Electroactive polymers for neural interfaces
Maria
Asplund
*a,
Tobias
Nyberg
a and
Olle
Inganäs
b
aSchool of Technology and Health, Royal Institute of Technology, Alfred Nobels Allé 10, SE-14152, Huddinge, Sweden. E-mail: maria.asplund@sth.kth.se; tobias.nyberg@sth.kth.se; Fax: +46 8 218368; Tel: +46 8 7904851
bBiomolecular and organic electronics, The Department of Physics Chemistry and Biology, Linköping University, Linköping, Sweden. E-mail: ois@ifm.liu.se
First published on 14th June 2010
Abstract
Development of electroactive conjugated polymers, for the purpose of recording and eliciting signals in the neural systems in humans, can be used to fashion the interfaces between the two signalling systems of electronics and neural systems. The design of desirable chemical, mechanical and electrical properties in the electroactive polymer electrodes, and the means of integration of these into biological systems, are here reviewed.
 Maria Asplund | Maria Asplund is a senior researcher at the Royal Institute of Technology, School of Technology and Health, Sweden. She received an MSc in Applied Physics and Electrical Engineering from Linköpings Universitet in 2003 and a PhD from the Royal Institute of Technology in 2009. Her main scientific interests include conducting polymer based neural interfaces and neural stimulation. |
 Tobias Nyberg | Tobias Nyberg received an MSc and a PhD in physics from Linköpings Universitet in 1997 and 2002 respectively. He has worked as a research scientist at the VTT Technical Research Centre of Finland before he carried out a postdoctoral scholarship at NTT basic Research Laboratories investigating neural stimulation using conducting polymer based electrodes. Since 2007 he is an assistant professor at the Royal Institute of Technology, School of Technology and Health. His main scientific interests include conducting polymer based neural interfaces and neural stimulation. |
 Olle Inganäs | Olle Inganäs is a professor of biomolecular and organic electronics, IFM, Linköpings Universitet, Sweden. He received an MSc in engineering physics from Chalmers University of Technology (1977), a BSc in philosophy and economics from Göteborg University (1978), and a PhD in applied physics at Linköping University (1984). He was appointed professor in 1999, and is presently director of a Center of Organic Electronics in Sweden. He has focused on studies of the class of conjugated polymers throughout areas of polymer physics, electrochemistry, electronics and optics. The uses of electronic polymers as interfaces to biological systems and organic photovoltaics are present topics of research. |
1. Introduction
Artificial communication with the nervous system is the objective for a genre of medical devices in the field of neuroprosthetics. Presently artificial hearing devices can be bought off the shelf in the shape of cochlear implants, and research is performed on brain machine interfaces and artificial vision. Even though this expansion of neuroprosthetic therapy into more fields of medicine is in progress, there still exist several weaknesses in current neuroprosthetic technology that complicates this development.The crucial part is the implanted electrode, translating signals between the electronic form in leads and ionic signals in tissue. This transition involves electrochemical processes which have to be carefully controlled not to be detrimental either to the electrode itself or to the surrounding tissue. Furthermore, in the absence of a rigid anatomical structure where the electrodes can be implanted, soft nervous tissue in general has to be in intimate contact with the electrode surface for the signal transition to be possible. The presence of the electrode implant evokes an immunological response aimed at removing the foreign body and if this fails, the response will be to encapsulate the implant in a cocoon of fibrous tissue. This reaction is a clear hindrance for the function of neural interfaces since these rely on close contact with neurons.
In the search for materials capable of meeting both the electrochemical and the biological requirements of the interface, conjugated polymers have emerged as a field of interest. Conjugated polymer coatings have hybrid charge transfer properties involving both electronic and ionic charge transports and are therefore highly effective for electron/ion transition over the solid/liquid boundary. Furthermore, the versatility of polymer materials when it comes to modifying, e.g. chemistry, microstructure and mechanical properties, widely exceeds that of a conventional metal or metal oxide. For a conjugated polymer, such properties can readily be altered still retaining its favourable electrochemical properties.
This review explores the possibilities conjugated polymers offer for improvement of the neural interface. The ambition is to present a comprehensive review on papers describing how conducting polymers (CPs) are used to solve problems within neural interfaces, and also to provide an elaborative background to the field. The involvement of both biological and electrochemical processes in the success or failure of the neural interface requires interdisciplinary efforts to solve a complex problem. Although the primary focus is neural interfaces, the importance of neighbouring fields such as drug delivery, biosensors and tissue engineering cannot be disregarded. Since development within these areas is more or less entwined, some papers not explicitly dealing with neural interfaces, but whose results have implications to this field, will also be included here.
From the perspective presented above it is near at hand to look at conjugated polymers simply as a coating material suitable for giving electrodes favourable electrochemical and biochemical properties. However, it is important to recognize that this new class of materials opens up for a wider range of solutions. The knowledge gained in other fields using conjugated polymers for optoelectronics, microactuators and printed circuitry can presumably be applied for the neural interface as well.
2. History of conducting polymers in biological systems
The use of conjugated polymers as interfaces to biological systems dates back to some early work on redox enzymes.1,2 In the 1980's Bull et al.3 reported that a common catalyst could be incorporated into polypyrrole (PPy) if present during electropolymerization. Encouraged by this finding, Umaña and Waller2 suggested that the same method could be applied using a biological catalyst, the enzyme glucose oxidase (GOx). Here the ambition was to use the electronic conductivity and malleability of polymers as a means to wire redox enzymes. GOx was incorporated as the counterion inside PPy, electrochemically generated from a solution of glucose oxidase, and electrical communication between the PPy matrix and the glucose oxidase was established, to allow measurements of glucose concentration in a solution contacted by the electrode. It was concluded that there was no adverse effect on the enzyme activity by the process, and that GOx was essentially entrapped in the formed material. Lifetime of the activity of the resulting film seemed to be governed by gradual leaking of GOx rather than enzyme denaturation.Successful incorporation of biological molecules was subsequently reported by many others, and some early examples are adenosine triphosphate (ATP),4 human serum albumin (HSA),5 heparin,6 anti-HSA7,8 and urease.9 The field of biosensors and analytical chemistry using conjugated polymers has experienced a considerable development during recent years.10–12
The possibility to use the polymer for electrically controllable delivery of incorporated ions is highly interesting for biomedical devices. Work on the controlled release of biologically relevant ions from PPy during redox was first published by Miller and Zinger,13 and later continued over the coming decades.14–16 This work has recently been taken up again,17,18 and also used to stimulate the neural systems in rodents, where release of glutamate by an ion pump helps to modify the neural response in the auditory system.19,20
Interfacing cells and conjugated polymers is a topic that has been developed by Langer et al.21,22 and Wallace23 over the last decades. The idea of using PPy versions as biomaterials emerged in the nineties.4,24,25 Williams and Doherty25 demonstrated the cytocompatibility of PPy in vitro and suggested that the conducting material could be used as a nerve guidance channel and as a material for carrying stimulation currents simultaneously.
Hodgson et al.24 showed that protein could be trapped within PPy thus forming a biofunctional and conducting material able to support cell growth. Many studies evaluating PPy/biomolecule materials for tissue engineering purposes would follow. For instance PPy with incorporated heparin was suggested as a suitable substrate for endothelial cells.23 Collier et al.26 incorporated hyaluronic acid (HA) into PPy material and showed that coated implants induced an increase in angiogenesis in tissue in comparison to controls.
In 2001, conducting polymers for the improvement of neural interfaces emerged as a field of interest.27,28 The same principles used to entrap biological molecules for biosensor and tissue engineering applications were proving their usefulness to this field. It was found that conducting polymer coatings on regular electrodes could be a route to immobilize important adhesion cues on the electrode surface without compromising the requirements on low interfacial impedance. As an additional benefit it was demonstrated that the polymer could act as a reservoir of charge allowing more efficient stimulation from miniaturized electrodes.30 Further developments of CPs relevant to neural interfaces (from 2001 and forward) are reviewed at appropriate location in coming sections.
3. Neural interfaces
The neuron is the basic unit of the nervous system. Neurons come in a plethora of varieties depending on their function and location in the central nervous system (CNS) or the peripherals nervous system (PNS). Typical morphological characteristics for the neuron are a cell body with processes, dendrites, for receiving input to the cell and one process, axon, that outputs signals from the cell or conducts sensory input (Fig. 1). The axon is a process that can vary considerably in its dimensions with a diameter ranging from sub-micrometre to tens of micrometres and a length of up to 1 metre in the peripheral nervous system. The main functions of the neuron are to transfer information and in conjunction with other neurons store and compute information. The ability to do this is dependent on the special property of neurons to momentarily change the voltage over the cell membrane to create an action potential. The voltage over the cell membrane, the membrane potential, is an electrochemical voltage originating from the ability of ion pumps in the membrane to create a concentration difference of ions between the inside and the outside of the cell. The opening of voltage gated channels in the cell membrane will transiently increase the permeability to ions and depolarise the cell before the channels are closed and the membrane potential is reconstituted.33 The typical time course for the action potential is around 1 ms (Fig. 1) during which currents on the order of mA cm−2 pass over the membrane.33 The resulting action potential is propagated through the axon and acts at the synapses to release neurotransmitters to excite the next neuron. | ||
| Fig. 1 Schematic representation of a neuron and an action potential of Hodgkin–Huxley type.33 The ca. 1 ms voltage excursion is due to sodium influx during the rising phase of the depolarisation and potassium efflux during the falling phase of repolarisation. The change in intracellular potential is greater than 100 mV, the extracellular signal declines with distance and is substantially smaller even for short distances between neuron and electrode (see Fig. 2). | ||
3.1 Artificial interfacing
The signal transduction in the nervous system is electrical and chemical. Interacting with either pathway opens up the possibility for artificial interfacing with the neuron. For electrical interfacing the electrodes record the electrical activity of the neurons and evoke action potentials by electrical stimuli. As electrical current is transported as ions in biological tissue and as electrons in the electrode material, the interface works as a transducer between ionic and electronic currents. The efficiency of this conversion is limited by the electrical impedance, with higher impedance leading to greater energy loss of the signal to be exchanged.To compensate for high impedance the applied potential can be increased in order to generate a satisfactory current density for stimulation. However, for high potentials, applied to the electrode, there are electrochemical considerations that have to be taken into account. Charge transfer reactions at the interface, due to elevated potentials when stimulating with the electrode, involve electrochemical oxidation or reduction of different species in the electrolyte leading to a change of chemical composition of the electrolyte, corrosion of the electrode, or even gas formation at the interface.29 Either of these effects may affect the biocompatibility of the electrode in a negative way which can be detrimental to the function of the interface. Reducing such deleterious electrochemical reactions is thus essential. Besides electrochemical charge transfer reactions, current may pass the interface as capacitive charging current. The capacitive charging will be present at potentials even too small to drive electrochemical reactions.30 The use of capacitive current thus circumvents electrochemical reactions and any negative impact these reactions would have on the electrode or tissue.
Besides impedance, a consideration that has to be taken into account for communication is the electrode–neuronal coupling. Insulating scar formation is commonly seen which increases distance and efficiently cut off communication between neuron and electrode.31,32 As an increase in distance will decrease the signal strength, the distance between the neuron and the electrode is a determinant for good recordings of neural activity. Distance is also an important parameter for stimulation as it is favourable to reduce required potential and thereby electrochemical side effects (Fig. 2a). Another aspect is that more neurons will come into view the farther away the electrode is acting, thus decreasing selectivity (Fig. 2b). The release of drugs or biomolecules that modulate the immune response or neuronal outgrowth and attachment are strategies for creating a good electrode–neuronal coupling.
 | ||
| Fig. 2 (a) The radial decline of the potential from the surface of a spherical electrode with unit diameter, x-axis scaling in electrode diameters. At a distance of one diameter from the electrode the potential has declined to a value less than 45% of the value at the electrode surface. (b) Increasing the stimulus output from α to β also increases the number of neurons affected by the stimulation, making the stimulus less selective. | ||
A feature of polymer electrodes is the possibility to controllably transport small molecules such as neurotransmitters in their ionic form.19 In the future the electrode could be used as an artificial synapse or as a means for neuromodulation. In these approaches the primary effect would be relying on a chemical interaction as opposed to influence of the electrical field. Chemical interfacing will also be dependent on the electrode–neuron distance as diffusion will have a major influence for larger distances.
4. Fabrication of polymer electrode coatings
The general method for applying conductive polymer coatings is electropolymerization. It is straightforward and simple to build adherent conducting polymer films on conducting substrates and the resulting material is insoluble in water. In short: current is driven through an electrode immersed in a solution containing the monomer and one or several counterions. As a result, monomers build up polymer chains at the working electrode incorporating the counterion (CI) in the process (Fig. 3). | ||
| Fig. 3 Polymerization of (a) PPy and (b) PEDOT from its respective monomer. The counterion (CI) is incorporated into the polymer film. | ||
Although there are numerous other routes besides electrochemical polymerization, this method has dominated the field of neural interfaces for several reasons. Compared to chemical polymerization this process has the advantage of limiting the coating to the underlying electrode, combining the fabrication and geometrical patterning step into one.27 The formed polymer is tightly bound to the underlying conducting surface. In addition, it provides convenient control of film topography and thickness through several process parameters.
The applied deposition charge, counterion concentration, solvent influence and electrodeposition method, galvanostatic or potentiostatic, influence physical parameters of the formed film.27,28,34–37 In general, the more charge that is applied during the process the more polymer growth is achieved and, up to a certain point, this can be correlated to an increase in surface roughness and a sharp decrease in impedance (Fig. 4). It has been shown that for extended deposition times the morphology of the film transforms from nodules to clusters27,28,37,38 leading to an increase in impedance at continued deposition. These transition stages have also been shown to correlate well with mechanical properties of the film, where the films providing the lowest impedance were those that were the most compliant.37
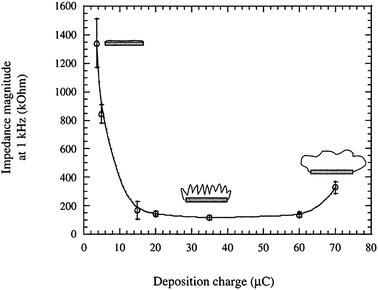 | ||
| Fig. 4 Impedance magnitude at 1 kHz of PPy electropolymerized with a synthetic protein polymer SLPF for increasing deposition charge. Cartoons show how the transformation of the surface from flat metal to a hairy morphology and eventually a smoothing effect as the growing film fills the pores, and its correlation to impedance. Cartoons can be compared to SEM images shown in Fig. 7. Reprinted from Cui et al.,28Journal of Biomedical Materials Research, 2001, by permission of John Wiley & Sons. | ||
A major coating process used is that of pyrrole electropolymerization (Fig. 3a)27,28,39 even though a shift to the use of ethylenedioxythiophene (EDOT) (Fig. 3b), due to its superior stability, can be noticed.30,34,40–42 Other materials used include EDOT derivatives43,44 and polyaniline.45
The nature of the counterion influences physical properties of the formed material e.g. softness, stability, surface structure, permeability and wettability.46–48 When the resulting material is intended for biomedical applications, with some exceptions, the undoubtedly most common solvent is water. If the solubility of the monomer in water is poor, as is the case with EDOT (below 0.01 M), surfactants are often suggested as counterions. Common surfactants are polystyrene sulfonate (PSS) and dodecylbenzenesulfonate (DBS) and it has been shown that their presence indeed facilitate electropolymerization from aqueous solutions.46,49,50 Furthermore, the incorporated counterion influences swelling at oxidation/reduction as described in detail by Jager et al.51
Apart from the physical functionality of the material, cellular interaction with the material surface is of equal importance for the neural interface. The incorporation of the counterion is in fact an excellent opportunity to build biological functionality into the polymer matrix, as will be described in detail in Section 5.
5. Conducting polymers for improved neuron contact
5.1 Biocompatibility aspects on conducting polymers
The most important property of polymer materials is that they can be tailored mechanically, chemically and biochemically to suit a specific application. Biomolecules can be attached to the polymer chains or mixed in during synthesis to create composite biomolecule/polymer materials. Mechanical properties can be engineered to anything from stiff and hard films to polymer gels, either biodegradable or aimed to be long term stable. The mechanical mismatch between silicon and tissue has been pointed out by many to be a contributing factor to more extensive foreign body reactions.52,53 It is feasible to produce polymer electrodes with mechanical properties similar to those of actual cells and it is hypothesised that such soft electrode coatings would act as mechanical buffering between stiff probes and soft tissue.54 Furthermore, an electrode coating allowing tight integration between tissue and implant can lower the strain resulting from micromotion.55Additionally, topography of the polymer surface can readily be altered and some examples on this will be given in Section 5.6. It has also been suggested that similarities between the chemical structure of PPy and naturally occurring hemoproteins and pigments like melanin give promise for future biocompatibility of conjugated polymers. In short, conducting polymers give the opportunity to create electrode materials mechanically and chemically more similar to neural tissue than metals and silicon, which indicates that they might also stand better chance to stably interface with neurons over time.
Biological tests in vitro and in vivo are essential parts of biomaterial development and testing. It has been demonstrated by many that relevant cell types can be successfully cultured on top of PEDOT and PPy films. Although such preliminary work, together with the many arguments stated in this section, speaks in favor of PPy and PEDOT biocompatibility, it is important to remember that more comprehensive biological testing still remains to be performed. Following is a short summary of the work presented this far, concerning in vitro and in vivo biocompatibility of PPy and PEDOT, in the context of neuroprosthetic applications.
A handful of studies describe extensive experimental work assessing PPy biocompatibility in tests relevant for neuroprosthetic applications. Williams et al.25 found that PPy inflicted minimal tissue response in situ (rat model, 4 weeks). Schmidt et al.22 investigated inflammatory tissue response to PPy upon intramuscular or subcutaneous implantation (rodent, up to 14 weeks of implantation) and found that inflammation was less severe compared to the response inflicted by poly(L-lactic acid). Collier et al.26 implanted PPy subcutaneously in rats and found no significant long-term inflammation after 6 weeks. Wang et al.56 investigated systemic toxicity of PPy through a battery of tests addressing acute and subacute toxicity, cytotoxicity, pyretogenesis, hemolysis, allergic reactions and a mutagenesis test, all according to standard methods described in ISO 10993 and ASTM F41748-82. All test results pointed out biocompatibility of PPy in these aspects. It was also shown that rodent sciatic nerve regeneration through a PPy/silicone guidance tube was slightly better than in a plain silicone tube further stressing its compatibility with neurons. Biomolecule-containing PPy has also been shown to promote neuron attachment to electrodes in guinea pig cortex.57
George et al.58 studied reactions surrounding stand-alone PPy films implanted in rodent cortex. Evaluation of tissue was performed after three and six weeks respectively and comparisons were made to Teflon implants of similar size and shape. Based on their results the authors stated that neural tissue had a high degree of tolerance for PPy and it was determined to be at least as biocompatible as Teflon. Furthermore, PPy has been shown to promote sciatic nerve regeneration for eight weeks in rats59 and the material caused “no acute or active chronic inflammatory infiltrate, or tissue damage in the surrounding tissues”.
Additional information on PPy compatibility with various cell types can be found in the numerous studies reporting successful cell culture on top of PPy surfaces. The most common cell line model system to evaluate neuron compatibility of these polymers has been the PC12 neuroendocrine tumor cell line, and many papers have confirmed attachment, viability and neurite extension of PC12 cells on different PPy surfaces (Table 1). PPy has also been shown to support cell growth of fibroblasts, endothelial cells, primary neurons, glial cells and neuroblastoma cell lines (Table 1).
| Study | Cell type |
|---|---|
| Pheochromocytoma cells | |
| Hodgson et al., 199424 | PC12 |
| Schmidt et al., 199722 | PC12 |
| Collier et al., 200026 | PC12 |
| Cen et al., 2004106 | PC12 |
| George et al., 200676 | PC12 |
| Gomez and Schmidt, 200767 | PC12 |
| Kim et al., 200765 | PC12 |
| Park et al., 200766 | PC12 |
| Fonner et al., 200847 | PC12 |
| Fibroblasts | |
| Williams et al., 199425 | L929 |
| Endothelial cells | |
| Garner et al., 199923 | Hum. umbilical vein endothelial cells |
| Wong et al., 199421 | Bovine aortic endothelial cells |
| Primary neurons | |
| Schmidt et al., 199722 | Chicken sciatic nerve explants |
| Evans et al., 200973 | Spiral ganglion neural explants |
| George et al., 200558 | Dissociated cortical neurons |
| Gomez et al., 200735 | Embryonic hippocampal neurons |
| Richardson et al., 200772 | Auditory neuron explants |
| Stauffer and Cui, 200664 | Rodent primary neurons |
| Thompson et al., 200974 | Cochlear neural explants |
| Neuroblastoma cells | |
| Cui et al., 200128 | SH-SY5Y |
| Williams and Doherty, 199425 | Neuro2a |
| Glial cells | |
| Cui et al., 200128 | C6 rodent glial |
| Huang et al., 2009107 | Primary rodent Schwann cells |
| Fonner et al., 200847 | Primary rodent Schwann cells |
The biological response to PEDOT is, to this date, not as extensively studied as PPy. A couple of studies describe biocompatibility of PEDOT in vivo. Luo et al.61 implanted PEDOT subcutaneously in mice and reported no inflammatory response after 1 week of implantation and only thin layers of tissue capsule after 28 days of implantation. Asplund et al.62 evaluated tissue reaction to PEDOT coated platinum pieces in rodent cortex. Response was similar to that of bare platinum but further testing would be needed to fully support this conclusion.
Numerous studies do, however, report cell cultures on top of various types of PEDOT films e.g. PC12 cells, fibroblasts, endothelial cells, neuroblastoma cells, glial cells and a cortical neuron cell line. Details can be found in Table 2. No reports of negative influence of the polymer on cell attachment or proliferation can be found in these studies. On the contrary, a common observation was that cells even selectively grew on the coated electrodes compared to bare surfaces when certain biomolecules were incorporated into the polymer.34,43,63
| Study | Cell type |
|---|---|
| Pheochromocytoma cells | |
| Xiao et al., 200869 | PC12 |
| Green et al., 200948 | PC12 |
| Green et al., 200963 | PC12 |
| Che et al., 200871 | PC12 |
| Kim et al., 200765 | PC12 |
| Fibroblasts | |
| Luo et al., 200861 | NIH3T3 |
| Asplund et al., 200962 | L929 |
| Epithelial cells | |
| Svennersten et al., 2009108 | MDCK |
| Cortical neuron cell line | |
| Isaksson et al., 200717 | HCN2 |
| Neuroblastoma cells | |
| Xiao et al., 200643 | SH-SY5Y |
| Asplund et al., 200962 | SH-SY5Y |
| Yang et al., 200536 | SH-SY5Y |
| Hepatocarcinoma cells | |
| Luo et al., 200861 | HepG2 |
| Glial cells | |
| Cui and Martin, 200334 | C6 rodent glial |
Evidently, there is a need for more work to confirm biocompatibility of conducting polymers in different situations. Especially there is a need for more information on PEDOT. It is also important to recognize that alterations in fabrication protocols will call for renewed assessments of the formed materials. For instance Fonner et al.47 presented a detailed study on how different deposition parameters influenced interaction of the polymer surface with cultured PC12 and Schwann cells. As will be shown in the following sections there are numerous degrees of freedom to vary the formed polymer, wherefore the material singled out for such extensive testing should be chosen wisely.
5.2 Incorporation of biomolecules at electro-polymerization
Cellular interaction with the electrode material is of great importance for the success of the neural interface. It is most likely not sufficient to confirm the absence of adverse effects on surrounding cells from the material, but the material preferably should be bioactive and encourage cells to grow close to, or even into the material. As described in the history Section 2 of this review, the discovery that biological catalysts, incorporated as counterions in PPy, retained their biological activity, opened up for a range of biomedical applications employing this technique to create biofunctional versions of the conducting material. This process has continuously proved its importance for the field of neural interfaces over the last decade.There are dual reasons why incorporation of bioactive species in the electrode material is of interest for the neural interface. Firstly, it comprises a route to immobilize molecules on the surface of the electrode that can e.g. attract and encourage attachment of neurons and discourage adverse immunological response. Secondly, the incorporated molecule can either passively leak from the material or actively be released through electrochemical activation enabling controlled delivery from the polymer bulk. For further details on the chemistry of the conducting polymer backbone the reader is referred to the review by Guimard et al.60 which provides a more thorough analysis of the conjugated polymer backbone and its interaction with dopants. More details can also be found in the review by Cosnier.10
Numerous studies concerning incorporation of biomolecules in PPy to provide a biocompatible and biofunctional electrode surface for the cells have followed. Stauffer and Cui64 continued the investigation of laminin fragments incorporated in PPy. They compared PPy electropolymerized with CDPGYIGSR and PPy:RNIAEIIKDI, the latter considered to be a neurite outgrowth promoting laminin domain, with conventional PPy:PSS. Neuron growth and neurite extension were clearly improved for primary cells cultured on the PPy:peptide coatings compared to PPy:PSS coatings. This effect was also more pronounced for PPy:RNIAEIIKDI. Furthermore, there was less astrocytic adhesion on PPy in general, compared to gold surfaces, which should be an additional benefit of the PPy surface.
Kim et al.65 used neural growth factor (NGF) and collagen as a co-dopant in the polymerization of PPy. An electrolyte of PBS or PSS was used together with the biomolecules to support polymerization. Films containing NGF did indeed induce differentiation of PC12 cells in culture indicating conserved bioactivity of NGF in the process and that incorporated NGF was accessible to cells. Cells did also adhere well to the PEDOT:PBS:collagen films but not to PEDOT:PBS controls which was taken as an indication of successful collagen incorporation.
Park et al.66 deposited PPy with heparin entrapped from aqueous solution containing only pyrrole and heparin salt. The formed material was tested with PC12 cell line and it was found that the heparin containing PPy more successfully attached PC12 cells and supported neurite extension, and stimulation through the PPy:heparin coating encouraged differentiation of cells further. In addition it was found that the amount of heparin available for reaction within the polymer increased with increasing deposition charge, and that the entrapped heparin was not released upon reduction of the material. Based on these findings the material was suggested as an interesting material for nerve guidance channels. The alternative strategy to entrapment of biomolecules is immobilization of biomolecules on top of the polymer surface. Gomez and Schmidt67 reported on immobilization of NGF on top of PPy films by a photolinker. The immobilized NGF was able to encourage neurite extension of PC12 cells and it was reported that conductivity was not affected.
A similar study to the work presented by Cui et al.,28,57 using the same peptide sequence (DCDPGYIGSR) paired with PEDOT, addressed the need for a more stable alternative to PPy.34 Although it was feasible to deposit PEDOT containing these fragments it was reported that the poor solubility of EDOT in water called for a modification of the original protocol for the process to be successful. A solvent mixture of acetonitrile and H2O was used instead. The bioactivity of the resulting polymer:peptide film was confirmed through cell culture studies and a comparison was made between cell attachment on PEDOT:DCDPGYIGSR and PEDOT:PSS, where the former had anchored significantly more cells than the latter.
PEDOT:DCDPGYIGSR deposited with a similar protocol has also been investigated by Green et al.48,63 as well as another peptide dopant, DEDEDYFQRYLI. They compared physical as well as biological properties of the polymer:peptide with bare platinum and conventional PEDOT:paratoluene sulfonate (PEDOT:pTS). The impact of an additional laminin coating applied on top of the polymer was also studied. Although cell attachment (PC12) was improved by all the polymer coatings, an unexpected finding was that cell attachment on PEDOT:pTS was superior to the polymer:peptide coatings. In contrast to this, neurite extension was improved by the polymer:peptides. Laminin coatings on top of surfaces increased cell attachment and neurite outgrowth further.
Whereas the ligand YIGSR improves cell attachment, the ligand YFQRYLI is expected to promote neurite outgrowth as well as mediate attachment. This relation was also reflected in their results indicating that the YIGSR containing films anchored more cells while YFQRYLI films gave a higher proportion of neurite outgrowth. These results point out that both peptides are accessible and active in their respective coatings but their concentration seems to be insufficient to fully encourage cell attachment and growth as shown by the improved results from an additional laminin coating.
Despite the solubility limitations of EDOT some studies show successful incorporation of biomolecules using only aqueous solution of biomolecule and EDOT.68,69 Asplund et al.68 showed that heparin, hyaluronic acid and fibrinogen could be used as counterions in the electropolymerization of PEDOT using only aqueous solution and no surfactants. The formed PEDOT:heparin had electrochemical performance similar to PEDOT:PSS and also PEDOT:HA showed similar impedance behaviour. Due to fast adsorption of fibrinogen to the metal surface, PEDOT:fibrinogen films were hard to form on bare platinum electrodes. A layering approach, where an electropolymerized coating of PEDOT:NaCl was applied prior to polymerization of PEDOT:fibrinogen, similar to the one described by Thompson et al.,70 allowed the formation of PEDOT films containing fibrinogen.
Xiao et al.69 reported successful incorporation of adenosine 5′-triphosphate (ATP) into PEDOT from aqueous monomer ATP solution. ATP release from the film to solution was confirmed for the first two days whereafter it declined. After a two week period no further release was seen indicating a stable ATP content in the film. Cultured PC12 cells responded positively to the incorporated ATP which provided better cell attachment than controls of PEDOT:LiClO4.
An ion exchange method to incorporate glutamate, a neurotransmitter in the central nervous system, into PEDOT was employed by Che et al.71 PEDOT:TSNa was initially deposited followed by strong reduction of the formed film in NaCl solution. In the final step the electrode was immersed in water containing Glu and oxidized leading to electrostatically bound Glu being incorporated into the polymer matrix. Cell spreading on the surface was confirmed and compared to cell attachment on PEDOT:TSNa coatings, with improved growth on the former.
5.3 Controlled release from neuroprosthetic electrodes
While many authors investigate immobilization of the biomolecule in the matrix, release of these molecules is of equal interest. It is not clear whether temporary improvements during the first month of implantation are sufficient to reduce glial scar formation and rescue neurons long term. If the events at the implant at an early stage are crucial also for long term performance, release of the right biomolecules initially might be an effective approach even though it would essentially mean depletion and thereby loss of biofunctionality of the surface over time.Thompson and co-workers have extensively investigated the release of neurotrophins from PPy coatings to improve the survival of SGNs after cochlear implant insertion. Incorporation and release of neurotrophin-3 (NT-3) were achieved through electropolymerization from a pTS–Py aqueous monomer solution with added NT-3.70 A primary layer of PPy:pTS was deposited to facilitate polymerization from the solution containing the biomolecule. Release behaviour of the formed films was studied over a 7 day period and it was found that an initial burst of NT-3 release over the first 24 h was followed by more modest release during the following 6 days. Stimulation through the polymer film increased the release rate more than threefold. Further studies by Richardson et al.72 confirmed that the bound NT-3 was able to interact with spiral ganglion neurons (SGNs) and promoted neurite outgrowth. The same protocol used for NT-3 incorporation was subsequently tested substituting NT-3 with brain derived neurotrophic factor (BDNF).73 These studies were followed by a paper74 concerning the effect of incorporating both NT-3 and BDNF through the same procedure described earlier.70 Release of both neurotrophins was confirmed although the presence of one neurotrophin significantly influenced the release dynamics of the other. The PPy/dual neurotrophin films encouraged neurite outgrowth from cochlear neural explants and presented an improvement from the PPy:single neurotrophin coatings studied in previous papers. Guinea pigs implanted with cochlear electrode arrays coated with the PPy/pTS/NT3, and stimulated through these electrodes over a two week period, exhibited lower electrically evoked brain stem response thresholds.18
The anti-inflammatory drug Dexamethasone (Dex) has been incorporated into PPy without the use of any concomitant dopant.75In vitro studies demonstrated the potential of the released drug from the film to reduce the number of reactive astrocytes without any detrimental effect on co-cultured neurons. In fact, the release of Dex in lower concentrations seemed to have a positive effect on neurite outgrowth.
An alternate route to incorporate the biomolecule directly is to use a dopant molecule that can in turn bind to the biomolecule after polymerization. George et al.76 describe a method where biotin is successfully incorporated as dopant into PPy. The PPy:biotin films were incubated with streptavidin yielding a surface readily accessible for attachment of a biotinylated compound. Incubation with biotinylated NGF resulted in a polymer film with immobilized NGF that could be released upon electrical activation. The bioactivity of NGF after release was confirmed through studying its effect on differentiation of PC12 cells. This approach should be applicable for a wide set of biomolecules making this protocol clearly attractive.
Another hybrid approach using entrapment and subsequent immobilization of biomolecules was presented by Song et al.77 Poly-L-glutamic acid (pGlu) was first incorporated in the PPy film through electropolymerization, thereby introducing carboxylic acid groups that can in turn be available for covalent immobilization of biomolecules. The method was tested for poly-L-lysine and laminin and it was shown that DRG neurons responded to the attached biomolecules. The method is described to be superior to simple entrapment since biomolecules are confined to, and strongly bound to, the surface of the material. It is also reported to give better control of the surface concentration of the bioactive molecules.
Ion-exchange of PPy:TSNa surfaces has been used to incorporate ATP for controlled release. Following deposition, two intermediate steps in NaCl using cyclic voltammetry and potential reduction were used to prepare the polymer for oxidation in the ATP solution. After ion-exchange, ATP remained stable in the film but was released upon stimulation.78
A sophisticated approach to incorporate Dex for controlled release was presented by Abidian et al.79 Nanofibers of poly(L-lactide) (PLLA) or poly(lactide-co-glycolide) (PLGA) applied prior to polymerization were used to grow tubular PEDOT structures (Fig. 5). Dex incorporated in the PLGA fibres could be released either by hydrolytic degradation of the fibres or controllably by contraction of the surrounding PEDOT shell through polymer reduction.79 Another intricate drug delivery surface was subsequently reported from the same group.80 Biodegradable nanofibres with drug incorporated were electrospun on top of the electrode array and dip coated with alginate gel. Electropolymerization of PEDOT:LiClO4 was performed through the fiber–gel layer. The delayed release of Dex to the polymer matrix, due to degradation dependent delivery from the fibres, inhibited the initial burst effect reported by others.70
 | ||
| Fig. 5 SEM images of PEDOT nanotubes at different magnifications. Nanotubes are produced by electropolymerization around PLGA nanofibres and subsequent dissolution of the core fibre leaving only the PEDOT shell. Reproduced from Abidian et al.,79 “Conducting-polymer nanotubes for controlled drug release”, Advanced Materials, 2006 (18). Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission. | ||
5.4 Effect of biomolecule entrapment on impedance characteristics
One important aspect of biomolecule incorporation through electropolymerization is how the procedure affects the physical properties of the resulting electrode material. Changes to the electrochemical behaviour of the film not only immediately after deposition but also after prolonged use must be taken into account. Besides electrochemical stability, mechanical integrity of the material and its adhesion to the substrate should be considered as will be discussed further in Section 7.Many authors use impedance spectroscopy to study the influence of incorporated biomolecules on electrochemical properties of the polymer. A common assumption is that signals in the range of 1 kHz are the most important for neural communication and discussions regarding impedance are therefore often focussed on impedance behaviour close to this frequency, commonly 300 Hz to 1 kHz. However, it should be noted that behaviour over the whole frequency range can give interesting clues to material permeability and porosity as well as composition.46
It is considered sufficient by many authors to confirm that the applied polymer layer lowers impedance as compared to the bare gold electrode. Thus, while many reports include impedance spectroscopy data relatively few provide a comparison to polymer films deposited using more conventional counterions. Green et al.48,63 reported that the incorporation of biomolecules (peptides and NGF) does not significantly alter impedance magnitude in the important high frequency range (300 Hz to 10 kHz) compared to polymer films deposited using pTS. Asplund et al.68 show similar results for PEDOT:HA and PEDOT:heparin compared to PEDOT:PSS. In contrast, PPy polymerized with SLPF,28 PEDOT–MeOH polymerized with peptide,44 and PEDOT with fibrinogen68 all had significantly higher impedance magnitude than the corresponding polymer deposited with PSS. Ion exchange from TSNa to ATP into PPy or TSNa to Glu into PEDOT did increase impedance magnitude.71,78 It should, however, be noted that optimal deposition charge for minimized impedance might differ for different counterions and direct comparisons might therefore not always be appropriate.65
Nyquist plots can be used to reveal differences in impedance characteristics (Fig. 6). An observation when larger counterions are used is the formation of a high frequency semi-circle in the Nyquist plot.28,64,68 Tentative explanations given are increased ionic resistance of the film and/or slow charge transfer over the polymer/electrolyte interface. It is plausible that some biomolecules are not incorporated into the polymer matrix through electrostatic interactions but rather as passive adsorption to the polymer surface during deposition. If that is the case, it can be expected that the formed polymer matrix will contain a non-conducting phase limiting charge transfer within the film.68
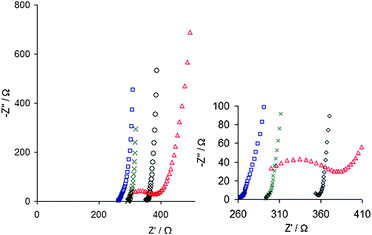 | ||
| Fig. 6 Nyquist plots revealing differences in impedance spectra between PEDOT polymerized with different counterions, PSS shown as (×), heparin (□), HA (○) and fibrinogen (△). A high frequency semi-circle can be seen for PEDOT:fibrinogen and also the beginning of such a circle for PEDOT:HA. Reprinted with permission from Asplund et al.,68 Copyright 2008, American Institute of Physics. | ||
5.5 Monomer functionalization for improved bioactive capabilities
One way to modify a polymer is to alter the composition of the monomer prior to polymerization. For instance, a modification to the pyrrole monomer into 1-(2-carboxyethyl)pyrrole has been shown to be a viable route to produce carboxylic acid-functionalized PPy (PPyCOOH) which can subsequently be used for the immobilization of important proteins.81 Following polymerization Arg-Gly-Asp (RGD), an adhesion protein sequence in the extracellular matrix, was attached to the PPyCOOH surface through a peptide bond. Cell culture (human umbilical vascular endothelial cells, HUVECs) confirmed that the RGD-grafted surface increased viability as compared to regular PPy.To enable polymer–peptide formation from aqueous solutions a modification to the EDOT molecule has been suggested44 according to a protocol previously described by Stéphan et al.82 A pendant hydroxymethyl group was added as side chain to the EDOT yielding a polar monomer with higher solubility in water, EDOT–MeOH.44 PEDOT–MeOH:PSS could readily be deposited from aqueous solution and yielded more uniform coatings than had previously been seen for regular PEDOT:PSS. It was also possible to deposit PEDOT–MeOH:CDPGYIGSR coatings from aqueous solutions without the introduction of additional solvents. A follow up study43 continued this work and showed that cells (SH-SY5Y cell line) preferentially grew on PEDOT–MeOH/CDPGYIGSR covered electrodes compared to uncoated gold sites.
5.6 Micro- and nano-templated polymer for improved cellular attachment
Standard electrodeposition in general produces microporous polymer surfaces, whose roughness is strongly influenced by deposition parameters and counterion used.27,28,34–36,47 The microstructure of a polymer film can have significant impact on its electrochemical properties as well as its ability to support cell adhesion.47 This implies that porosity of the formed surface must be taken into account when designing polymer electrodes for the neural interface.Varying deposition time is one way to control morphology. The polymer growth follows a time course where an initial increase of roughness is followed by a smoothing when these cavities are in turn filled by the growing polymer (Fig. 4 and 7).28,36,78 In addition, if the process is driven galvanostatically or potentiostatically as well as the concentration of the counterion used and the applied current can have a dramatic effect on film topography.34,35 Film growth conditions are therefore important factors to take into account.
 | ||
| Fig. 7 SEM images of PPy/SLPF-coated electrodes for increasing deposition time from (a) to (d). Reprinted from Cui et al.,28Journal of Biomedical Materials Research, 2001, by permission of John Wiley & Sons. | ||
The nature of the counterion can also be a way to tailor surface morphology. For instance TSNa has been shown to yield PPy coatings with nano-tentacle morphology under the right circumstances.78 The inclusion of large biological molecules like SLPF in the film can, besides being a route to biofunctionalization, provide the surface with a fibrillar morphology as can be seen in Fig. 7.28
Control of topography through e-beam lithography of the underlying conducting substrate has been suggested as a possible route to create conducting polymer patterns in the size range 1–2 µm.35 Produced patterns were found to have a significant influence on the growth of embryonic hippocampal neurons.
Apart from alterations in deposition parameters there is also a range of techniques described where different templates are used to create polymer films with well defined nano- and microscale features. For instance PEDOT and PPy with micropores have been produced through electrochemical deposition around self-assembled layers of latex spheres. Spheres in the range 100–485 nm in diameter were used and the latex templates could subsequently be dissolved, leaving a highly porous polymer material (Fig. 8).83,84
 | ||
| Fig. 8 Microporous PEDOT:LiCLO4 films produced through deposition around polystyrene latex sphere templates. Spheres were subsequently removed leaving a highly porous PEDOT film. Morphology for increasing deposition charge as follows: (a) 0 µC; (b) 0.36 µC; (c) 0.6 µC; (d) 1.2 µC; (e) 1.8 µC and (f) 3.6 µC. Reprinted from Sensors and Actuators, B: Chemical, 101, Yang and Martin,83 “Microporous conducting polymers on neural microelectrode arrays: I electrochemical deposition”, Copyright (2004), with permission from Elsevier. | ||
Polymerization of PEDOT from a gel containing monomer, counterion and the surfactant poly(oxyethylene)10–oelyl ether has been shown to provide highly ordered PEDOT films on neural probes.36 The cytotoxic surfactant was removed after polymerization through several washing steps. The surfactant templated film showed improved stability and higher charge capacity compared to PEDOT deposited without surfactant template. Cell culture (SH-SY5Y) on top of the films was possible although bare platinum surfaces provided a better substrate for cell adhesion and proliferation. Possible explanations for the less cell friendly surface provided by the polymer could be microstructural changes and failure to remove residual surfactants from the film.
The same procedure used to produce PEDOT nanotubes for controlled delivery (see Section 5.5) has also been employed as a templating method. Electropolymerization around PLLA nanofibres produced highly fibrillar polymer films and the template fibres could easily be removed. EIS and equivalent circuit modelling showed that electrolyte diffusion in the film was improved by the open morphology created by the template.85 Such nanofibrous PEDOT has also recently proven its usefulness long term (7 weeks) in vivo, where electrode sites covered with PEDOT nanotubes registered high quality unit activity more often (30% more sites) than uncoated gold sites.86
An innovative approach to templated electropolymerization was described by Richardson-Burns et al.,87 where PEDOT was allowed to grow around living neural cells cultured on the electrode site. Cells could be removed from the polymer and left a surface with cell shaped cavities. In vitro studies showed that freshly added cells preferred the cell templated films compared to conventional PEDOT and that some cells indeed repopulated the cavities. Although not shown in the paper, the authors hope that the cytomimetic topology might encourage cell growth into the cavities upon implantation.
These are just some examples of how templates can be used to control film morphology. It should, however, be kept in mind that subsequently treating the polymer films with solvents to remove templates reduces the possibility of retaining viable biomolecules in the films simultaneously.78 Templates not requiring aggressive solvents for dissolution are therefore preferable for applications where incorporated biomolecules should be part of the material design.
6. Conducting polymers for improved signal transduction
One route by which conducting polymer coatings can be used to decrease impedance is by increasing the surface roughness and the true surface area of a planar metal electrode. The true surface area is defined by the area accessible to the electrolyte, which is dependent on the roughness of the surface, and may be greater than the simple geometrical dimensions of the electrode. The coating is electropolymerised using a counterion of synthetic and/or biological origin. Pyrrole27,28 and EDOT34 are the most common starting monomers for preparing the coating. Atomic force measurements of surface roughness have been used to corroborate the impedance dependence on surface roughness down to the resolution limits of the atomic force microscope.27 Using this coating method to increase surface roughness there will be an impedance minimum for a certain thickness of the coating. This has been attributed to a change in film morphology during deposition starting with a smooth film that becomes rougher with deposition time until a certain thickness whereafter it starts to become smoother.27,28Another approach to increase surface roughness is by depositing polystyrene latex spheres, used as a template for polymer growth, and dissolving the spheres after polymerization (Fig. 8).83,84 Access of the electrolyte to the conducting polymer framework is thus greatly enhanced. In an analogous manner carbon nanofibers have been used as the template for coating with PPy.39 In this case the template is not removed but used as a hairy framework, with inherently large surface area, for the polymer coating.
An additional route for decreasing the impedance is to increase the ionic permeability of the polymer material and thus provides access to the internal surface area of the electrode bulk. In this manner the height of the electrode becomes a parameter for decreasing impedance. One strategy used is to grow the polymer in a pre-deposited non-conducting hydrogel network on the electrode.54 Another approach to grow a conducting hydrogel electrode is by trapping dispersed particles of PEDOT–PSS in a conducting scaffold of PPy or PEDOT.30,40 In the latter case the extent of the hydrogel electrode is fully defined by the electropolymerization process.
6.1 Recording
Conducting polymer coatings have been used to record in vitro39,40 and in vivo.27,28,34,41,54,57,86,88 Decreasing the impedance will increase signal quality through an increase in signal strength and lower noise.41 For implanted electrodes an initial quality increase of the coated electrodes compared to uncoated electrodes was noticed.41 This is concomitant with what would be expected from the lower impedance of the coated electrodes. However, the immune system's natural response to a foreign body that cannot be degraded is to encapsulate it. The capsule formation during the first two weeks results in a migration of neurons away from the surface of the electrode reducing the efficacy of the electrode to pick up signals.41,57 After the initial insertion trauma of two weeks an increase in active recording sites associated with a return of the neurons could be noticed. The resulting overall response for coated electrodes was on average better in both the number of active recording sites and the signal quality for up to six weeks.41Recently, conducting polymer hydrogel electrodes have been used in acute experiments as a means of increasing signal quality of electrodes coated with hydrogels.88 The distance between the electrode material and the neuronal signal source is increased by the hydrogel layer as the layer is deposited on the electrode in this case. The benefits of the hydrogel layer may be as a depository for drugs that can be released, especially anti-inflammatory agents to decrease capsule formation, or as a means of tailoring the mechanical modulus of the probe. For both polymer coated and especially hydrogel coated electrodes the distance to the neural signal source is of paramount importance. Even though low impedance is beneficial for recording purposes it cannot outweigh the importance of keeping the neurons close to the electrode.
6.2 Stimulation
Input of information, from an external sensor, to the nervous system is fundamental to all major sensory neuroprosthetic applications to give e.g. auditory, visual, tactile, and proprioceptive feedback. When using electrical stimulation to input information in the nervous system it is necessary to keep the applied electrode voltage low in order to reduce unwanted electrochemical reactions. To high voltages may corrode or degrade the electrode and drive electrochemical reactions that create noxious by-products. Reducing the impedance of the electrode, by for example a conducting polymer coating,30,39,40,42,89 reduces the voltage needed to output a stimulating current, adequate to evoke neural responses, and thus decrease the probability of unwanted electrochemical reactions.Comparisons of conducting polymer coatings to conventional electrode materials of platinum and iridium oxide place the polymer coatings comparable or better at lowering the impedance of neural electrodes at 1 kHz.30,42,89 A concern, for all materials intended as long-term stimulation electrodes, is the stability of the coatings when subjected to repeated stimulation pulses. As the electrode part of a neuroprosthetic device is implanted in intimate contact with nervous tissue, and there may be a risk of damaging the tissue upon implantation or replacement of broken electrodes, a long lifespan of the electrodes is preferable, years or decades. The stability of PEDOT coatings has been reported to surpass that of iridium oxide for physiologically relevant stimulation.89 However, more extensive tests have shown a tendency for thicker PEDOT films to crack and delaminate after continuous stimulation cycling for days.42 Inherent stress in the film, increasing with film thickness, has been pointed out as one of the failure mechanisms,42 volume changes due to ion transport could also be factor behind delamination.97 Conducting polymer coatings have been successfully used to stimulate neurons in neural cell cultures40 and brain slices.39 Polymer hydrogel coated indium tin oxide (ITO) electrodes showed a clear increase in evoked potentials from cultured cerebral neural networks as compared to non-coated ITO electrodes for stimulus voltages less than |0.5| volts40 (Fig. 9). The low stimulus voltages minimize the contribution of the electrochemical reactions at the interface to the total current, and instead counterions are pushed in and out of the film as a response to the changing charge on the polymer backbone. In this in vitro application 4 months of interfacing were reported possible.
 | ||
| Fig. 9 Polymer hydrogel electrodes used for evaluating the stimulation efficacy of cultured neurons. The round electrodes to the left were higher than the square electrodes on the right resulting in neurons preferentially following the circumference of the electrode as opposed to the square electrodes where processes grow on top of the electrode. Reprinted from Journal of Neuroscience Methods, 160, Nyberg et al.,40 “Ion conducting polymer microelectrodes for interfacing with neural networks”, Copyright (2007), with permission from Elsevier. | ||
An interesting combination is the coating of carbon nanofibres with PPy for interfacing with hippocampal brain slices.39 In this case the electrodes consist of brushes of nanofibres, with inherently large surface area, that are electrochemically coated with PPy. The brain slice was placed on top of the electrode array. Comparison with uncoated nanofibres showed a decrease of the smallest voltage necessary to evoke a neural response of field potentials for the coated fibres and a stronger short time latency response.39
For in vivo use the combined effect of polymer coated electrodes for stimulation of the cochlea with release of NT3 was studied by Richardson et al.18 Platinum arrays for cochlear stimulation were coated with Ppy/pTS/NT3 and implanted for two weeks. Electrical stimulation combined with the release of NT3 promoted neuronal survival and gave a low threshold for stimulation which is beneficial for reducing stimulation voltages. The superiority of this system compared to other drug-eluting electrodes is that the conducting polymer coating does not impede current flow from the electrode.18
6.3 Chemical interfacing
The chemical communication between neurons is governed by neurotransmitters such as glutamine, γ-aminobutyric acid and acetylcholine to mention just a few. For neural prosthesis applications, electrodes that are able to release neurotransmitters in a controlled manner could be used as a chemical mode of communication.13 The polymer coating can only store a finite amount of neurotransmitter which has to be replenished when drained. NT3 loaded electrodes had a practical use of a few weeks before being depleted of neurotrophin,18 this short time span would obviously limit the use for long term neural interfaces. Isaksson et al. introduced an external ion reservoir and an electrophoretic ion pump to control ion homeostasis in neurons.17 This concept was further developed for the controlled release of glutamate and acetylcholine to modulate in vivo sensory function.19 If refillable reservoirs could be implemented in vivo this would obviously offer a long term solution for the supply of neurotransmitters.If a neural interface for evoking action potentials is to be realized using chemical interfacing the requirement is that the target neurons are in close proximity to the chemical electrode as the transport through diffusion and electrical field driven transport is short on the timescale appropriate for neural signalling. Preferably the neuronal tissue should have synapses on the electrode to allow for efficient communication; however, an intricate strategy of biochemical tailoring of the electrode would be needed to induce the neurons to synapse with the electrode.
7. Stability of conducting polymers for neuroprosthetic electrodes
Whereas stability is not crucial for tissue engineering applications, and in some cases biodegradability is even a desired property,90,91 neuroprosthetic electrodes are in general aimed for long term function. To warrant the risk and cost of surgery, it is reasonable to require that the material remains functional for several years. However, not many studies cover long term stability of conducting polymers in the biological environment. While many authors claim PEDOT to be sufficiently stable, few studies provide data on polymer stability exceeding one week of experiments. Although the most vulnerable interface is at the location of the neural electrode/biological tissue, this problem might be reduced only then to see that the polymer electrode still does not survive for long enough in this environment. It is therefore essential to evaluate the stability of the polymer material to gain information on how the electrode might change with time.Many parameters can be expected to influence polymer stability, which also complicates direct comparison of data from different studies. The counterions used, the procedure used for polymer deposition, the method of evaluation as well as the chosen test environment vary widely. Predicting survival time in vivo based on data from in vitro simulations is also not trivial. A short summary of what is reported on PPy and PEDOT stability within the field of neural electrodes can be found in Table 3 and the results are discussed in detail in the following section.
| Study | Additional load | Material | Evaluation method | Duration | Results |
|---|---|---|---|---|---|
| a Estimated from figure presented in paper. b Deduced from sweep rate used. c ST = surface templated. | |||||
| PBS or saline, no load | |||||
| Fonner et al., 200847 | PPy:PSS | Conductance meas. | 2 weeks | ∼80% conduct. losta | |
| PPy:ToS | ∼100% conduct. losta | ||||
| PPy:Cl | ∼100% conduct. losta | ||||
| Xiao et al., 200869 | PEDOT:ATP | EIS | 18 days | ∼10% increase of Za | |
| Thaning et al., 201092 | 37 °C | PEDOT:PSS | CV and light spectroscopy | 5–6 weeks | 83% electroact. remaining |
| PEDOT:heparin | 76% electroact. remaining | ||||
| PBS or saline with load | |||||
| Yang and Martin, 200484 | CV −0.9 to 0.5 V vs. SCE | PPy:LiClO4 microporous | CV | 120 sweeps, <1 hb | 30% electroact. lost |
| PEDOT:LiClO4 microporous | 5% electroact. lost | ||||
| Green et al., 200963 | CV −0.8 to 0.6 V vs. Ag:AgCl | PEDOT:pTS | CV | 400 sweeps, ∼1.5 hb | 12% electroact. lost |
| PEDOT:peptide | 17–19% electroact. lost | ||||
| Green et al., 200948 | −0.8 to 0.6 V vs. Ag:AgCl | PEDOT:pTS:NGF | CV | 400 sweeps, ∼1.5 hb | 12% electroact. losta |
| PEDOT:peptide:NGF | 39–51% electroact. losta | ||||
| Wilks et al., 200989 | 5 mC cm−2, pulse 100 Hz | PEDOT:PSS | CV | 2 h | 5% electroact. lost |
| Jan et al., 200995 | CV −0.9 to 0.5 V vs. Ag:AgCl | PEDOT | CV | 300 sweeps, <3 hb | 15% electroact. lost |
| Cui and Martin, 200334 | CV −0.9 to 0.5 V vs. SCE | PPy:PSS | CV | 400 sweeps, > 3 hb | All electroact. lost |
| PEDOT:PSS | Stable after first 3 cycles | ||||
| Yang et al., 200536 | CV −0.9 to 0.5 V vs. SCE | PEDOT:TEAP | CV | 1000 cycles, ∼8 hb | 15% electroact. lost |
| STcPEDOT:TEAP | 10% electroact. lost | ||||
| Yamato et al., 199594 | Const.polar. 0.4 V vs. Ag:AgCl | PPy:PSS | CV | 16 h | 5% electroact. remain |
| PEDOT:PSS | 80 h | 76% electroact. remain | |||
| Cui and Zhou, 200742 | 1 mC cm−2, pulse 50 Hz | PEDOT:PSS | Microscopy and EIS | 2 weeks | Cracks/delamin. 5−9 days |
| Thaning et al., 201092 | ±0.2 V, 50 ms per pulse | PEDOT:PSS | CV and light spectroscopy | 11 days | 94% electroact. remain |
| PEDOT:heparin | 86% electroact. remain | ||||
| GSH 10 mM | |||||
| Xiao et al., 200738 | PPy:TSNa | Reduction potential change | 6–9 days | 20 mV per day deterioration | |
| PEDOT:TSNa | 5 mV per day deterioration | ||||
| Che et al., 200871 | PEDOT:glutamate | CV | 14 days | 12% of electroact. lost | |
| Xiao et al., 200869 | PPy:ATP | EIS | 11 days | ∼150% increase of Za | |
| PEDOT:ATP | 18 days | ∼40% increase of Za | |||
| H 2 O2 10 mM | |||||
| Thaning et al., 201092 | 37 °C | PEDOT:PSS | CV and light spectroscopy | 18 days | All electroact. lost |
| PEDOT:Heparin | 18 days | All electroact. lost | |||
7.1 Stability in saline solutions
Many authors study how immersion in PBS or similar saline solutions affects material, either without applied voltage load47,69,92 or with additional load like constant polarization,93,94 pulsing42,89,92 or repeated oxidation and reduction like CV.34,36,48,63,84,95 The majority of these studies are relatively short term with exposure times in the range 1–8 h, and repetitive CV cycling34,36,48,63,84,95 or pulsing89 during that time period. The instability of PPy is evident and even after relatively short time periods it is reported that 30%84 (120 cycles, <1 h) or even 100%34 (400 cycles, ∼3 h) of the original electroactivity is lost. In contrast, large part of the electroactivity of PEDOT is preserved under similar conditions, with reported losses of electroactivity in the range 5–15%.36,48,63,84,89,95 It is also frequently reported that the loss is greater during the first 100–150 sweeps and electroactivity subsequently stabilizes.A few long term (80 h to 6 weeks) studies of polymer electroactivity in saline have also been published. Yamato et al.94 investigated electroactivity of PPy:PSS and PEDOT:PSS after constant polarization for up to 80 h. Whereas 76% of the original electroactivity was preserved for PEDOT:PSS after 80 h, only 5% of PPy:PSS electroactivity remained after 16 h.
Two long term studies consider repetitive pulsing of PEDOT electrodes in PBS over a two week period.42,92 Cui and Zhou reported that cracks and delamination of film occur within 5–9 days and this also correlated with a large increase in impedance. However, Thaning et al.92 reported that pulsing for eleven days did not have a significant impact on electroactivity. Different substrates (platinum vs. gold) and different pulsing protocols are probable reasons for the discrepancy in results.
Three studies describe changes upon immersion in saline (PBS or NaCl) without any additional voltage load.47,69,92 From data presented in Fonner et al.47 it can be seen that 80–100% of the original conductance of PPy is lost after 14 days of immersion in PBS and most is lost within the first three days. An estimated impedance increase of ∼10%† over 18 days is seen for PEDOT69 and data in Thaning et al.92 show that ∼80% of electroactivity remains after five weeks. The latter study also used elevated temperature (37 °C) which should give a better approximation to the real situation in vivo.
7.2 Stability upon exposure to oxidants and reducing agents
While the above mentioned results describe polymer performance in relatively harmless saline solutions, the environment upon implantation can be expected to contain both oxidants and reducing agents and therefore be more aggressive to the polymer.Several authors have studied the influence of the biologically relevant reducing agent glutathione (GSH) at a concentration of 10 mM. Xiao et al.38 studied the change in reduction potential of PPy:TSNa and PEDOT:TSNa. The reduction potential of PPy deteriorated 20 mV per day whereas the corresponding value for PEDOT was 5 mV per day. In a later study Xiao et al.69 found that PPy:ATP lost its conducting properties completely within 18 days of passive exposure. Even PEDOT was not resistant to GSH reduction, and although more stable than PPy, impedance increased an estimated 40% after 18 days† to be compared with 10% in pure saline. Che et al.71 reported that PEDOT:glutamate had lost 12% of its electroactivity after 12 days.
Thaning et al.92 addressed the influence of the oxidant H2O2 which can be expected to be present at the implant site in the case of inflammatory responses. Complete loss of electroactivity was seen within 22 days of passive exposure to 10 mM H2O2 for PEDOT. It should be noted that this concentration was about ten times of what could be expected in the physiological setting so these data present an accelerated degradation.
7.3 Indications on polymer stability from biological experiments
A couple of studies that do not directly address stability should also be considered since they report on the use of polymer electrodes in practice rather than simulated environments.Nyberg et al.40 successfully used PEDOT:PSS hydrogel electrodes for stimulation and recording from neural networks in vitro for four months. Cui et al.57 used PPy:peptide electrodes to record signals from rat cortex for two weeks but reported an increase in impedance after the first week. In a 6 weeks study by Ludwig et al.,41 surfactant templated PEDOT:TEAP was proven adequate for signal recording in rodent cortex.
The longest time period reported for successful use of polymer electrodes in vivo is PEDOT nanotube electrodes implanted in rodent cortex. Electrodes were used for effective recording of neural signals over 49 days.86 This information is valuable since in vivo methods take into account the effect of immune system responses on the degradation of the material as well as protein adsorption to the material surface.
7.4 The influence of biological counterions on stability
A wide range of counterions are used in the above mentioned studies. However, few of these papers provide comparative data showing how the choice of biological counterions affects stability compared to the more conventional non-biological counterions.Green et al.48,63 addressed the question of how biomolecule incorporation affected electrochemical and mechanical stabilities of the polymer. Their investigations focused on PEDOT polymerized in the presence of large peptide sequences, DEDEDYFQRYLI or DCDPGYIGSR, and compared properties like stability, morphology, hardness, film adhesion and impedance to conventional PEDOT:pTS. Among their findings the counterion appeared to have a strong influence on the mechanical performance of the polymer. The peptide doped films were softer and also clearly less adherent. Furthermore, they showed that the introduction of the second biomolecule, NGF, into the film further deteriorated mechanical performance.48
Electrochemical stability was also reduced upon the inclusion of biomolecules and especially when both peptide and NGF were used. Decline of electroactivity in saline over 400 CV sweeps was 12% for the non-biological counterion pTS, to be compared with 17% and 19% for the peptide counterions DCDPGYIGSR and DEDEDYFQRYLI.63 If NGF was included in the films the corresponding decline was 12%, 39% and 51% respectively.48 The suggested mechanism is that with a smaller dopant, like pTS, the PEDOT matrix can still form efficiently, whereas a larger dopant like the peptides limits this formation making the PEDOT matrix more vulnerable to the influence of a second large non-doping molecule like NGF. Smaller co-dopants, biological or inorganic, are therefore suggested as a possible way to optimize film performance in this aspect.48,63 This will be especially important to consider if the material should contain multiple biomolecules. It should be noted that the inclusion of dual biomolecules did not alter impedance data significantly in the high frequency region (above 300 Hz).
Thaning et al.92 performed a thorough study on PEDOT stability explicitly comparing performance of PEDOT:heparin to PEDOT:PSS during a 6 week period upon immersion in deionized water, PBS and H2O2, and samples in PBS with additional voltage pulsing over a two week period. The biomolecule doped PEDOT performed less stable than conventional PEDOT:PSS, both upon immersion in PBS and in diluted hydrogen peroxide. It should be noted that partial loss of electroactivity was seen for both types of films and although clearly statistically significant, there was not a large difference between the two materials (17% and 24% decline in PBS for PEDOT:PSS and PEDOT:heparin respectively).
7.5 Implications for future work
The introduction of PEDOT dramatically increased the stability of polymer electrodes. Whereas PPy in simulated biological environments loses conductivity within days to a few weeks, the vast majority of PEDOT conductivity remain after more than a month. It is, however, evident that also PEDOT loses part of its electroactivity over time and the time course of this decline needs to be investigated further. While many authors describe that most of the electroactivity loss occurs at an early stage and performance thereafter is relatively stable, studies covering longer time periods still show continued decline over time, even in fairly non-aggressive environments.92 Thaning et al.92 state that if electroactivity loss continues to follow a linear time course, the half-life for electroactivity of PEDOT films could be expected to be in the range 90–200 days, even in the absence of further voltage load.These kinds of estimations are naturally very imprecise. Firstly, simulations are only crude approximations of the in vivo milieu. There is reason to believe that the materials would show both smaller and greater stability in an in vivo setting. Degrading attacks from the immune system speak for the former while deposition of serum albumin on the material could have protective ability, as already shown for platinum in protein solution.96
Not solely the electrochemical stability needs further investigation. The problems with polymer adhesion reported by several authors42,48,63,92 can be a substantial challenge for polymer electrode implants. Hence, understanding and improving film adhesion might be a crucial part in future developments. Cui and Martin showed that electrodeposited gold, providing a more roughened surface for the polymer to attach to, could be one way to improve adhesion.97
On the other hand, some applications may not be depending on the polymer to function long term. The conducting polymer can still be an attractive way to immobilize important biomolecules without compromising low impedance. In that case a polymer designed to be biodegradable might be a suitable choice to ensure that no reminiscent film is left to insulate the electrode once conductivity is lost.
8. Discussion and future focus
8.1 Future for conducting polymer coatings on neuroprosthetic devices
It is obvious that conducting polymers possess several properties that are very attractive for the neural interface. Coatings of a conducting polymer can be used to decrease interfacial impedance and increase the charge per pulse that can be injected reversibly from the electrode surface. Furthermore, functionalization of the polymer film with biomolecules can easily be performed and their mechanical and morphological properties can be tuned at synthesis. The possibility to tailor interaction with surrounding tissue is an important benefit considering it is highly likely that every effort will be needed to rescue neurons and mediate the glial scar formation. Some concerns have also been pointed out in the previous sections, and also by others,109 and additional work is needed.Firstly, long term biocompatibility of specific conducting polymer coatings remains to be proven. Such work is time consuming but is a prerequisite for future use of conducting polymers in clinical applications. The majority of the work published this far evaluates material compatibility with cultured cells but more studies are needed that take interaction with the immune system into account. Possible negative consequences on surrounding tissue from debris and degradation products also need to be evaluated. For an implant to be considered safe it is important that possible risks concerning implant related infections have been evaluated. A porous surface might be desirable from the electrochemical and tissue interaction perspective, but can also be a surface with increased risk of bacterial infection and proliferation. It is well known that dead spaces, not readily accessible for blood borne immune cells, are a risk factor for such infections. It is therefore possible that the most relevant biomolecules to incorporate in the material are antimicrobial and not growth factors.
Secondly, although PEDOT is reported to be the most stable electrically conducting polymer available,98 its electroactivity can be expected to have a limited lifetime in vivo. Long term benefits of the implant must be of substantial impact to motivate the risk taken by the surgical procedure. The intention with introducing conducting polymers to the field of neural electrodes is to improve stability of the neuron/electrode coupling through increasing charge delivery capacity, enhancing compatibility with neurons and reduce glial scar formation. If, on the other hand, the favourable properties of the polymer do not persist for the expected lifetime of the implant, this must be taken into consideration when designing the device. Replacing one stability issue with another is hardly the way to proceed.
Either conducting polymer stability must be improved or the consequences of degradation must be dealt with accordingly. It is not clear whether temporary improvements during the first month of implantation are sufficient to reduce glial scar formation and rescue neurons long term. If that is the case, a conducting polymer coat, designed to enhance biocompatibility at an early stage and subsequently be degraded, could be an acceptable solution. If so the remaining function of the implant, after the electroactivity of the conducting polymer is lost, must still be sufficient for device functionality.
8.2 PPy vs. PEDOT for neural interfaces
The vast majority of papers describing conducting polymers for neuroprosthetic applications focus on PPy. Several arguments in favor of PPy exist. For biomedical applications in general, PPy is at present the most thoroughly studied conducting polymer. There is more experience of PPy from neighbouring fields like tissue engineering, biomedical sensors and drug delivery systems that the neuroprosthetic researcher can rely on. Several extensive studies have been performed to confirm the biocompatibility of PPy whereas the polymers based on EDOT are, although preliminary results are promising, not as thoroughly tested. Electrochemical polymerization from aqueous solutions is also more conveniently performed for PPy than PEDOT due to the higher solubility of the pyrrole monomer in water. The formed polymer has a more mechanically rigid structure making is fairly easy to produce stand-alone films of PPy, whereas PEDOT is more brittle. PPy also provides a better structure for controlled delivery of incorporated substances.However, one issue of considerable importance remains that clearly speaks in favor of using PEDOT. Most neuroprosthetic devices are aimed for long term function and the conductivity of PPy can be expected to diminish rather quickly in the biological environment.
8.3 Carbon nanotube conducting polymer composites for improved electrochemical properties
One study has recently been published addressing the problem of long term stability through the introduction of conducting polymer carbon nanotube (CNT) composites. Multi-walled carbon nanotubes (MWNTs) were incorporated in PPy films through either codeposition or layering, the former a procedure similar to biomolecule entrapment (Sections 5.3 and 5.4) and the latter including an intermediate step drying the MWNTs on top of the electrodes prior to electropolymerization.99 The authors found that incorporation of MWNTs improved stability of the films with at its best an estimated 29% loss of electroactivity after 400 oxidation–reduction cycles to be compared with 61% for PPy:PSS without MWNTs. In addition to this, films produced with the layering approach showed an increase in conductivity compared to PPy without MWNTs. Others report on how MWNT–polyelectrolyte materials are more efficient neural interface materials in the respect that they reduced impedance more, displayed higher charge storage capacity and retained more of its charge storage capacity after repeated cycling (300 cycles) than PEDOT.95Similar studies exist and, although these do not explicitly evaluate the effect on stability, they report on other benefits of nanotube incorporation. Forests of carbon nanofibres grown vertically on electrodes create a three dimensional conductive surface highly accessible to the electrolyte. These vertically aligned carbon nanofibres (VACNFs) can in turn be embedded in a conducting polymer (Fig. 10). Nguyen-Vu et al.100 reported that a PPy shell did not only provide a dramatic increase of specific capacitance but also supported the freestanding structure preventing the VACNFs from collapsing into bundles. The difference in topography between the microbundles and the stabilized fibres that remained vertically aligned greatly influenced morphology of PC12 cell networks cultured on top.101 Such PPy-coated VACNFs were also found superior for the stimulation of hippocampal brain slices in comparison to platinum electrodes or VACNFs without PPy.39
 | ||
| Fig. 10 SEM images of VACNF arrays (a) as-grown VACNF, (b) same type as shown in (a) but after soaking and drying, (c and d) VACNFs embedded in PPy. (a) and (c) are 45° perspective views, scale bars of 500 nm, (b) and (d) show top views, scale bars of 1.0 µm. Reprinted from Nguyen-Vu et al.100, Vertically Aligned Carbon Nanofiber Arrays: An Advance Toward Electrical-Neural Interfaces, Small, 2006. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission. | ||
There is to date little data on long term performance of such CNT conducting polymer composite films but there is a possibility that such hybrids can provide neural interfaces with the best of two worlds. Conducting polymers would function as a conductive material for improved biological interaction in terms of softness and biofunctionalization as well as stabilization of the CNTs, while the CNTs would create an integrated conducting structure in the material less vulnerable to degrading attacks from surrounding tissue.
8.4 Electrode implants that can be modified in situ
One crucial question to this field is if bioactive conducting polymer coatings will suffice to solve the problem of functional loss due to tissue reactions? Even with the clear benefits given by lower impedance and significantly improved charge delivery, it might be necessary to apply other strategies as well. On the other hand, it is possible that conducting polymers can contribute to neural interfaces beyond their use as a coating material. A glance at other fields of research where conducting polymers are used in diverse applications like solar cells, printed electronics and microactuators can be an inspirational source to find out how the full potential of these materials can be exploited. In the following section some papers are discussed that introduce novel ideas on how conducting polymers can contribute to neural interfaces.In 2007, a publication from the University of Michigan103 showed that it is possible to electropolymerize PEDOT within living tissue forming a conductive polymer network infiltrating the surrounding tissue (Fig. 11). It was further shown that cells in the tissue remained intact throughout polymerization and at least three hours thereafter. These results open up intriguing possibilities to modify an electrode after implantation. The authors suggested this could be a possible route to bypass the glial scar and bring the electrode closer to viable neurons. This also implies that the polymer electrodes can be refilled in situ which could be a new way to address the problem of polymer stability. The idea of being able to modify the implant even after implantation is therefore exciting for multiple reasons.
 | ||
| Fig. 11 PEDOT electropolymerized into a mouse brain slice. (a) Electrochemical setup with slice embedded in gelatin for stability, (b) brain slice with PEDOT electropolymerized in three zones representing three different deposition charges, (c) higher magnification of image in (b), and (d) optical micrograph revealing structure of formed PEDOT microfilaments. Reprinted from Journal of Neural Engineering, Richardson-Burns et al.,103 “Electrochemical polymerization of conducting polymers in living neural tissue”, (2007), by permission of IOP Publishing. | ||
One example on how polymer actuators can indeed be useful within this field is described in Zhou et al.105 A PPy bilayer-based actuator was applied to facilitate curling of a common cochlear electrode implant. Electrically controllable curling of the implant is expected to facilitate navigation inside the spiralling cochlea. Presumably, this could be made of use for other neuroprosthetic implants as well. For instance one problem that would be interesting to address using electroactive polymers is the need for an implant with switchable mechanical properties. To cause minimal volume displacement of tissue the implant should be as thin as possible and, to be able to penetrate the tissue, the inserted material must therefore be rather rigid. Once inserted, it is on the other hand desirable to have an implant with flexibility in the same range as the surrounding tissue. Through successful implementation of a material with electronically controllable stiffness, it might be possible to replace the stiffer silicon arrays with truly flexible polymer arrays in the future. Whether conducting polymers can contribute to the solution to this problem is an open question.
9. Conclusions
Many preliminary studies have shown promising results on the potential of both PPy and PEDOT to significantly improve both recording and stimulation properties of the neural interface. These advantages are very clear in terms of the reduced impedance, the enhanced charge capacity and the time response. Some of these advantages may persist also when more knowledge has been gained on the long term properties of materials and devices in contact with real biological systems.The cost for these improvements is less experience of long term performance of these materials in vivo compared to already established biomaterials. One might argue that the most important feature of the polymer coating is to entrap essential biomolecules at the electrode surface. In that respect, the binding of biomolecules inside the conducting polymer matrix is more beneficial than binding molecules in insulating material risking to distort or diminish the signal. The prospect of releasing entrapped biomolecules and drugs to mitigate the immune response and attract neurons to the interface presents a potential tool for realizing stable interfaces.
Notes and references
- N. C. Foulds and C. R. Lowe, J. Chem. Soc., Faraday Trans. 1, 1986, 82, 1259–1264 RSC.
- M. Umaña and J. Waller, Anal. Chem., 1986, 58, 2979–2983 CrossRef CAS.
- R. A. Bull, F. R. Fan and A. J. Bard, J. Electrochem. Soc., 1983, 130, 1636–1638 CAS.
- A. Boyle, E. Genies and M. Fouletier, J. Electroanal. Chem., 1990, 279, 179–186 CrossRef CAS.
- R. John, M. Spencer, G. G. Wallace and M. R. Smyth, Anal. Chim. Acta, 1991, 249, 381–385 CrossRef CAS.
- H. Naarmann, Angew. Makromol. Chem., 1988, 162, 1–17 CrossRef CAS.
- A. J. Hodgson, M. J. Spencer and G. G. Wallace, React. Polym., 1992, 18, 77–85 CrossRef CAS.
- O. A. Sadik and G. G. Wallace, Anal. Chim. Acta, 1993, 279, 209–212 CrossRef CAS.
- S. B. Adeloju, S. J. Shaw and G. G. Wallace, Anal. Chim. Acta, 1993, 281, 620.
- S. Cosnier, Biosens. Bioelectron., 1999, 14, 443–456 CrossRef CAS.
- G. G. Wallace and L. A. P. Kane-Maguire, Adv. Mater., 2002, 14, 953–960 CrossRef CAS.
- S. Cosnier, Anal. Lett., 2007, 40, 1260–1279 CrossRef CAS.
- B. Zinger and L. L. Miller, J. Am. Chem. Soc., 1984, 106, 6861–6863 CrossRef CAS.
- L. A. Prezyna, Y. J. Qiu, J. R. Reynolds and G. E. Wnek, Macromolecules, 1991, 24, 5283–5287 CrossRef CAS.
- M. Pyo, G. Maeder, R. T. Kennedy and J. R. Reynolds, J. Electroanal. Chem., 1994, 368, 329–332 CrossRef CAS.
- M. Pyo and J. R. Reynolds, Chem. Mater., 1996, 8, 128–133 CrossRef CAS.
- J. Isaksson, P. Kjall, D. Nilsson, N. D. Robinson, M. Berggren and A. Richter-Dahlfors, Nat. Mater., 2007, 6, 673–679 CrossRef CAS.
- R. T. Richardson, A. K. Wise, B. C. Thompson, B. O. Flynn, P. J. Atkinson, N. J. Fretwell, J. B. Fallon, G. G. Wallace, R. K. Shepherd, G. M. Clark and S. J. O'Leary, Biomaterials, 2009, 30, 2614–2624 CrossRef CAS.
- D. T. Simon, S. Kurup, K. C. Larsson, R. Hori, K. Tybrandt, M. Goiny, E. H. Jager, M. Berggren, B. Canlon and A. Richter-Dahlfors, Nat. Mater., 2009, 8, 742–746 CrossRef CAS.
- K. Tybrandt, K. C. Larsson, S. Kurup, D. T. Simon, P. Kjall, J. Isaksson, M. Sandberg, E. W. H. Jager, A. Richter-Dahlfors and M. Berggren, Adv. Mater., 2009, 21, 4442–4446 CrossRef CAS.
- J. Y. Wong, R. Langer and D. E. Ingber, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 3201–3204 CrossRef CAS.
- C. E. Schmidt, V. R. Shastri, J. P. Vacanti and R. Langer, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 8948–8953 CrossRef CAS.
- B. Garner, A. Georgevich, A. J. Hodgson, L. Liu and G. G. Wallace, J. Biomed. Mater. Res., 1999, 44, 121–129 CrossRef CAS.
- A. J. Hodgson, K. Gilmore, C. Small, G. G. Wallace, I. L. Mackenzie, T. Aoki and N. Ogata, Supramol. Sci., 1994, 1, 77–83 CAS.
- R. L. Williams and P. J. Doherty, J. Mater. Sci.: Mater. Med., 1994, 5, 429–433 CrossRef CAS.
- J. H. Collier, J. P. Camp, T. W. Hudson and C. E. Schmidt, J. Biomed. Mater. Res., 2000, 50, 574–584 CrossRef CAS.
- X. Y. Cui, J. F. Hetke, J. A. Wiler, D. J. Anderson and D. C. Martin, Sens. Actuators, A, 2001, 93, 8–18 CrossRef.
- X. Y. Cui, V. A. Lee, Y. Raphael, J. A. Wiler, J. F. Hetke, D. J. Anderson and D. C. Martin, J. Biomed. Mater. Res., 2001, 56, 261–272 CrossRef CAS.
- S. B. Brummer and M. J. Turner, Bioelectrochem. Bioenerg., 1975, 2, 13–25 CrossRef CAS.
- T. Nyberg, O. Inganas and H. Jerregard, Biomed. Microdevices, 2002, 4, 43–52 CrossRef CAS.
- W. M. Grill and J. T. Mortimer, Ann. Biomed. Eng., 1994, 22, 23–33 CrossRef CAS.
- D. H. Szarowski, M. D. Andersen, S. Retterer, A. J. Spence, M. Isaacson, H. G. Craighead, J. N. Turner and W. Shain, Brain Res., 2003, 983, 23–35 CrossRef CAS.
- A. L. Hodgkin and A. F. Huxley, J. Physiol., 1952, 117, 500–544 CAS.
- X. Y. Cui and D. C. Martin, Sens. Actuators, B, 2003, 89, 92–102 CrossRef.
- N. Gomez, J. Y. Lee, J. D. Nickels and C. E. Schmidt, Adv. Funct. Mater., 2007, 17, 1645–1653 CrossRef CAS.
- J. Y. Yang, D. H. Kim, J. L. Hendricks, M. Leach, R. Northey and D. C. Martin, Acta Biomater., 2005, 1, 125–136 CrossRef.
- J. Y. Yang and D. C. Martin, J. Mater. Res., 2006, 21, 1124–1132 CrossRef CAS.
- Y. H. Xiao, C. M. Li, S. C. Yu, Q. Zhou, V. S. Lee and S. M. Moochhala, Talanta, 2007, 72, 532–538 CrossRef CAS.
- E. D. de Asis, T. D. B. Nguyen-Vu, P. U. Arumugam, H. Chen, A. M. Cassell, R. J. Andrews, C. Y. Yang and J. Li, Biomed. Microdevices, 2009, 11, 801–808 CrossRef CAS.
- T. Nyberg, A. Shimada and K. Torimitsu, J. Neurosci. Methods, 2007, 160, 16–25 CrossRef CAS.
- K. A. Ludwig, J. D. Uram, J. Y. Yang, D. C. Martin and D. R. Kipke, J. Neural Eng., 2006, 3, 59–70 Search PubMed.
- X. T. Cui and D. D. Zhou, IEEE Trans. Neural Syst. Rehabil. Eng., 2007, 15, 502–508 CrossRef.
- Y. H. Xiao, D. C. Martin, X. Y. Cui and M. Shenai, Appl. Biochem. Biotechnol., 2006, 128, 117–129 CrossRef CAS.
- Y. H. Xiao, X. Y. Cui, J. M. Hancock, M. B. Bouguettaya, J. R. Reynolds and D. C. Martin, Sens. Actuators, B, 2004, 99, 437–443 CrossRef.
- D. K. Cullen, A. R. Patel, J. F. Doorish, D. H. Smith and B. J. Pfister, J. Neural Eng., 2008, 5, 374–384 Search PubMed.
- J. Bobacka, A. Lewenstam and A. Ivaska, J. Electroanal. Chem., 2000, 489, 17–27 CrossRef CAS.
- J. M. Fonner, L. Forciniti, H. Nguyen, J. D. Byrne, Y. F. Kou, J. Syeda- Nawaz and C. E. Schmidt, Biomed. Mater., 2008, 3, 034124 Search PubMed.
- R. A. Green, N. H. Lovell and L. A. Poole-Warren, Acta Biomater., 2010, 6, 63–71 CrossRef CAS.
- A. Lima, P. Schottland, S. Sadki and C. Chevrot, Synth. Met., 1998, 93, 33–41 CrossRef CAS.
- N. Sakmeche, S. Aeiyach, J. J. Aaron, M. Jouini, J. C. Lacroix and P. C. Lacaze, Langmuir, 1999, 15, 2566–2574 CrossRef CAS.
- F. E. W. H. Jager, E. Smela and O. Inganas, Science, 2000, 290, 1540–1545 CrossRef CAS.
- J. Subbaroyan, D. C. Martin and D. R. Kipke, J. Neural Eng., 2005, 2, 103–113 Search PubMed.
- A. Gilletti and J. Muthuswamy, J. Neural Eng., 2006, 3, 189–195 Search PubMed.
- D. H. Kim, M. Abidian and D. C. Martin, J. Biomed. Mater. Res., 2004, 71a, 577–585 Search PubMed.
- H. Lee, R. V. Bellamkonda, W. Sun and M. E. Levenston, J. Neural Eng., 2005, 2, 81–89 Search PubMed.
- X. D. Wang, X. S. Gu, C. W. Yuan, S. J. Chen, P. Y. Zhang, T. Y. Zhang, J. Yao, F. Chen and G. Chen, J. Biomed. Mater. Res., 2004, 68a, 411–422 Search PubMed.
- X. Y. Cui, J. Wiler, M. Dzaman, R. A. Altschuler and D. C. Martin, Biomaterials, 2003, 24, 777–787 CrossRef CAS.
- P. M. George, A. W. Lyckman, D. A. LaVan, A. Hegde, Y. Leung, R. Avasare, C. Testa, P. M. Alexander, R. Langer and M. Sur, Biomaterials, 2005, 26, 3511–3519 CrossRef CAS.
- P. M. George, R. Saigal, M. W. Lawlor, M. J. Moore, D. A. Lavan, R. P. Marini, M. Selig, M. Makhni, J. A. Burdick, R. Langer and D. S. Kohane, J. Biomed. Mater. Res., Part A, 2009, 91a, 519–527 CrossRef CAS.
- N. K. Guimard, N. Gomez and C. E. Schmidt, Prog. Polym. Sci., 2007, 32, 876–921 CrossRef CAS.
- S. C. Luo, E. M. Ali, N. C. Tansil, H. H. Yu, S. Gao, E. A. B. Kantchev and J. Y. Ying, Langmuir, 2008, 24, 8071–8077 CrossRef CAS.
- M. Asplund, E. Thaning, J. Lundberg, A. C. Sandberg-Nordqvist, B. Kostyszyn, O. Inganas and H. von Holst, Biomed. Mater. (Bristol, U. K.), 2009, 4, 045009 Search PubMed.
- R. A. Green, N. H. Lovell and L. A. Poole-Warren, Biomaterials, 2009, 30, 3637–3644 CrossRef CAS.
- W. R. Stauffer and X. T. Cui, Biomaterials, 2006, 27, 2405–2413 CrossRef CAS.
- D. H. Kim, S. M. Richardson-Burns, J. L. Hendricks, C. Sequera and D. C. Martin, Adv. Funct. Mater., 2007, 17, 79–86 CrossRef CAS.
- K. H. Park, E. A. Jo and K. Na, Biotechnol. Bioprocess Eng., 2007, 12, 463–469 Search PubMed.
- N. Gomez and C. E. Schmidt, J. Biomed. Mater. Res., Part A, 2007, 81a, 135–149 CrossRef CAS.
- M. Asplund, H. von Holst and O. Inganas, Biointerphases, 2008, 3, 83–93 Search PubMed.
- Y. H. Xiao, C. M. Li, M. L. Toh and R. Xue, J. Appl. Electrochem., 2008, 38, 1735–1741 CrossRef CAS.
- B. C. Thompson, S. E. Moulton, J. Ding, R. Richardson, A. Cameron, S. O'Leary, G. G. Wallace and G. M. Clark, J. Controlled Release, 2006, 116, 285–294 CrossRef CAS.
- J. F. Che, Y. H. Xiao, X. F. Zhu and X. J. Sun, Polym. Int., 2008, 57, 750–755 CrossRef CAS.
- R. T. Richardson, B. Thompson, S. Moulton, C. Newbold, M. G. Lum, A. Cameron, G. Wallace, R. Kapsa, G. Clark and S. O'Leary, Biomaterials, 2007, 28, 513–523 CrossRef CAS.
- A. J. Evans, B. C. Thompson, G. G. Wallace, R. Millard, S. J. O'Leary, G. M. Clark, R. K. Shepherd and R. T. Richardson, J. Biomed. Mater. Res., Part A, 2009, 91a, 241–250 CrossRef CAS.
- B. C. Thompson, R. T. Richardson, S. E. Moulton, A. J. Evans, S. O'Leary, G. M. Clark and G. G. Wallace, J. Controlled Release, 2010, 141, 161–167 CrossRef CAS.
- R. Wadhwa, C. F. Lagenaur and X. T. Cui, J. Controlled Release, 2006, 110, 531–541 CrossRef CAS.
- P. M. George, D. A. LaVan, J. A. Burdick, C. Y. Chen, E. Liang and R. Langer, Adv. Mater., 2006, 18, 577–581 CrossRef CAS.
- H. K. Song, B. Toste, K. Ahmann, D. Hoffman-Kim and G. T. R. Palmore, Biomaterials, 2006, 27, 473–484 CrossRef CAS.
- Y. H. Xiao, J. F. Che, C. M. Li, C. Q. Sun, Y. T. Chua, V. S. Lee and J. H. T. Luong, J. Biomed. Mater. Res., Part A, 2007, 80a, 925–931 CrossRef CAS.
- M. R. Abidian, D. H. Kim and D. C. Martin, Adv. Mater., 2006, 18, 405–409 CrossRef CAS.
- M. R. Abidian and D. C. Martin, Adv. Funct. Mater., 2009, 19, 573–585 CrossRef CAS.
- J. W. Lee, F. Serna, J. Nickels and C. E. Schmidt, Biomacromolecules, 2006, 7, 1692–1695 CrossRef CAS.
- O. Stéphan, P. Schottland, P. Y. Le Gall, C. Chevrot, C. Mariet and M. Carrier, J. Electroanal. Chem., 1998, 443, 217–226 CrossRef CAS.
- J. Y. Yang and D. C. Martin, Sens. Actuators, B, 2004, 101, 133–142.
- J. Y. Yang and D. C. Martin, Sens. Actuators, A, 2004, 113, 204–211 CrossRef.
- M. R. Abidian and D. C. Martin, Biomaterials, 2008, 29, 1273–1283 CrossRef CAS.
- M. R. Abidian, K. A. Ludwig, T. C. Marzullo, D. C. Martin and D. R. Kipke, Adv. Mater., 2009, 21, 3764–3770 CrossRef CAS.
- S. M. Richardson-Burns, J. L. Hendricks, B. Foster, L. K. Povlich, D. H. Kim and D. C. Martin, Biomaterials, 2007, 28, 1539–1552 CrossRef CAS.
- D. H. Kim, J. A. Wiler, D. J. Anderson, D. R. Kipke and D. C. Martin, Acta Biomater., 2010, 6, 57–62 CrossRef CAS.
- S. J. Wilks, S. M. Richardson-Burns, J. L. Hendricks, D. C. Martin and K. J. Otto, Front. Neuroeng., 2009, 2, 7 Search PubMed.
- T. J. Rivers, T. W. Hudson and C. E. Schmidt, Adv. Funct. Mater., 2002, 12, 33–37 CrossRef CAS.
- Q. S. Zhang, Y. H. Yan, S. P. Li and T. Feng, Biomed. Mater., 2009, 4, 035008 Search PubMed.
- E. M. Thaning, M. L. Asplund, T. A. Nyberg, O. W. Inganas and H. von Holst, J. Biomed. Mater. Res., Part B, 2010, 93b, 407–415 CAS.
- A. Kros, N. Sommerdijk and R. J. M. Nolte, Sens. Actuators, B, 2005, 106, 289–295 CrossRef.
- H. Yamato, M. Ohwa and W. Wernet, J. Electroanal. Chem., 1995, 397, 163–170 CrossRef CAS.
- E. Jan, J. L. Hendricks, V. Husaini, S. M. Richardson-Burns, A. Sereno, D. C. Martin and N. A. Kotov, Nano Lett., 2009, 9, 4012–4018 CrossRef CAS.
- L. S. Robblee, J. McHardy, J. M. Marston and S. B. Brummer, Biomaterials, 1980, 1, 135–139 CrossRef CAS.
- X. Y. Cui and D. C. Martin, Sens. Actuators, A, 2003, 103, 384–394 CrossRef.
- G. C. Li and P. G. Pickup, Phys. Chem. Chem. Phys., 2000, 2, 1255–1260 RSC.
- R. A. Green, C. M. Williams, N. H. Lovell and L. A. Poole-Warren, J. Mater. Sci., 2008, 19, 1625–1629 CAS.
- T. D. B. Nguyen-Vu, H. Chen, A. M. Cassell, R. Andrews, M. Meyyappan and J. Li, Small, 2006, 2, 89–94 CrossRef CAS.
- T. D. B. Nguyen-Vu, H. Chen, A. M. Cassell, R. J. Andrews, M. Meyyappan and J. Li, IEEE Trans. Biomed. Eng., 2007, 54, 1121–1128 CrossRef.
- E. Khor, H. C. Li and A. Wee, Biomaterials, 1995, 16, 657–661 CrossRef CAS.
- S. M. Richardson-Burns, J. L. Hendricks and D. C. Martin, J. Neural Eng., 2007, 4, L6–L13 Search PubMed.
- J. E. W. H. Jager, O. Inganas and I. Lundstrom, Science, 2000, 288, 2335–2338 CrossRef CAS.
- D. Zhou, G. G. Wallace, G. M. Spinks, L. Liu, R. Cowan, E. Saunders and C. Newbold, Synth. Met., 2003, 135, 39–40 CrossRef.
- L. Cen, K. G. Neoh, Y. Li and E. T. Kang, Biomacromolecules, 2004, 5, 2238–2246 CrossRef CAS.
- J. Huang, X. Hu, L. Lu, Z. Ye, Q. Zhang and Z. Luo, J. Biomed. Mater. Res., Part A, 2010, 93A, 164–174 CAS.
- K. Svennersten, M. H. Bolin, E. W. H. Jager, M. Berggren and A. Richter-Dahlfors, Biomaterials, 2009, 30, 6257–6264 CrossRef CAS.
- R. A. Green, N. H. Lovell, G. G. Wallace and L. A. Poole-Warren, Biomaterials, 2008, 29, 3393–3399 CrossRef CAS.
Footnote |
| † Estimated from figure presented in paper. |
| This journal is © The Royal Society of Chemistry 2010 |