolit OP1230 and OP1311 of Clariant, Germany) compared to ‘classical’ FRs such as ammonium polyphosphate (APP) and melamine polyphosphate (MP200).18 LOI of the flame retarded PLA increases as a function of FR loading and jumps from 21 vol% to 28 vol% (PLA/OP1230; OP1230 is an aluminium phosphinate) until 34 vol% (PLA/APP) (Fig. 1-a). In addition to this, all formulations exhibit V-2 rating at the UL-94 test (3.2 mm) from 10 wt% loading and V-0 rating (3.2 mm) is achieved for 20 wt% of MP200 and 25 wt% of OP1311 (OP1311 is the commercial combination of OP1230 and MP200 in the weight ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The peak of heat release rate (PHRR) determined by cone calorimetry is only decreased by 33% for MP200 while for the other FRs the reduction is poor (Fig. 1-b). The ignition time is only improved using MP200 and APP (100 s vs. 50 s). Those results show that the FR performance remains relatively poor in PLA. Visually it is observed that PLA and FR PLA drip easily permitting to reach high LOI but only UL-94 V-2 rating. The poor reduction of PHRR was assigned to the low viscosity of the FR PLA upon heating. The charred material formed by reaction between the additives and the polymer is not viscous enough to be expanded by the fuel consisting of the degradation gases and then to make a protective coating.
:1). The peak of heat release rate (PHRR) determined by cone calorimetry is only decreased by 33% for MP200 while for the other FRs the reduction is poor (Fig. 1-b). The ignition time is only improved using MP200 and APP (100 s vs. 50 s). Those results show that the FR performance remains relatively poor in PLA. Visually it is observed that PLA and FR PLA drip easily permitting to reach high LOI but only UL-94 V-2 rating. The poor reduction of PHRR was assigned to the low viscosity of the FR PLA upon heating. The charred material formed by reaction between the additives and the polymer is not viscous enough to be expanded by the fuel consisting of the degradation gases and then to make a protective coating.
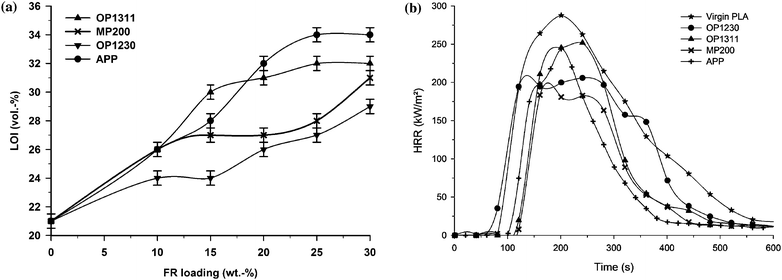 | ||
| Fig. 1 LOI as a function of FR amount in PLA (a) and HRR (heat release rate) as a function of time of flame retarded PLA at 30 wt% FR loading (external heat flux = 35 kW m−2) (b) (from ref. 18). | ||
(c) Use of nanoparticles
Of particular interest is the developed nanocomposite technology consisting of a polymer and nanoparticles because they often exhibit remarkably improved mechanical and various other properties as compared with those of virgin polymer at loading as low as 3–5 wt%.40 In PLA nanocomposites it was reported that this family of composites exhibits improved properties including a high storage modulus both in the solid and melt states, an increased flexural properties, a decrease in gas permeability, an increased heat distortion temperature, an increase in the rate of biodegradability of pure PLA, etc…41Adding a small amount of clay has been shown to reduce PHRR for numerous polymer nanocomposites42–44 including PLA.45Fig. 2 shows significant reduction in PHRR in PLA when dispersing at the nanoscale 3 wt% of montmorillonite (MMT) platelets. PHRR is decreased by 40%. The suggested mechanism by which clay nanocomposites function involves the formation of a char that serves as a potential barrier to both mass and energy transport. It is reported that a smooth char layer is formed at the surface of the nanocomposite leaving a black char residue at the end of the experiment. In the case of pure PLA, vigorous bubbling is observed when the polymer is burning and there is no residue left at the end of the experiment.
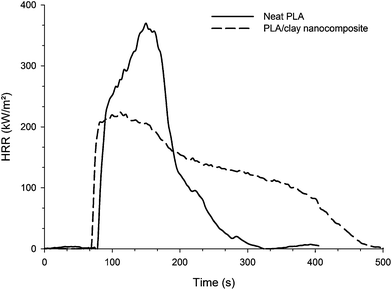 | ||
| Fig. 2 HRR as a function of time of pure PLA and PLA/clay nanocomposite (external heat flux = 35 kW m−2) (fromref. 45). | ||
Similar results have been observed with fabrics made of PLA knitted multifilaments.46 4 wt% of organomodified clay were incorporated in PLA which were meltspun afterward. The authors reports that a plasticizer must be added to be able to meltspin PLA/clay. The knitted fabric made with the PLA/clay nanocomposite exhibits 38% reduction in PHRR compared to the virgin one. Similar mechanism of action is suggested.
In addition to organomodified clay, multi-walled carbon nanotubes (MWNT) are also known to significantly improve the flame retardancy of polymers when MWNT is nanodispersed in the polymeric matrix.18,47,48 PLA/MWNT was evaluated in terms of flame retardancy but very limited enhancement was observed during cone calorimetry because of the bad dispersion of MWNT in PLA (5% reduction of PHRR).45 Nevertheless, the flame spread of PLA/MWNT compared to virgin PLA is much lower when testing PLA strip in vertical position (Fig. 3).49
 | ||
| Fig. 3 Flammability test of virgin PLA and PLA/MWNT at the ignition (a) and after 15 s (b) (from ref. 49). | ||
The issue was then to evenly disperse MWNT in the polymeric matrix to get the best flame retardancy properties.50 The method was to chemically functionalize the carbon nanotubes to enhance the interfacial adhesion between the nanotubes and the polymer.51 As melamine-based compounds are known to be flame retardants, the strategy was to graft melamine onto the surface of nanotube. MWNT and the functionalized MWNT (f-MWNT) were incorporated in PLA via melt blending. Transmission electron microscopy (TEM) reveals that a high level of nanodispersion is achieved in PLA with f-MWNT while the dispersion is poor with virgin MWNT (Fig. 4). Hence the PLA nanocomposite containing f-MWNT exhibits significant reduction of PHRR (28% reduction of PHRR) (Fig. 5). It is reported that in the final residues, the char of PLA/f-MWNT is relatively compact and cohesive while that with MWNT exhibits islands of char without any cohesion. The mechanism of action is the formation of a charred layer limiting mass transfer. The cohesion of the layer formed from PLA/f-MWNT permits to reach higher performance.
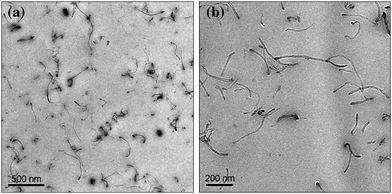 | ||
| Fig. 4 TEM pictures of PLA/f-MWNT at low magnification (a) and at high magnification (b). | ||
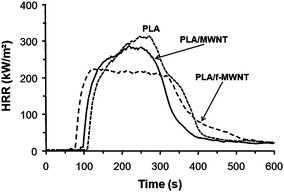 | ||
| Fig. 5 HRR curves as a function of time of PLA/MWNT and PLA/f-MWNT compared to virgin PLA (external heat flux = 35 kW m−2). | ||
Clay is a two dimensional plate like filler and graphite has also the similar planar structure with several characteristics.52 If graphite can be dispersed in polymeric matrix in the nanosize range, it can be expected it could improve overall properties of the system as clay does.53,54 In addition, it can provide its own characteristic properties of thermal and electrical conductivity as well. Graphite has already shown great potential when incorporated in polymers.55 Expanded graphite (EG) was incorporated at different loading into PLA via melt blending and the resulting materials were evaluated measuring the burning rate in horizontal position (Table 1).56 PLA burns easily with drips and without charring while PLA/EG nanocomposites burns with a decreasing number of drops when increasing the EG loading until to reach 0 drop at 6 wt% and a limited burning rate at 12 wt%.
| Entry | Composition (%, by weight) | Mean burning rate, mm min−1 | Classificationa | Remarks |
|---|---|---|---|---|
| a Thickness of specimens ∼3.1mm; a burning rate less than 40 mm min−1 allows the classification HB. | ||||
| 1 | PLA | 20 (±1) | HB | Burning with drips |
| 2 | PLA-3% EG | 21 (±2) | HB | Burning with drips, low charring |
| 3 | PLA-6% EG | 25 (±1) | HB | Burning without drips, charring |
| 3 | PLA-12% EG | 18 (±1) | HB | Burning without drips, charring |
In typical cone calorimetry experiment done at an external heat flux of 35 kW m−2, PHRR is decreased by 30% in PLA containing 6% EG compared to virgin PLA. During burning, the PLA/EG swells to form a porous foamed carbonaceous mass that acts as a barrier to limit the combustion (Fig. 6).
 | ||
| Fig. 6 The cone calorimeter residue of PLA/6%EG (external heat flux = 35 kW m−2) (from ref. 56). | ||
The high amount of char formed during burning (intumescence behavior) is assigned to additional expansion of graphite nanolayers provoked by both the heat and gases/products of combustion. It was evidenced by TEM observations on the residue after burning in which images revealed the presence of exfoliated graphite nanolayers of different dimension (Fig. 7).
 | ||
| Fig. 7 A TEM picture of cone calorimeter residue of PLA/EG (external heat flux = 35 kW m−2) (fromref. 56). | ||
(d) Intumescence
The word “intumescence” comes from Latin “intumescere” which means “to swell up”. It describes the behavior of an intumescent material well. When heating beyond a critical temperature, it begins to swell and then to expand. The result of this process is a foamed cellular charred layer on the surface which protects the underlying material from the action of the heat flux or the flame.13,57 This concept was applied for flame retarding polyolefins, polyamides and polyesters and was proven to be efficient for those polymers.Typically the ingredients of intumescence are mainly composed of an inorganic acid or a material yielding acidic species upon heating, of a char former and of a component that decomposes at the right temperature and at the right time to enable the blowing of the system. The compounds used as an acid source are acids, ammonium salts, and phosphates. Char formers are mainly hydroxyl-containing compounds, such as polyols and starch, and blowing agents are compounds which can evolve gases upon heating, in the temperature range corresponding to the development of the intumescence process. Melamine, urea, and urea–formaldehyde resins are typical examples of blowing agents. A typical example of intumescent system is the combination of ammonium polyphosphate (APP) with pentaerythritol (PER) where APP plays the role of both acid source and blowing agent (evolution of ammonia during the degradation of APP) and PER is the char former.58 As PER comes from petrochemistry, Reti et al. investigated the substitution of PER by lignin (LIG) or starch (ST) issue from renewable resources in the APP/char former system.59 The basic idea was to make an intumescent PLA with the highest percentage of bio-based ingredients. At 40 wt% loading of APP/char former (PER, LIG or ST), high LOI values are observed (up to 58 vol% with PER) and in all cases an intumescent protective coating was formed at the surface of the material. The interesting feature of the ‘bio-based systems’ are V-0 classification at the UL-94 test (3.2 mm) while PLA-APP/PER is only V-2 rated. By cone calorimetry (external heat flux of 35 kW m−2), those systems exhibits a reduction of PHRR by 65% for APP/PER and by 40% for APP/LIG and APP/ST. The mode of action is the formation of an intumescent layer protecting the underlying material limiting heat and mass transfer. The formulations were then optimized by an experimental design in order to have the highest amount of renewable resources (LIG and ST) while keeping acceptable fire performance (LOI higher than 30 vol% and V-0 rating at the UL-94 test). The authors showed that the amount of APP can be decreased at 12 wt% in APP/ST and at 28 wt% in APP/LIG (total loading was kept constant at 40 wt%). A promising application of this was to make PLA-APP/ST and PLA-APP/LIG films (150 μm thick) to be applied to nonwovens.60 The resulting materials exhibit superior flame retardancy performance at horizontal and vertical flame spread tests as well as by cone calorimetry (Fig. 8).
 | ||
| Fig. 8 Horizontal spread test according to FMVSS of virgin hemp nonwoven, virgin PLA film applied on hemp nonwoven and intumescent PLA film applied on hemp nonwoven and intumescent (from the left to the right respectively). | ||
Fibers can be incorporated in polymers in order to reinforce them and in particular in terms of mechanical properties. Shumao et al.61 used natural fibers (ramie fibers) combined with APP and they made an intumescent system for PLA. At 40 wt% loading LOI reaches 35 vol% and the formulation exhibits V-0 rating. Thermal analysis and visual observation show that the char yield of the intumescent PLA is greatly enhanced compared to the virgin polymer. The authors also report an intumescent behavior of the material. They suggest that APP releases phosphoric acid, polyphosphoric acid and non-flammable gases when exposed to flame; the resulting acid contributes to intramolecular or intermolecular ramie dehydration, then dehydrogenizing, charring and rupture of chemical bonds. Although it was not formally evidenced this mechanism is probably true because it is the typical intumescent mechanism.
An intumescent flame retardant (hereafter called SPDPM) was synthesized from spirocyclic pentaerythritol bisphosphorate disphosphoryl chloride (SPDPC) and melamine by Zhan et al.62 (Fig. 9). SPDPM was evaluated at different loadings in PLA. LOI reaches 38 vol% at 25 wt% loading and it is V-0 rated. At lower loading, the intumescent PLA is no longer V-0 rated (V-2 at 5 and 15 wt%) but LOI values are still quite high (28 and 33 vol% at 5 and 15 wt%, respectively). Visual observation shows an intumescent behavior with a high amount of char on the top of the material. The fire performance is confirmed by microscale combustion calorimetry (MCC) where PHRR is decreased by 33% at 25 wt% loading. It is reported that the flame retardant mechanism of SPDPM in PLA was not only due to the intumescent protective charred layer but also for the change of the degradation process of PLA. It makes sense because PHRR in the MCC experiment is significantly decreased, indicating that the degradation pathway of the intumescent PLA is modified compared to the virgin PLA (in a typical MCC experiment only few milligrams of sample are used heat and mass transfer in an intumescent system are not predominant and evolving gases entering into the combustor and burning with lower energy evidence different degradation pathway).
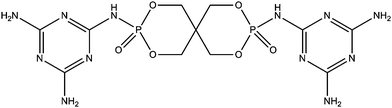 | ||
| Fig. 9 An intumescent flame retardant (SPDPM) synthesized from spirocyclic pentaerythritol bisphosphorate disphosphoryl chloride and melamine. | ||
(e) Combination of fillers with nanoparticles
Polymer nanocomposite exhibits low flammability in terms of cone calorimetry but it fails to other tests, in particular those with samples in vertical position (e.g. LOI, UL-94). The mechanism of protection involves the formation of char layer covering the entire sample surface acting as insulative barrier and reducing volatiles escaping to the flame. The formation of such a layer which does not form cracks when burning is critical to obtain low heat release rate from nanocomposites.9 It is the reason why, nanoparticles have been combined with conventional flame retardants in order to create a synergistic effect.In the section above, we have shown that intumescence is a method to limit the flammability of PLA. Fontaine and Bourbigot63 reported the combination of APP with melamine (MEL) in an appropriate ratio (5 to 1 respectively) as efficient intumescent for PLA at a loading as low as 10 wt%. The incorporation of an additional nanofiller such as organomodified montmorillonite (C30B) or multiwall carbon nanotubes (MWNT) shows large effects on reaction to fire of the materials. A large synergistic effect is observed using C30B (Fig. 10-a) while it is antagonistic using MWNT (Fig. 10-b). It can be observed that the intumescent PLA combined with C30B does not burn and the small PHRR is only due at a very short ignition of the material. At 30 wt% extremely high LOI values were measured (up to 52 vol%) and V-0 rating (3.2 mm) was achieved. This jump of performance was partially explained by the increase of the thermal stability of the formulation with C30B compared to that without. It might be justified according to previous works of our group64,65 that C30B could react with APP to form alumino- and silicophosphate stabilizing the intumescent structure at high temperature. On the contrary, the incorporation of MWNT leads to an antagonistic effect which was not explained in the paper. According to the expertise of our group, it might be assigned to the accumulation of MWNT in the intumescent char leading to the percolation threshold. If it is the case, the apparent heat conductivity increases a lot and does not permit the intumescent coating to play its role of heat barrier.
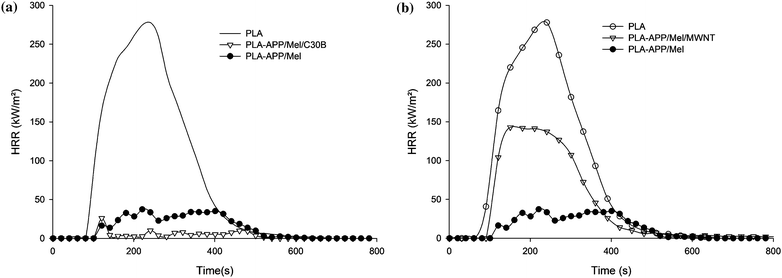 | ||
| Fig. 10 Heat release rate (RHR) curves as a function of time of PLA/APP/Mel/Nanoparticles (C30B (a) or MWNT (b)) loaded at 30 wt% compared to virgin PLA. | ||
The intumescent PLA was also evaluated with a total loading of 10 wt%. This loading is still sufficient to get relatively high LOI (33 vol%) and a synergistic effect is observed with C30B (35 vol%) while LOI falls down when MWNT is incorporated in the formulation (24 vol%). It is noteworthy that V-0 at UL-94 is achieved for the formulations with and without C30B. As a conclusion, this kind of intumescent formulation offers an interesting way for designing flame retarded PLA.
Starting from gypsum, a by-product of lactic acid fabrication process, PLA composites were prepared by melt blending. The novelty of this approach is based on the selection of gypsum. It was made after a previous specific dehydration to obtain anhydrite II (AII) because it was shown that it permitted the preservation of PLA molecular weights and was favorable key properties for the final composite products (the AI form makes the contrary).66 In order to obtain PLA-AII composites characterized by specific end-use flame retardant properties, the addition of selected organo-modified layered silicates (OMLS) was considered.67 Co-addition of AII and OMLS leads to PLA nanocomposites characterized by good (nano)filler dispersion, thermal stability and adequate mechanical resistance. The flame retardant properties as shown by cone calorimetry showed significant increase in the ignition time compared to neat PLA and a substantial decrease, i.e., ca. 40%, of PHRR, whereas the UL94 HB test was successfully passed revealing non-dripping effect and extensive char formation.
Discussion
Thermogravimetric analysis shows that PLA degrades in a main apparent step taking place between 300 and 400 °C (highest degradation rate is observed at 350 °C in pyrolysis conditions at 10 °C min−1 as heating ramp) (Fig. 11).68 Using infrared spectroscopy, gaseous products of cyclic oligomers, lactide, acetaldehyde, carbon monoxide and carbon dioxide are detected which was attributed to the thermal degradation of PLA based upon a hydroxyl end-initiated ester interchange process and chain homolysis. Acetaldehyde and carbon dioxide exist in the products until the end of the experiments, whereas carbon monoxide gradually decreases above the peak temperature.68,69 In the condensed phase, char yield is quite low (2 wt%) and aromatic-based materials were identified.68 Based on those measurements, flame retardants should then act before the degradation of PLA (T < 300 °C) and/or modify the degradation pathway of PLA to yield char and to avoid (or at least to decrease) the evolution of flammable products (acetaldehyde, lactide, methane etc.).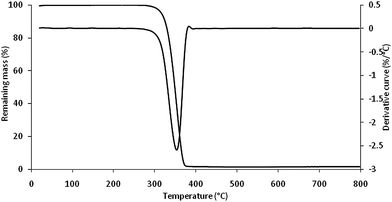 | ||
| Fig. 11 Thermogravimetry curve of PLA in pyrolysis conditions (N2 flow, 10 °C min−1) (from ref. 68). | ||
According to the literature on flame retardancy (see e.g.ref. 9 and references therein), the methods to flame retard PLA reviewed in the previous section suggest in all cases that the mechanisms of action takes place via the condensed phase. The exception might be the mode of action of phosphinates which can act both in gaseous and condensed phases as shown by Braun et al. in polyamide-6,6.70 It has not been elucidated for PLA yet. The incorporation of nanoparticles in PLA probably involves a classical mechanism in which the formation of physical barrier formed by the accumulation of particles at the surface and promotion of charring provides reduction of HRR limiting mass transfer of flammable gases to the flame. Some authors reported the modification of the degradation pathway of the polymer because of the presence of the nanoparticles reinforcing their action.71 It is not clear in the case of PLA, but recent work of our group using pyrolysis combustion flow calorimetry (PCFC also called MCC) suggest that the chemical degradation pathway could be modified by the incorporation of MWNT perfectly dispersed in PLA (functionalized MWNT or f-MWNT as described in the previous section). Our assumption is based on the particular design of PCFC in which 2 mg of sample is put in a pan and undergoes its thermal degradation at high heating rate like in TGA and where the evolving gases are then burnt in a combustor measuring HRR. In such a design, we do not believe that an efficient physical barrier could be formed and the modification of HRR can only be due to the chemical changes of the evolving gases. If it is so, we should admit that the degradation pathway of the material is modified. HRR of PLA/f-MWNT is decreased by 25% compared to virgin PLA suggesting that the degradation pathway of PLA is changed when containing MWNT (Fig. 12). No indication is given on the chemical nature of the evolving products and it should be investigated further. Similar results are observed when incorporating different loading of expanded graphite in PLA (unpublished results); HRR measured with the PCFC is decreased by 20% when 9 wt% EG is incorporated in PLA. No data is available for organomodified clay but we may reasonably assume that the incorporation of nanoparticles in PLA can modify the degradation pathway of the materials. According to the results it is beneficial for flame retardancy but the main mode of action remains the formation of a charred physical barrier at the surface of the material.
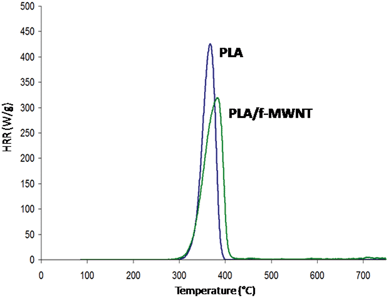 | ||
| Fig. 12 HRR curves from PCFC experiments for PLA and PLA/f-MWNT (unpublished results). | ||
The incorporation of intumescent formulation in PLA involves a mechanism in the condensed phase leading to the formation of carbonaceous layer acting mainly as heat barrier. Reti investigates the action of the intumescent system APP/ST in PLA in detail and she found a mechanism similar to that described in the first section of this review.59,68 Besides the gaseous phase, PLA-APP/ST compared to PLA is not significantly modified except for the detection of a small amount of ammonia because of the degradation of APP. In the case of PLA-APP/Mel-C30B, the action of the synergist is to reinforce the char and to improve the thermal stability of the material. Recently, we have observed that the intumescent PLA exhibits a reduction of HRR of 25% in PCFC experiment (Fig. 13) as SPDPM did in similar experiment (see the previous section). It suggests that the gaseous phase is modified probably because of the chemical reactions involving the intumescent additives and because of the formation of more stable material. Nevertheless it is not the main mode of action as mentioned above, because in a typical cone calorimetry experiment, PHRR is decreased by 90% by the formation of an expanded charred layer.
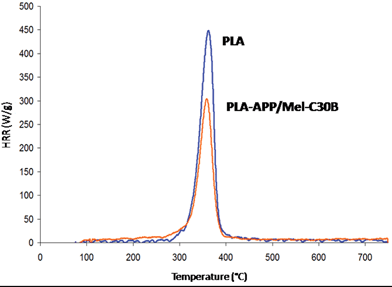 | ||
| Fig. 13 HRR curves from PCFC experiments for PLA and PLA-APP/Mel-C30B (unpublished results). | ||
As a conclusion of this discussion, we have shown that the mechanism of action remains similar to those observed in other polymers when incorporating the same kind of additives. Nevertheless, no comprehensive study has been published to investigate the specific behavior of the thermal and fire degradation of flame retarded PLA. It should be done in the near future in order to develop more appropriate methods to flame retard PLA.
Conclusion
This paper has reviewed the flame retardancy of PLA. After reminding the reader of the basics of flame retardancy for polymeric materials, an overview of the methods providing flame retardancy to PLA was commented. Based on this, the main conclusion of this work is the same approaches as for other polymers have been applied to PLA. Very high performance can be reached in certain cases (e.g. intumescence). The patent literature is increasing because PLA was mainly used for non durable applications but it is turning to durable applications. Because of the growing market for PLA for durable applications where flame retardancy is required, we may expect more and more scientific papers exploring specifically the flame retardancy of PLA and its mechanisms.Acknowledgements
This work was supported by the European project Interreg IV ‘NANOLAC’ and by the North Region of France (Région Nord - Pas de Calais): they are gratefully acknowledged. The UpTex competitive cluster is also acknowledged for administrative assistance in this project.References
- L. Shen, J. Haufe and M. K. Patel, Product Overview and Market Projection of Emerging Bio-based Plastics, Copernicus Institute for Sustainable Development and Innovation, Utrecht University, Utrecht, 2009 Search PubMed.
- L. T. Lim, R. Auras and M. Rubino, Prog. Polym. Sci., 2008, 33, 820–852 CrossRef CAS.
- R. Auras, B. Harte and S. Selke, Macromol. Biosci., 2004, 4, 835–864 CrossRef CAS.
- B. Gupta, N. Revagade and J. Hilborn, Prog. Polym. Sci., 2007, 32, 455–482 CrossRef CAS.
- E. T. H. Vink, K. R. Rábago, D. A. Glassner, B. Springs, R. P. O'Connor, J. Kolstad and P. R. Gruber, Macromol. Biosci., 2004, 4, 551–564 CrossRef CAS.
- E. T. H. Vink, K. R. Rábago, D. A. Glassner and P. R. Gruber, Polym. Degrad. Stab., 2003, 80, 403–419 CrossRef CAS.
- K. Kimura and Y. Horikoshi, Fujitsu Sci. Tech. J., 2005, 41, 173–180 Search PubMed.
- S. Bourbigot, M. Le Bras and J. Troitzsch, in Flammability Handbook, ed. J. Troitzsch, Hanser Verlag, Munich, 2003, pp. 3–7 Search PubMed.
- S. Bourbigot and S. Duquesne, J. Mater. Chem., 2007, 17, 2283–2300 RSC.
- S. Bourbigot and X. Flambard, Fire Mater., 2002, 26, 155–168 CrossRef CAS.
- J. Choi, A. F. Yee and R. M. Laine, Macromolecules, 2003, 36, 5666–5682 CrossRef CAS.
- D. Q. Chen, Y. Z. Wang, X. P. Hu, D. Y. Wang, M. H. Qu and B. Yang, Polym. Degrad. Stab., 2005, 88, 349–356 CrossRef CAS.
- S. Bourbigot, M. Le Bras, S. Duquesne and M. Rochery, Macromol. Mater. Eng., 2004, 289, 499–511 CrossRef CAS.
- M. Jimenez, S. Duquesne and S. Bourbigot, Thermochim. Acta, 2006, 449, 16–26 CrossRef CAS.
- S. Bourbigot, M. Le Bras, R. Leeuwendal, K. K. Shen and D. Schubert, Polym. Degrad. Stab., 1999, 64, 419–425 CrossRef CAS.
- M. Lewin, Polym. Degrad. Stab., 2005, 88, 13–19 CrossRef CAS.
- S. Bourbigot, M. Le Bras, P. Bréant, J. M. Trémillon and R. Delobel, Fire Mater., 1996, 20, 145–154 CrossRef CAS.
- S. Bourbigot, S. Duquesne, G. Fontaine, S. Bellayer, T. Turf and F. Samyn, Mol. Cryst. Liq. Cryst., 2008, 486, 325–339 CrossRef CAS.
- A. Vannier, S. Duquesne, S. Bourbigot, A. Castrovinci, G. Camino and R. Delobel, Polym. Degrad. Stab., 2008, 93, 818–826 CrossRef CAS.
- M. Jimenez, S. Duquesne and S. Bourbigot, Ind. Eng. Chem. Res., 2006, 45, 7475–7481 CrossRef CAS.
- S. Bourbigot, C. Jama, M. Le Bras, R. Delobel, O. Dessaux and P. Goudmand, Polym. Degrad. Stab., 1999, 66, 153–155 CrossRef CAS.
- A. R. Horrocks, M. Y. Wang, M. E. Hall, F. Sunmonu and J. S. Pearson, Polym. Int., 2000, 49, 1079–1091 CrossRef CAS.
- J. H. Koo, W. Wootan, W. K. Chow, H. W. Au Yeung and S. Venumbaka, 2001, vol. 797, pp. pp. 361–374.
- A. Quédé, J. Cardoso, M. Le Bras, R. Delobel, P. Goudmand, O. Dessaux and C. Jama, J. Mater. Sci., 2002, 37, 1395–1399 CrossRef CAS.
- A. Quédé, C. Jama, P. Supiot, M. Le Bras, R. Delobel, O. Dessaux and P. Goudmand, Surf. Coat. Technol., 2002, 151–152, 424–428 CrossRef CAS.
- I. Errifai, C. Jama, R. Delobel, R. De Jaeger and A. Mazzah, Mol. Cryst. Liq. Cryst., 2008, 486, 316–324 CrossRef CAS.
- I. Errifai, C. Jama, M. Le Bras, R. Delobel, L. Gengembre, A. Mazzah and R. De Jaeger, Surf. Coat. Technol., 2004, 180–181, 297–301 CrossRef CAS.
- M. J. Tsafack and J. Levalois-Grützmacher, Surf. Coat. Technol., 2006, 201, 2599–2610 CrossRef CAS.
- M. J. Tsafack and J. Levalois-Grützmacher, Surf. Coat. Technol., 2006, 200, 3503–3510 CrossRef CAS.
- M. Jimenez, S. Duquesne and S. Bourbigot, Surf. Coat. Technol., 2006, 201, 979–987 CrossRef CAS.
- M. Jimenez, S. Duquesne and S. Bourbigot, Ind. Eng. Chem. Res., 2006, 45, 4500–4508 CrossRef CAS.
- S. Duquesne, N. Renaut, P. Bardollet, C. Jama, M. Traisnel and R. Delobel, in Fire and Polymers V - Materials and Concepts for Fire Retardancy, ed. C. A. Wilkie, A. B. Morgan and G. L. Nelson, American Chemical Society, Washington, DC, 2009, ch. 12, pp. 192–204 Search PubMed.
- A. Nodera and Y. Hayata, Compatibilization and Characteristics of PC/PLA alloy, Nagoya, 2006 Search PubMed.
- K. Iga, T. Ueno, K. Mizuno, T. Ishikawa and K. Takeda, Kobunshi Ronbunshu, 2007, 64, 561–567 CrossRef CAS.
- H. Nishida, Y. Fan, T. Mori, N. Oyagi, Y. Shirai and T. Endo, Ind. Eng. Chem. Res., 2005, 44, 1433–1437 CrossRef CAS.
- T. Yanagisawa, Y. Kiuchi and M. Iji, Kobunshi Ronbunshu, 2009, 66, 49–54 CrossRef CAS.
- H. Kubokawa and T. Hatakeyama, Sen'i Gakkaishi, 1999, 55, 349–355 Search PubMed.
- H. Kubokawa, M. Ohta and T. Hatakeyama, Sen'i Gakkaishi, 1999, 55, 290–297 Search PubMed.
- H. Kubokawa and H. Komatsu, Sen'i Gakkaishi, 2000, 56, 497–500 Search PubMed.
- M. Alexandre and P. Dubois, Mater. Sci. Eng., R, 2000, 28, 1–63 CrossRef.
- S. Sinha Ray and M. Okamoto, Macromol. Rapid Commun., 2003, 24, 815–840 CrossRef CAS.
- J. W. Gilman, C. L. Jackson, A. B. Morgan, R. Harris Jr, E. Manias, E. P. Giannelis, M. Wuthenow, D. Hilton and S. H. Phillips, Chem. Mater., 2000, 12, 1866–1873 CrossRef CAS.
- S. Bourbigot, D. L. Vanderhart, J. W. Gilman, S. Bellayer, H. Stretz and D. R. Paul, Polymer, 2004, 45, 7627–7638 CrossRef CAS.
- X. Zheng and C. A. Wilkie, Polym. Degrad. Stab., 2003, 82, 441–450 CrossRef CAS.
- S. Bourbigot, G. Fontaine, S. Duquesne and R. Delobel, Int. J. Nanotechnol., 2008, 5, 683–692 Search PubMed.
- S. Solarski, F. Mahjoubi, M. Ferreira, E. Devaux, P. Bachelet, S. Bourbigot, R. Delobel, P. Coszach, M. Murariu, A. Da Silva Ferreira, M. Alexandre, P. Degee and P. Dubois, J. Mater. Sci., 2007, 42, 5105–5117 CrossRef CAS.
- T. Kashiwagi, E. Grulke, J. Hilding, R. Harris, W. Awad and J. Douglas, Macromol. Rapid Commun., 2002, 23, 761–765 CrossRef CAS.
- T. Kashiwagi, F. Du, J. F. Douglas, K. I. Winey, R. H. Harris Jr and J. R. Shields, Nat. Mater., 2005, 4, 928–933 CrossRef CAS.
- S. Bourbigot, F. Fontaine and A. Gallos, Polym. Adv. Technol., 2010 DOI:10.1002/pat.1715.
- S. Bourbigot, S. Duquesne and C. Jama, Macromol. Symp., 2006, 233, 180–190 CrossRef CAS.
- S. Bourbigot, G. Fontaine, A. Gallos, C. Gérard and S. Bellayer, in Fire and Polymers V - Materials and Concepts for Fire Retardancy ed. C. A. Wilkie, A. B. Morgan and G. L. Nelson, American Chemical Society, Washington, DC, 2009, vol. 1013, pp. 25–34 Search PubMed.
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132–145 CrossRef CAS.
- S. Wang, M. Tambraparni, J. Qiu, J. Tipton and D. Dean, Macromolecules, 2009, 42, 5251–5255 CrossRef CAS.
- P. Steurer, R. Wissert, R. Thomann and R. Mülhaupt, Macromol. Rapid Commun., 2009, 30, 316–327 CrossRef CAS.
- J. J. George and A. K. Bhowmick, J. Mater. Sci., 2008, 43, 702–708 CrossRef CAS.
- M. Murariu, A. L. Dechief, L. Bonnaud, Y. Paint, A. Gallos, G. Fontaine, S. Bourbigot and P. Dubois, Polym. Degrad. Stab., 2010, 95(5), 889–900 CrossRef CAS.
- H. J. Vandersall, J. Fire Flammability, 1971, 2, 97–140 Search PubMed.
- S. Bourbigot, M. Le Bras and R. Delobel, Carbon, 1993, 31, 1219–1230 CrossRef CAS.
- C. Réti, M. Casetta, S. Duquesne, S. Bourbigot and R. Delobel, Polym. Adv. Technol., 2008, 19, 628–635 CrossRef CAS.
- C. Reti, M. Casetta, S. Duquesne, R. Delobel, J. Soulestin and S. Bourbigot, J. Eng. Fibers Fabr., 2009, 4, 33–39 Search PubMed.
- L. Shumao, R. Jie, Y. Hua, Y. Tao and Y. Weizhong, Polymer Int., 2010, 59, 242–248.
- J. Zhan, L. Song, S. Nie and Y. Hu, Polym. Degrad. Stab., 2009, 94, 291–296 CrossRef CAS.
- G. Fontaine and S. Bourbigot, J. Appl. Polym. Sci., 2009, 113, 3860–3865 CrossRef CAS.
- S. Bourbigot, M. Le Bras, R. Delobel, R. Decressain and J. P. Amoureux, J. Chem. Soc., Faraday Trans., 1996, 92, 149–158 RSC.
- S. Bourbigot, M. Le Bras, F. Dabrowski, J. W. Gilman and T. Kashiwagi, Fire Mater., 2000, 24, 201–208 CrossRef CAS.
- M. Murariu, A. Da Silva Ferreira, L. Bonnaud and P. Dubois, Compos. Interfaces, 2009, 16, 65–84 Search PubMed.
- M. Murariu, L. Bonnaud, P. Yoann, G. Fontaine, S. Bourbigot and P. Dubois, Polym. Degrad. Stab., 2010, 95(3), 374–381 CrossRef CAS.
- C. Reti, PhD Thesis, Université des Sciences et Technologies, University of Lille, France, 2009.
- H. Zou, C. Yi, L. Wang, H. Liu and W. Xu, J. Therm. Anal. Calorim., 2009, 97, 929–935 CrossRef CAS.
- U. Braun, B. Schartel, M. A. Fichera and C. Jäger, Polym. Degrad. Stab., 2007, 92, 1528–1545 CrossRef CAS.
- B. N. Jang and C. A. Wilkie, Polymer, 2005, 46, 2933–2942 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |