Efficient synthesis of photoresponsive azobenzene-containing side-chain liquid crystalline polymers with high molecular weights by click chemistry†
Zhibin
Li
,
Ying
Zhang
,
Lirong
Zhu
,
Tao
Shen
and
Huiqi
Zhang
*
Key Laboratory of Functional Polymer Materials (Nankai University), Ministry of Education, Department of Chemistry, Nankai University, Tianjin, 300071, P. R. China. E-mail: zhanghuiqi@nankai.edu.cn; Fax: +86-2223507193; Tel: +86-2223507193
First published on 16th August 2010
Abstract
A new and highly efficient approach to obtaining high molecular weight (up to 303000) azobenzene (azo)-containing side-chain liquid crystalline polymers by copper(I)-catalyzed polymer analogous click chemistry is described. A series of azo compounds with an azido end group and different flexible spacers were successfully attached onto a polymer bearing pendant acetylene groups (i.e., poly(propargyl methacrylate)) in rather high functionalization efficiency (≥97%). The chemical structures, phase transition behaviors, and photoresponsivity of the obtained polymers were characterized, and they were also compared with those of the corresponding low molecular weight azo polymers (prepared via conventional free radical polymerization) to study the effects of the largely different molecular weights on the properties of the polymers. Both the high and low molecular weight azo polymers with a flexible spacer = (CH2)10 showed smectic C liquid crystallinity and an increase in the molecular weight led to a broader range of the liquid crystalline phase, which is positive for potential applications. Furthermore, the highly reversible photoisomerization of the polymer solutions was proven to be hardly affected by the increase of the molecular weights.
Introduction
Azobenzene (azo)-containing polymers have attracted significant interest in recent years due to their potential in various optical and optoelectronic applications such as optical data storage, liquid-crystal displays, molecular switches, nonlinear optical devices, and photomechanical systems.1–10 The fast and reversible trans-cis photoisomerization of the azo groups can provide azo polymers with interesting photoresponsive properties and trigger significant changes in their physicochemical properties upon exposure to UV or visible light. Liquid crystalline polymers (LCPs) with azo groups in the side chains are among the most extensively studied azo polymers because of their large photoinduced anisotropy and photochemical phase transition. So far, many side-chain LCPs bearing pendant azo mesogens with different substituting groups such as alkyl,11 perfluoroalkyl,12 alkyloxy,13 chiral alkyloxy,14 nitro,15 cyano,16 carboxyl,17 cinnamoyl,18 and N-hydroxysuccinimide carboxylate19 have been prepared for different purposes.The azo-containing side-chain LCPs have been mostly prepared via free radical polymerization because of its tolerance to a wide range of functional groups and mild reaction conditions. Unfortunately, the molecular weights of the resulting polymers are generally rather low (normally ranging from a few to tens of thousands) due to the large steric hindrance of the azo monomers, which results in their poor mechanical properties and thus severely limits their application for fabricating robust optical and optoelectronic devices. Therefore, an effective approach to generating high molecular weight azo-containing side-chain LCPs is highly desirable. In this respect, the polymer analogous reaction approach appears to be a useful solution to this problem, which involves the first preparation of a precursor polymer with reactive groups and its subsequent coupling with an azo compound bearing a corresponding reactive group. The flexibility in the preparation of high molecular weight precursor polymers via free radical polymerization (or other polymerization techniques), together with the availability of many different types of efficient coupling reactions,20 makes the polymer analogous reaction approach a highly versatile route for obtaining high molecular weight polymers with desired functional groups. So far, many relatively high molecular weight azo polymers have been successfully prepared by attaching azo mesogens to the preformed polymer backbones.21–26 The polymer analogous esterification reaction between a polymer bearing carboxyl chloride (or hydroxyl) groups and an azo compound with a hydroxyl (or carboxyl chloride) group appears to be one of the most efficient methods in terms of the high functionalization efficiencies.23,24 However, the use of carboxyl chloride in the above system requires fully anhydrous reaction conditions in order to achieve a functionalization degree close to 100%, which prevents it from being applied in aqueous media. Therefore, new and versatile synthetic strategies still need to be explored.
Recent years have witnessed an ever-increasing interest in the “click” chemistry because of its remarkable features such as its tolerance to a large number of functional groups, mild reaction conditions (the presence of water is allowed), and nearly quantitative yields.27–29 In particular, the copper(I)-catalyzed Huisgen 1,3-dipolar cycloaddition reaction between azides and alkynes has proven to be one of the most powerful click reactions and it has been successfully applied in the field of polymer science, affording a variety of functional polymer materials such as block polymers, dendrimers, and many other complex macromolecular structures.30,31 Very recently, a number of azo-containing functional polymers have been prepared via copper(I)-catalyzed click chemistry.32–41 Haddleton and coworkers prepared a series of azo group end-capped polymers by the polymer analogous click reaction between azido terminally functionalized poly(methyl methacrylate)s and an azo compound bearing an alkyne group.32 Liu and coworkers reported the successful synthesis of triazole-linked azo dendrons via repetitive click reactions between a diazido functionalized azobenzene and a monoalkyne functionalized azobenzene with two chloroethyl groups.33 Li and coworkers prepared a series of azo-containing nonlinear optical polymers via copper(I)-catalyzed polymer analogous click chemistry.34–36 Zhu and coworkers described the preparation of some main-chain azo polymers via step-growth polymerization based on click chemistry.37,38 Kannan and coworkers reported the synthesis of a series of side-chain LCPs with triazole-substituted azo groups by conventional free radical polymerization of the corresponding azo monomers (which were prepared via click chemistry).39 The groups of Oriol and Sánchez reported the preparation of a series of azo-containing linear-dendritic diblock copolymers by click chemistry.40,41 To the best of our knowledge, however, there have been no reports on the synthesis of azo-containing side-chain LCPs via copper(I)-catalyzed polymer analogous click chemistry up to now.
In this paper, we present the efficient synthesis of photoresponsive azo-containing side-chain LCPs with high molecular weights (up to 303000) by copper(I)-catalyzed polymer analogous click reaction (Scheme 1, the right side). A model system was chosen here to show the proof-of-principle, where a series of azo compounds with an azido end group and different flexible spacers were successfully attached onto a polymer with pendant acetylene groups (i.e., poly(propargyl methacrylate), poly(PgMA)), leading to azo-containing side-chain LCPs with high functionalization efficiencies (≥97%). As far as we know, this is also the first report on the synthesis of azo-containing side-chain LCPs with a triazole ring between the polymer backbone and the azo mesogenic moiety. Their chemical structures, phase transition behaviors, and UV/visible light-induced photoisomerization in solutions were studied in detail, and they were also compared with those of the corresponding low molecular weight azo polymers with same structures prepared via the conventional free radical polymerization of the methacrylate azo monomers (prepared via click chemsitry).
 | ||
| Scheme 1 The synthetic route to the azo polymers Pn-C and Pn-F (n = 4, 6, 10) by copper(I)-catalyzed polymer analogous click chemistry (the right side) and conventional free radical polymerization (the left side), respectively. | ||
Experimental
Materials
Tetrahydrofuran (THF, Tianjin Jiangtian Chemicals, China, 99%) was refluxed over sodium and then distilled. N,N-Dimethylformamide (DMF, Tianjin Jiangtian Chemicals, 99.5%) was dried with anhydrous magnesium sulfate (MgSO4) and then distilled under vacuum. Triethylamine (Tianjin Jiangtian Chemicals, 99%) was dried with anhydrous sodium sulfate (Na2SO4) and then distilled. Azobisisobutyronitrile (AIBN, Tianjin Jiangtian Chemicals, Chemical purity (CP)) was recrystallized from ethanol prior to use. Copper(I) bromide (CuBr, Aldrich, 98%) was stirred with acetic acid for 12 h, washed with ethanol and diethyl ether successively, and then dried under vacuum at 75 °C for 3 days. The purified CuBr was stored in an argon atmosphere. Methacrylic acid (Tianjin Jiangtian Chemicals, 99%) was firstly dried with anhydrous Na2SO4 and then distilled under vacuum. Thionyl chloride (Tianjin Jiangtian Chemicals, 99.5%) was purified by distillation just prior to use. Methacrylic chloride was prepared by the reaction between methacrylic acid and thionyl chloride. Propargyl methacrylate (PgMA, Scheme 1) was prepared by reacting methacrylic chloride and propargyl alcohol (Shanghai Jingchun Chemical Company, China, 99%) in the presence of triethylamine. 4-(4-Nitrophenylazo)phenol, 1-bromo-n-(4-nitroazobenzene-4′-oxy)butane (B4), 1-bromo-n-(4-nitroazobenzene-4′-oxy)hexane (B6), and 1-bromo-n-(4-nitroazobenzene-4′-oxy)decane (B10) were synthesized according to a previously reported procedure.42N-Hexyl-2-pyridylmethanimine (NHPMI) was synthesized by the condensation of pyridine-2-carboxaldehyde (Acros, 99%) and n-hexylamine (Acros, 99%).43 All the other reagents were commercially available and used without further purification.Synthesis of 1-azido-n-(4-nitroazobenzene-4′-oxy)hexane (N6, Scheme 1)
Sodium azide (0.17 g, 2.52 mmol) was added to a solution of B6 (0.51 g, 1.26 mmol) in DMF (6 mL). The reaction mixture was bubbled with argon for 5 min and then stirred at 100 °C for 12 h, which was subsequently poured into a large amount of deionized water (60 mL) and then stirred for 2 h. Finally, the precipitate was filtered, washed with deionized water several times, and then dried under vacuum at ambient temperature for 24 h to provide the desired pure product (0.43 g, yield: 93%). δH(400 MHz; CDCl3; Me4Si) 8.40–8.33 (d, J = 8.8 Hz, 2H, Ar–H), 8.02–7.93 (m, 4H, Ar–H), 7.07–7.00 (d, J = 9.2 Hz, 2H, Ar–H), 4.12–4.03 (t, J = 6.4 Hz, 2H, –OCH2–), 3.34–3.26 (t, J = 6.8 Hz, 2H, –CH2N3) and 1.91–1.43 (m, 8H, –(CH2)4–).Synthesis of 1-azido-n-(4-nitroazobenzene-4′-oxy)butane (N4)
Prepared as for N6 (yield: 96%). δH(400 MHz; CDCl3; Me4Si) 8.40–8.33 (d, J = 8.8 Hz, 2H, Ar–H), 8.01–7.94 (m, 4H, Ar–H), 7.06–7.00 (d, J = 8.8 Hz, 2H, Ar–H), 4.15–4.07 (t, J = 6.0 Hz, 2H, –OCH2–), 3.44–3.36 (t, J = 6.6 Hz, 2H, –CH2N3) and 1.99–1.79 (m, 4H, –(CH2)2–).Synthesis of 1-azido-n-(4-nitroazobenzene-4′-oxy)decane (N10)
Prepared as for N6 (yield: 95%). δH(400 MHz; CDCl3; Me4Si) 8.39–8.33 (d, J = 8.8 Hz, 2H, Ar–H), 8.02–7.93 (m, 4H, Ar–H), 7.06–6.99 (d, J = 9.2 Hz, 2H, Ar–H), 4.10–4.03 (t, J = 6.6 Hz, 2H, –OCH2–), 3.30–3.22 (t, J = 6.8 Hz, 2H, –CH2N3) and 1.88–1.27 (m, 16H, –(CH2)8–).Synthesis of triazole-containing methacrylate azo monomer M6 (Scheme 1)
A round-bottom flask was charged with N6 (0.37 g, 1.00 mmol), PgMA (0.15 g, 1.21 mmol), DMF (4 mL), triethylamine (16.8 μL, 0.12 mmol), and CuBr (3.6 mg, 0.025 mmol). After the above solution was bubbled with argon for 5 min, the ligand NHPMI (8.7 mg, 0.046 mmol) was added. The reaction mixture was further bubbled with argon for 15 min and then stirred at 60 °C for 12 h, which was purified by passing through a neutral aluminium oxide column and then subsequently poured into a large amount of water (200 mL). Finally, the precipitate was filtered, washed with n-hexane several times, and then dried under vacuum at ambient temperature for 24 h to provide a pure product (0.48 g, yield: 97%). mp 142–144 °C (determined by polarizing optical microscopy (POM) with a heating rate of 10 °C min−1); UV-vis (DMF): λmax/nm (ε/dm3 mol−1 cm−1) = 380 (25600), around 480 (ε is not available due to the overlap of the absorption bands); δH (400 MHz; CDCl3; Me4Si) 8.40–8.32 (d, J = 8.8 Hz, 2H, Ar–H), 8.02–7.93 (m, 4H, Ar–H), 7.62 (s, 1H, –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–N–), 7.05–6.98 (d, J = 9.2 Hz, 2H, Ar–H), 6.14 (s, 1H, CHC–), 5.58 (s, 1H, CHC–), 5.30 (s, 2H, –COOCH2–), 4.43–4.34 (t, J = 7.2 Hz, 2H, –CH2O–Ar–), 4.09–4.02 (t, J = 6.4 Hz, 2H, –NCH2–) and 2.03–1.37 (m, 11H, –(CH2)4–, –CH3).
CH–N–), 7.05–6.98 (d, J = 9.2 Hz, 2H, Ar–H), 6.14 (s, 1H, CHC–), 5.58 (s, 1H, CHC–), 5.30 (s, 2H, –COOCH2–), 4.43–4.34 (t, J = 7.2 Hz, 2H, –CH2O–Ar–), 4.09–4.02 (t, J = 6.4 Hz, 2H, –NCH2–) and 2.03–1.37 (m, 11H, –(CH2)4–, –CH3).
Synthesis of triazole-containing methacrylate azo monomer M4
Prepared as for M6 (yield: 95%). mp 152–154 °C (by POM, heating rate: 10 °C min−1); UV-vis (DMF): λmax/nm (ε/dm3 mol−1 cm−1) = 378 (24820), around 480 (ε is not available due to the overlap of the absorption bands); δH (400 MHz; CDCl3; Me4Si) 8.40–8.33 (d, J = 8.8 Hz, 2H, Ar–H), 8.02–7.93 (m, 4H, Ar–H), 7.67 (s, 1H, –CCH–N–), 7.05–6.97 (d, J = 9.2 Hz, 2H, Ar–H), 6.14 (s, 1H, CHC–). 5.59 (s, 1H, CHC–), 5.31 (s, 2H, –COOCH2–), 4.51–4.44 (t, J = 7.0 Hz, 2H, –CH2O–Ar–), 4.14–4.07 (t, J = 6.0 Hz, 2H, –NCH2–) and 2.23–1.84 (m, 7H, –CH2CH2–, –CH3).
Synthesis of triazole-containing methacrylate azo monomer M10
Prepared as for M6 (yield: 96%). mp 116–118 °C (by POM, heating rate: 10 °C min−1); UV-vis (DMF): λmax/nm (ε/dm3 mol−1 cm−1) = 380 (26000), around 480 (ε is not available due to the overlap of the absorption bands); δH(400 MHz; CDCl3; Me4Si) 8.39–8.33 (d, J = 8.8 Hz, 2H, Ar–H), 8.02–7.93 (m, 4H, Ar–H), 7.60 (s, 1H, –C = CH–N–), 7.05–6.99 (d, J = 9.2 Hz, 2H, Ar–H), 6.13 (s, 1H, CHC–), 5.58 (s, 1H, CHC–), 5.30 (s, 2H, –COOCH2–), 4.37–4.30 (t, J = 7.2 Hz, 2H, –CH2O–Ar–), 4.10–4.03 (t, J = 6.4 Hz, 2H, –NCH2–) and 1.97–1.26 (m, 19H, –(CH2)8–, –CH3).
Synthesis of low molecular weight azo polymers [Pn-F (n = 4, 6, 10) (Scheme 1), F refers to free radical polymerization] via the conventional free radical polymerization of triazole-containing methacrylate azo monomers Mn (n = 4, 6, 10)
The conventional free radical polymerization of Mn (n = 4, 6, 10) was performed in DMF with AIBN as the initiator. A typical polymerization procedure for M6 is presented as follows: M6 (0.45 g, 0.91 mmol), AIBN (3.0 mg, 0.018 mmol), and the freshly distilled DMF (4.5 mL) were added into a one-neck round-bottom flask (25 mL) successively. The reaction mixture was degassed via three freeze-thaw-pump cycles and the flask was then sealed and immersed into a thermostated oil bath at 60 °C. After 48 h of polymerization with stirring, the reaction mixture was cooled down to room temperature and it was added dropwise into methanol (50 mL). The precipitate was filtered and the obtained orange solid was washed thoroughly with warm methanol until no monomer was detected with thin layer chromatography. Finally, the product was dried at 60 °C under vacuum for 48 h to provide the desired polymer P6-F (0.34 g, yield: 76%).The polymerization of M4 and M10 was performed similarly as for M6, respectively, leading to their corresponding homopolymers (i.e., P4-F and P10-F) in a yield of 70 and 74%, respectively.
Synthesis of poly(propargyl methacrylate) (i.e., poly(PgMA))
Poly(PgMA) was prepared following a literature procedure,44 but with some modification in the polymerization time: PgMA (12.42 g, 100.05 mmol), AIBN (0.164 g, 1.00 mmol), and dioxane (40 mL) were added into a one-neck round-bottom flask (100 mL) successively. The reaction mixture was bubbled with argon for 30 min and the flask was then sealed and immersed into a thermostated oil bath at 65 °C. After 45 min of polymerization with stirring, the reaction mixture was cooled down to room temperature and it was added dropwise into a large amount of methanol (400 mL). The precipitate was filtered and the obtained white solid was redissolved into THF and precipitated into methanol again. The obtained solid was then dried under vacuum at ambient temperature for 72 h to provide the desired product (1.52 g, yield: 12%).Synthesis of high molecular weight azo polymers [Pn-C (n = 4, 6, 10) (Scheme 1), C refers to click chemistry] via copper(I)-catalyzed polymer analogous click chemistry between poly(PgMA) and Nn (n = 4, 6, 10)
A typical procedure for the synthesis of P6-C is presented as follows: a round-bottom flask (100 mL) was charged with poly(PgMA) (0.15 g, 1.21 mmol of acetylene groups), N6 (0.53 g, 1.44 mmol), DMF (30 mL), triethylamine (33.6 μL, 0.23 mmol), and CuBr (7.2 mg, 0.05 mmol). After the solution was bubbled with argon for 5 min, the ligand NHPMI (16.8 μL, 0.09 mmol) was added. The reaction mixture was further bubbled with argon for 30 min and the flask was then sealed and immersed into a thermostated oil bath at 60 °C. After being stirred for 24 h, the reaction solution was cooled down to room temperature and it was then added dropwise into a large amount of methanol (300 mL). The precipitate was filtered and washed with warm methanol several times until no N6 could be detected in the washing solution by thin layer chromatography. The obtained solid was dried under vacuum at 50 °C for 48 h to provide the desired product (0.54 g, yield: 90%).The polymer analogous click reaction between poly(PgMA) and N4 or between poly(PgMA) and N10 was performed similarly following the above procedure, leading to the corresponding functionalized polymers (i.e., P4-C and P10-C) in a yield of 93 and 91%, respectively.
Characterization
All 1H NMR spectra were recorded on a Varian Unity plus-400 spectrometer (400 MHz). Fourier transform infrared (FT-IR) spectra were measured with a Bio-Rad FTS-6000 spectrometer. The number-average molecular weights (Mn,SEC) and polydispersity indices (PDI) of poly(PgMA) and the low molecular weight azo polymers Pn-F (n = 4, 6, 10) were determined by size exclusion chromatography (SEC) with a gel permeation chromatograph (GPC) equipped with a Waters 717 autosampler, a Waters 1525 HPLC pump, three Waters UltraStyragel columns with 5 K–600 K, 500–30 K, and 100–10 K molecular ranges, and a Waters 2414 refractive index detector. THF was used as the eluent at a flow rate of 1.0 mL min−1. The calibration curve was obtained by using polystyrene (PS) standards. The Mn,SEC and PDI of the high molecular weight azo polymers Pn-C (n = 4, 6) were determined by SEC with a GPC equipped with an Agilent 1200 series manual injector, an Agilent 1200 HPLC pump, an Agilent 1200 refractive index detector, and two Waters Styragel HT columns (HT5 and HT4) with 50 K–4000 K and 5 K–600 K molecular ranges (the temperature of the column oven: 50 °C). DMF containing 0.5 wt% LiBr was used as the eluent at a flow rate of 1.0 mL min−1. The calibration curve was also obtained by using PS standards. Differential scanning calorimetry (DSC, Netzsch 204) was utilized to study the phase transitions of the polymers at a heating/cooling rate of 10 °C min−1 in a nitrogen atmosphere. The temperature and heat flow scale were calibrated with standard materials including indium (70–190 °C), tin (150–270 °C), zinc (350–450 °C), bismuth (190–310 °C), and mercury (−100–0 °C) in different temperature ranges. The glass transition temperatures (Tg) of the polymers were determined as the midpoints of the step changes of the heat capacities, while the phase transition temperatures were measured from the maximum/minimum of the endothermic/exothermic peaks. The melting points of the azo monomers and the liquid crystalline textures of the polymers were observed by using an Olympus BX51 polarizing optical microscope (POM) equipped with a Linksys 32 THMSE600 hot stage and a digital camera (Micropublisher 5.0 RTV). Small angle X-ray scattering (SAXS) measurements were carried out on a Bruker NanoSTAR SAXS system by using Cu Ka (λ = 1.542 Å) as the radiation source. The working voltage and current were 40 kV and 35 mA, respectively. The distance between the samples and the detector was 26.55 cm. A temperature control unit in conjunction with the instrument was utilized to study the structure evolutions of liquid crystalline mesophases by heating the samples to the desired temperatures. An UV-vis scanning spectrophotometer (TU1900, Beijing Purkinje General Instrument Co., Ltd) was utilized to obtain the UV-vis spectra of the monomer and polymer solutions at 25 °C. The photochemical isomerization of the polymer samples was investigated by irradiating them firstly with a 365 nm UV lamp (12 W) until the photostationary state was reached and then with a 450 nm visible light lamp (18 W, wavelength range: 400–550 nm, λmax = 450 nm, a filter was put between the samples and the lamp during the study in order to block the light with wavelength λ < 430 nm). The photoisomerization of the polymer solutions was also studied by utilizing cycles of UV and visible light irradiation.Results and discussion
Synthesis of Pn-C by copper(I)-catalyzed polymer analogous click chemistry
Copper(I)-catalyzed click chemistry has proven to be highly versatile for the synthesis of functional polymer materials because of its excellent functional group compatibility, high efficiency and specificity under mild reaction conditions (in both aqueous and organic media), and its broad applicability. Herein, we report the first successful application of copper(I)-catalyzed polymer analogous click chemistry in the preparation of photoresponsive azo-containing side-chain LCPs with high molecular weights. In principle, the copper(I)-catalyzed polymer analogous click reaction can be performed via the azide-alkyne coupling either between a polymer bearing pendant acetylene groups and an azo compound with an azido group or between a polymer bearing pendant azido groups and an azo compound with an acetylene group. In this work, the former strategy was chosen to demonstrate the proof-of-principle, which utilized poly(PgMA) as the polymer bearing pendant acetylene groups and Nn (n = 4, 6, 10) as the azo compound with an azido group, as shown in Scheme 1 (the right side).Poly(PgMA) was firstly prepared via the conventional free radical polymerization of PgMA in dixoane at 65 °C following a literature procedure,44 but with a much shorter polymerization time. It is known that the free radical polymerization of PgMA is rather difficult to control due to the presence of the reactive acetylene groups, which usually leads to crosslinked polymer networks.45 Therefore, the polymerization condition should be carefully controlled to avoid the occurrence of the possible crosslinking. A poly(PgMA) with a Mn,SEC = 69000 and PDI = 2.10 was readily obtained by using a very short reaction time (45 min) (Table 1, Fig. S1 in the ESI†), which proved to be soluble in common organic solvents such as chloroform, THF, and DMF. The obtained poly(PgMA) was characterized with both 1H NMR (Fig. 1a) and FT-IR (Fig. 2d). The presence of both the proton signal around 2.51 ppm in its 1H NMR spectrum and the absorption peaks around 2129 and 3292 cm−1 in its FT-IR spectrum confirmed the existence of acetylene groups in the polymer. Furthermore, the chemical shifts and the peak integrations of all the protons in the polymer were well consistent with its expected structure.
| Sample | M n,SEC (×10−4) | PDIa | UV absorb. (AU) b | f (%) | M n,UV-vis (×10−5) | Thermal transitioneT/°C | △Hi/J g−1 |
|---|---|---|---|---|---|---|---|
| a The number-average molecular weights (Mn,SEC) and polydispersity indices (PDI) of the polymers determined by SEC with THF (for poly(PgMA), P4-F, P6-F, and P10-F) or DMF containing 0.5 wt% LiBr (for P4-C and P6-C) as the eluent and PS standards. b UV absorbance (at 380 nm, the average values of three measurements) of the azo polymer solutions in DMF (for P4-F, P6-F, P4-C, and P6-C) or in N-methyl pyrrolidone (for P10-F and P10-C) (Cpolymer = 5 × 10−5 mol L−1 azo mesogen). c f refers to the azo functionalization degrees of the polymers, and the f values of the azo polymers Pn-C (prepared via polymer analogous click reaction) were determined by comparing their UV-vis absorbance with that of their low molecular weight counterparts Pn-F. d The number-average molecular weights of the azo polymers Pn-C determined by UV-vis. e G, glassy; SC, smectic C; I, isotropic. f Determined with DSC at the second heating scan under nitrogen (10 °C min−1). g Determined with DSC at the first cooling scan under nitrogen (−10 °C min−1). h No Tg was detectable. i △H represents the enthalpy of the phase transition. | |||||||
| Poly(PgMA) | 6.90 | 2.10 | 0 | ||||
| P4-F | 3.21 | 2.07 | 1.18 | 100 | G 100 f | ||
| G 89 g | |||||||
| P6-F | 2.51 | 2.02 | 1.23 | 100 | G-SC 120 I fh | 3.4 | |
| I 109 SC-G gh | −3.4 | ||||||
| P10-F | 2.77 | 1.90 | 1.21 | 100 | G-SC 114 I fh | 3.2 | |
| I 109 SC-G gh | −2.9 | ||||||
| P4-C | 51.30 | 1.53 | 1.14 | 97 | 2.53 | G 100 f | |
| G 89 g | |||||||
| P6-C | 52.50 | 1.53 | 1.19 | 97 | 2.68 | G 80 f | |
| G 71g | |||||||
| P10-C | 1.20 | 99 | 3.03 | G-SC 125 I fh | 3.8 | ||
| I 119 SC-G gh | −3.2 |
 | ||
| Fig. 1 1H NMR spectra of poly(PgMA) in CDCl3 (a), P6-C in DMF-d7 (b), P6-F in CDCl3 (c), and M6 in CDCl3 (d). The peaks from the solvents and water are marked with symbol * and #, respectively. | ||
 | ||
| Fig. 2 FT-IR spectra of P4-C (a), P6-C (b), P10-C (c), and poly(PgMA) (d). | ||
A series of azo compounds with an azido end group and different flexible spacers (Nn, n = 4, 6, 10, Scheme 1) were also readily prepared by reacting 1-bromo-n-(4-nitroazobenzene-4′-oxy)alkanes (Bn, n = 4, 6, 10) with an excessive amount of sodium azide. All of them could be obtained in quite good yields, and their purity was satisfactory, as revealed by the thin layer chromatography and 1H NMR techniques.
In the next step, different reaction parameters including the utilized solvent, copper catalyst, reaction temperature, and reaction time were explored in order to provide the optimal reaction conditions for the polymer analogous click reaction. It is known that CuBr/N-alkyl-2-pyridylmethanimine is a highly versatile catalyst for atom transfer radical polymerization.43,46 Recently, it has also proven to be an efficient catalyst for the Huisgen-type cycloadditions.47 Herein, we further checked whether this copper catalyst was also an active catalyst for our study. A model click reaction system between PgMA and Nn (n = 4, 6, 10) was chosen for this purpose (Scheme 1). A series of triazole-containing methacrylate azo monomers (Mn, n = 4, 6, 10) were readily obtained in high yields by performing the reactions in DMF at 60 °C for 12 h with CuBr/NHPMI as the catalyst in the presence of an excess of PgMA. Fig. 1d shows the 1H NMR spectrum of one representative triazole-containing methacrylate azo monomer M6, and the presence of only a single peak at 7.62 ppm for the chemical shift of the triazole ring proton reveals the exclusive formation of the 1,4-disubstituted adduct.
With the starting materials (i.e., poly(PgMA) and Nn (n = 4, 6, 10)) and the optimal click reaction conditions in hand, we tried to perform the polymer analogous click chemistry between poly(PgMA) and Nn (n = 4, 6, 10) (Scheme 1, the right side). The reactions were carried out in DMF at 60 °C in the presence of an excess of Nn with CuBr/NHPMI as the catalyst. The reaction time was extended to 24 h in order to assure a higher functionalization efficiency because a much larger volume of DMF was used in these cases in order to dissolve poly(PgMA) and polymer analogous reactions normally proceed more slowly than the reactions between small molecules. After the coupling reactions, the excess Nn was completely removed from the obtained polymers by washing them with warm methanol, as confirmed with thin layer chromatography. The polymers obtained via the click coupling reactions between poly(PgMA) and Nn (n = 4, 6) (i.e., P4-C, P6-C) were proven to be fully soluble in DMF, while that prepared via the click reaction between poly(PgMA) and N10 (i.e., P10-C) turned out to be only partially soluble in DMF (but it was found to be easily soluble in N-methyl pyrrolidone). The obtained polymers were characterized with FT-IR and 1H NMR. The efficient occurrence of click reactions was demonstrated by the disappearance of the specific IR bands for acetylene groups at 2129 and 3292 cm−1 and the appearance of some new absorption bands associated with the asymmetric (1341 cm−1) and symmetric (1520 cm−1) stretching vibrations of the nitro groups and the ring-stretching mode of the phenyl groups (around 1600 cm−1) (Fig. 2). The 1H NMR spectrum of one representative azo polymer P6-C in deuterated DMF is shown in Fig. 1b, and the chemical shifts and the peak integrations of all the protons in the polymer are in good agreement with its expected structure.
Synthesis of Pn-F by conventional free radical polymerization
To accurately determine the azo functionalization degrees of Pn-C (n = 4, 6, 10), a series of low molecular weight reference polymers (namely Pn-F, n = 4, 6, 10) were also prepared by the conventional free radical polymerization of triazole-containing methacrylate azo monomers (Mn, n = 4, 6, 10) (Scheme 1, the left side), which have almost same chemical structures as Pn-C (n = 4, 6, 10) and possess an azo functionalization degree of 100% (Table 1). By assuming that the molar extinction coefficient of the azo chromophore in Pn-F is the same as that of the azo chromophore in Pn-C, the azo functionalization degrees of Pn-C could be readily deduced by comparing the UV-vis absorbance of the Pn-C solution with that of the Pn-F solution.The obtained polymers Pn-F (n = 4, 6, 10) were proved to be soluble in common organic solvents such as chloroform, THF, and DMF. Their number-average molecular weights determined by SEC (Mn,SEC) were found to range from 25100 to 32100 and their PDI were around 2 (Table 1), which are reasonable for the azo polymers prepared via conventional free radical polymerization. Fig. 1c shows the 1H NMR spectrum of one representative azo polymer P6-F in deuterated chloroform, which agrees well with its chemical structure. In addition, it can be seen clearly that the 1H NMR spectrum of P6-F is quite similar with that of P6-C although they were determined in different deuterated solvents, further indicating that the obtained P6-C indeed has the desired chemical structure.
Determination of the azo functionalization degrees and molecular weights of Pn-C
As can be seen from Fig. 3, the UV-vis spectra of one representative high molecular weight azo-containing polymer P6-C and its low molecular weight counterpart P6-F in DMF are also rather similar. They exhibit one strong absorption band around 380 nm and another very weak one around 480 nm, which are typical for the azo compounds and can be ascribed to the π → π* and n → π* electron transitions of the –NN– bond, respectively.48 By comparing their UV absorption at 380 nm (i.e., AP6-C,380nm and AP6-F,380nm), an azo functionalization degree (f) of 97% was determined for P6-C by using the equation f = (AP6-C,380nm/AP6-F,380nm)×100% (Table 1). It is important to point out here that this approach is facile and efficient for the determination of azo functionalization degrees of the azo polymers because there exists very good linear relationship between the UV absorbance of the azo polymers and their concentrations, even in the cases of rather high concentrations of the azo chromophores (Fig. S2 in the ESI†). Similarly, an azo functionalization degree of 97 and 99% was also obtained for P4-C and P10-C, respectively. These results strongly demonstrated that the copper(I)-catalyzed polymer analogous click reactions between poly(PgMA) and Nn (n = 4, 6, 10) were highly efficient under the studied reaction conditions.
 | ||
Fig. 3 UV-vis spectra of the P6-F solution (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ) and P6-C solution ( ) and P6-C solution (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) in DMF (CP6-F = CP6-C = 0.025 mg mL−1 = 5.0 × 10−5 mol L−1 azo mesogens). ) in DMF (CP6-F = CP6-C = 0.025 mg mL−1 = 5.0 × 10−5 mol L−1 azo mesogens). | ||
On the basis of the above-obtained azo functionalization degrees for Pn-C (n = 4, 6, 10) and the molecular weight of poly(PgMA) (Mn,poly(PgMA), which is assumed to be equal to Mn,SEC,poly(PgMA), i.e., 69000), the number-average molecular weights of Pn-C determined by UV-vis (i.e., Mn,UV-vis) can be readily derived by using the following equation:
| Mn,UV-vis = Mn,SEC,poly(PgMA) + x × f × MNn |
P4-C and P6-C were also characterized with SEC by using DMF containing 0.5 wt% LiBr as the eluent. The results showed that P4-C and P6-C had a Mn,SEC of 513000 and 525000, respectively, and both of their PDI were 1.53 (Table 1 and Fig. S3 in the ESI†). The difference between the Mn,SEC and Mn,UV-vis values of the azo polymers might be assigned to the calibration of the SEC on the basis of polystyrene standards.
Phase transition behaviors of the azo polymers
The phase transition behaviors of the obtained polymers Pn-C (n = 4, 6, 10) were firstly analyzed with a combination of DSC and POM. The DSC curves of P10-C exhibited one phase transition peak in both the second heating and the first cooling scans, while there existed no transition peak in those of P4-C and P6-C (Fig. 4). On the other hand, only a glass transition was observed for P4-C and P6-C in both the DSC second heating and the first cooling scans, and their Tg values in the DSC second heating scan were determined to be 100 and 80 °C, respectively, indicating that the Tg values of the polymers decreased with an increase in the spacer length. This effect has often been observed in side-chain LCPs and has proven to be caused by the increased internal plasticization action with the increase in the spacer length.14 | ||
| Fig. 4 DSC curves of the azo polymers Pn-C (n = 4, 6, 10) from the first cooling scan (a) and from the second heating scan (b) (±10 °C min−1). | ||
POM study showed that when P10-C was cooled from its isotropic state, a large number of anisotropic entities appeared from the dark background of the isotropic liquid. However, it was impossible to observe any characteristic liquid crystalline texture although the sample was annealed in its liquid crystalline phase for a rather long time (Fig. 5a), probably due to the high polymer viscosity. It has been well-established that the high viscosity of the polymer melts can hinder the formation of well-defined liquid crystalline textures.49 This is especially true for smectic liquid crystalline polymers where the development of two-dimensional positional order promotes a further increase in viscosity.50,51 Nevertheless, the phase transition of P10-C proved to be fully reversible. Its birefringence disappeared completely at 130 °C upon heating (10 °C min−1), and it appeared again at 125 °C upon cooling from the isotropic state at a rate of 10 °C min−1, which suggested that the transition peaks in the DSC curves of P10-C represented the transition between the liquid crystalline phase and isotropic phase and the mesomorphism was enantiotropic. The liquid crystalline textures of the polymer were similar for both the heating and cooling scans (the exact nature of the liquid crystalline phase of P10-C was assigned with the aid of SAXS, as shown in the following part). In contrast to P10-C, no obvious birefringence was observed for both P6-C and P4-C, which is likely to be due to their relatively shorter spacers, thus limiting the decoupling effects between the pendant mesogens and the polymer backbones.52
 | ||
| Fig. 5 The POM images upon cooling from the isotropic phases: P10-C at 118 °C (annealed for 70 min) (a), P10-F at 108 °C (annealed for 60 min) (b), and P6-F at 108 °C (annealed for 60 min) (c). | ||
The phase transition behaviors of Pn-C were also compared with those of Pn-F to study the influence of the largely different molecular weights. As shown in Fig. 6, the DSC curves of both P10-F and P6-F exhibited one phase transition peak in both the second heating and the first cooling scans, while there existed no transition peak in those of P4-F. In addition, a glass transition at 100 and 89 °C was observed for P4-F in the DSC second heating and the first cooling scan, respectively, whereas no obvious glass transition was detected for P6-F and P10-F. Note that the Tg value of P4-F is almost the same as that of P4-C although their molecular weights are considerably different, which suggests that the Tg of the studied azo polymers have reached their maximum values at the molecular weights of the low molecular weight polymers and they become leveled off with the further increase in the molecular weights. Similar phenomenon was also reported for other polymer systems previously.53 In contrast, the molecular weights showed much influence on the phase transition temperatures (Ti) of the azo polymers with a flexible spacer =(CH2)10. The Ti value of P10-F was found to be about 10 °C lower than that of P10-C in both the second heating and the first cooling scans (±10 °C min−1) (Table 1), which confirmed the high molecular weight of P10-C and demonstrated that P10-C had a broader liquid crystalline temperature range than its low molecular weight counterpart P10-F.
 | ||
| Fig. 6 DSC curves of the azo polymers Pn-F (n = 4, 6, 10) from the first cooling scan (a) and from the second heating scan (b) (±10 °C min−1). | ||
POM observation showed that there existed obvious liquid crystalline textures in both the heating and cooling processes of P10-F and P6-F (Fig. 5b,c). The phase transition peaks in their DSC curves were proven to represent the transition between the liquid crystalline phases and isotropic phases and the mesomorphism was enantiotropic. Similar to P10-C, no well-defined liquid crystalline texture was obtained for P10-F. On the contrary, P6-F showed a batonnet texture of the smectic C mesophase after careful annealing of the sample in its liquid crystalline phase.51 In contrast to P6-F and P10-F, no obvious birefringence was observed for P4-F, which is similar as P4-C. In this context, it is worth mentioning that the presence of the liquid crystalline phase in P6-F instead of P6-C is probably due to their largely different molecular weights. The much lower molecular weight of P6-F might significantly reduce its viscosity above its Tg and allow its mesogenic azo groups to move more freely, thus leading to their more ordered self-assembled molecular structures.
It is well known that SAXS can provide useful information concerning molecular arrangement, packing mode of the mesogen, and type of order in the mesophase of a liquid crystalline polymer and it has thus been widely utilized for accurately assigning the exact nature of the liquid crystalline phases.19,54 Therefore, the liquid crystalline mesophases of both P10-C and P10-F were further studied with in situ variable temperature SAXS. Fig. 7 shows the SAXS patterns of P10-C and P10-F, which were obtained at their liquid crystalline temperatures (with a certain time of annealing) upon cooling from the isotropic states. The results showed that both P10-C and P10-F exhibited one scattering peak at q = 2.95 nm−1, suggesting the existence of an ordered smectic lamellar structure with a layer spacing d = 2.13 nm (d = 2π/q, Fig. 8).54 This d value is smaller than the calculated molecular length (l) for the fully extended side-chain liquid crystalline unit of P10-F or P10-C (l = 3.11 nm), revealing a fully interdigitated smectic C structure (i.e., near to 100% overlap of the side-chain units) with a tilted angle of 46.8° relative to the normal of the polymer backbone, as schematically shown in Fig. 8.
 | ||
| Fig. 7 SAXS patterns of P10-C at 118 °C with an annealing time of 80 min (scan time: 60 min) (a) and P10-F at 108 °C with an annealing time of 60 min (scan time: 50 min) (b) upon cooling from the isotropic states. | ||
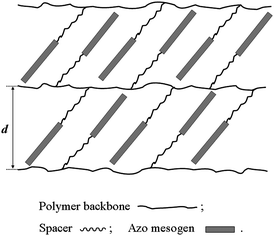 | ||
| Fig. 8 Proposed liquid crystalline structure for P10-C and P10-F. | ||
Photoisomerization behaviors of the azo polymers
The photoresponsive behaviors of Pn-C were also studied, and one representative polymer P6-C was chosen for this purpose. By being irradiated with 365 nm UV light, the studied polymer solution in DMF underwent trans to cis photoisomerization until a photostationary state was eventually reached in 210 s (Fig. 9a). The intensity of the π → π* transition band around 380 nm decreased, whereas that of the n → π* transition band around 480 nm slightly increased. The existence of the isobestic points at 328 and 452 nm was characteristic for the presence of two distinct absorbing species in equilibrium with each other and at the same time proved that no side reaction took place during the photoisomerization process in the range studied.48 | ||
| Fig. 9 UV-vis spectral changes in dependence of time for the P6-C solution in DMF (C = 0.025 mg mL−1) at 25 °C upon irradiation with 365 nm light (a) and upon irradiating the polymer solution at the photostationary state with λ > 430 nm visible light (b). | ||
Fig. 9b shows the UV-vis spectral changes of the P6-C solution in DMF with time under irradiation with λ > 430 nm visible light, where the polymer solution had been irradiated with 365 nm UV light for 210 s prior to the visible light irradiation. It can be seen clearly that the polymer solution underwent cis to trans back-isomerization upon visible light irradiation, but the finally recovered absorbance of trans-azobenzene was lower than that before UV irradiation with the recovery of the trans-isomer being 92%. Similar phenomena were also observed previously.19,55,56 Its cause might be that, at the same wavelength where the cis to trans photochemical back-isomerization was performed, there was also a weak absorption from the trans-isomer, which eventually led to an equilibrium with cis to trans and trans to cis isomerizations taking place under the same visible light after most of the cis-isomer returned to the trans-isomer.13 Nevertheless, the photochemical isomerization became completely reversible upon the subsequent cycles of UV and visible light irradiation (Fig. 10).
 | ||
| Fig. 10 UV and visible light-induced photoisomerization cycles of the P6-C solution in DMF (C = 0.025 mg mL−1). In each cycle, the polymer solution was irradiated firstly with UV light for 210 s and then with visible light for 120 s, respectively. | ||
The photochemical isomerization of the P6-C solution in DMF was also compared with that of its low molecular weight counterpart P6-F solution. As expected, the P6-F solution in DMF also underwent reversible trans to cis and cis to trans photoisomerization under the irradiation of UV and visible light, respectively (Fig. 11a and b). The time dependence of their relative absorbance (At/A0, where At refers to the absorbance of the polymer solution at 380 nm at time t upon UV or visible light irradiation and A0 the original absorbance of the polymer solution at 380 nm without UV light irradiation) showed that the photoinduced isomerization rate of the P6-F solution was close to that of the P6-C solution under the irradiation of 365 nm UV light (Fig. 12). However, the relative absorbance of the trans-azobenzene in the photostationary state finally reached for the P6-C solution proved to be somehow larger than that of the P6-F solution. Its real cause is not very clear yet at this point and further investigation is ongoing to provide a reasonable explanation for this phenomenon. In comparison, the photochemical back-isomerization processes of the polymer solutions under the irradiation of visible light were more or less the same, suggesting that the molecular weights of the azo polymers had little influence on their visible light-driven back-isomerization processes.
 | ||
| Fig. 11 UV-vis spectral changes in dependence of time for the P6-F solution in DMF (C = 0.025 mg mL−1) at 25 °C upon irradiation with 365 nm light (a) and upon irradiating the polymer solution at the photostationary state with λ > 430 nm visible light (b). | ||
 | ||
| Fig. 12 Dependence of the relative absorbance (At/A0) on time upon irradiating with 365 nm UV light (filled symbols) or with λ > 430 nm visible light (empty symbols) for the P6-C solution (cycle) and P6-F solution (triangle) in DMF (C = 0.025 mg mL−1) at 25 °C, respectively. | ||
Conclusions
We have demonstrated that copper(I)-catalyzed polymer analogous click chemistry is a highly efficient approach to generating photoresponsive azo-containing side-chain LCPs with rather high molecular weights (up to 303000). A series of azo compounds with an azido end group and different flexible spacers were successfully attached onto poly(PgMA) with CuBr/NHPMI as the catalyst. The azo functionalization degrees of the obtained polymers were determined to be ≥ 97% by comparing their UV-vis spectra with those of the corresponding low molecular weight counterparts (which were prepared via the conventional free radical polymerization of triazole-containing methacrylate azo monomers). The largely different molecular weights of the azo polymers proved to have significant influence on their phase transition behaviors. Both the high and low molecular weight azo polymers with a flexible spacer = (CH2)10 showed liquid crystallinity with their mesophases being smectic C (as revealed by SAXS) and an increase in the molecular weight resulted in a broader temperature range of the liquid crystalline phase, which is highly useful for the potential applications. In addition, the photoresponsivity of the polymer solutions was confirmed by their occurrence of the UV and visible light-induced reversible trans-cis photoisomerization and the highly reversible photoisomerization of the polymer solutions was proved to be hardly affected by the increase of the molecular weights. Considering the great versatility of copper(I)-catalyzed click chemistry and the easy accessibility of many different kinds of precursor polymers with either acetylene or azido functional groups (in particular those well-defined ones prepared via controlled radical polymerization techniques), we believe that copper(I)-catalyzed polymer analogous click reaction represents a facile, general, and efficient route to obtaining novel advanced functional polymer materials bearing either azo or various other pendant groups with both high molecular weights and high functionalization degrees. Furthermore, we also foresee that such high molecular weight azo-containing side-chain LCPs will find broad applications in the fabrication of various optical and optoelectronic devices with good mechanical properties.57 Work in this direction is currently in progress.Acknowledgements
The authors thank the financial support from National Natural Science Foundation of China (20974048), Natural Science Foundation of Tianjin (06YFMJC15100), and a Start-up Foundation from Nankai University.References
- H. Zhang, C. Li, W. Huang and B. He, Chin. Polym. Bull., 1998, 63–69 Search PubMed.
- N. K. Viswanathan, D. Y. Kim, S. Bian, J. Williams, W. Liu, L. Li, L. Samuelson, J. Kumar and S. K. Tripathy, J. Mater. Chem., 1999, 9, 1941–1955 RSC.
- A. Natansohn and P. Rochon, Chem. Rev., 2002, 102, 4139–4175 CrossRef CAS.
- T. Ikeda, J. Mater. Chem., 2003, 13, 2037–2057 RSC.
- Y. Yu, M. Nakano and T. Ikeda, Nature, 2003, 425, 145 CrossRef CAS.
- V. Shibaev, A. Bobrovsky and N. Boiko, Prog. Polym. Sci., 2003, 28, 729–836 CrossRef CAS.
- Y. Zhao, Pure Appl. Chem., 2004, 76, 1499–1508 CrossRef CAS.
- P. Xie and R. Zhang, J. Mater. Chem., 2005, 15, 2529–2550 RSC.
- Y. Li, Y. He, X. Tong and X. Wang, J. Am. Chem. Soc., 2005, 127, 2402–2403 CrossRef CAS.
- T. Ikeda, J. I. Mamiya and Y. Yu, Angew. Chem., Int. Ed., 2007, 46, 506–528 CrossRef CAS.
- Q. Bo, A. Yavrian, T. Galstian and Y. Zhao, Macromolecules, 2005, 38, 3079–3086 CrossRef CAS.
- F. You, M. Y. Paik, M. Häckel, L. Kador, D. Kropp, H. W. Schmidt and C. K. Ober, Adv. Funct. Mater., 2006, 16, 1577–1581 CrossRef CAS.
- M. H. Li, P. Keller, B. Li, X. Wang and M. Brunet, Adv. Mater., 2003, 15, 569–572 CrossRef CAS.
- Z. Zheng, J. Xu, Y. Sun, J. Zhou, B. Chen, Q. Zhang and K. Wang, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 3210–3219 CrossRef CAS.
- H. Zhang, W. Huang, C. Li and B. He, Eur. Polym. J., 1998, 34, 1521–1529 CrossRef CAS.
- H. Yu, T. Iyoda and T. Ikeda, J. Am. Chem. Soc., 2006, 128, 11010–11011 CrossRef CAS.
- H. J. Haitjema, R. Buruma, G. O. R. Alberda van Ekenstein, Y. Y. Tan and G. Challa, Eur. Polym. J., 1996, 32, 1447–1455 CrossRef CAS.
- H. W. Gu, P. Xie, P. F. Fu, T. Y. Zhang and R. B. Zhang, Adv. Funct. Mater., 2005, 15, 125–130 CrossRef CAS.
- X. Li, R. Wen, Y. Zhang, L. Zhu, B. Zhang and H. Zhang, J. Mater. Chem., 2009, 19, 236–245 RSC.
- M. A. Gauthier, M. I. Gibson and H. A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS.
- M. Ahlheim and F. Lehr, Macromol. Chem. Phys., 1994, 195, 361–373 CrossRef CAS.
- C. Samyn, T. Verbiest, E. Kesters, K. Van den Broeck, M. Van Beylen and A. Persoons, Polymer, 2000, 41, 6049–6054 CrossRef CAS.
- C. Frenz, A. Fuchs, H. W. Schmidt, U. Theissen and D. Haarer, Macromol. Chem. Phys., 2004, 205, 1246–1258 CrossRef CAS.
- T. Breiner, K. Kreger, R. Hagen, M. Häckel, L. Kador, A. H. E. Müller, E. J. Kramer and H. W. Schmidt, Macromolecules, 2007, 40, 2100–2108 CrossRef.
- M. Murali, P. Sudhakar, B. S. Kumar and A. B. Samui, J. Appl. Polym. Sci., 2009, 111, 2562–2573 CrossRef CAS.
- F. D. Jochum and P. Theato, Polymer, 2009, 50, 3079–3085 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- M. Meldal, Macromol. Rapid Commun., 2008, 29, 1016–1051 CrossRef CAS.
- C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900–4908 CrossRef CAS.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15–54 CrossRef CAS.
- J. F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef CAS.
- G. Mantovani, V. Ladmiral, L. Tao and D. M. Haddleton, Chem. Commun., 2005, 2089–2091 RSC.
- X. Shen, H. Liu, Y. Li and S. Liu, Macromolecules, 2008, 41, 2421–2425 CrossRef CAS.
- Q. Zeng, Z. A. Li, Z. Li, C. Ye, J. Qin and B. Z. Tang, Macromolecules, 2007, 40, 5634–5637 CrossRef CAS.
- Z. A. Li, G. Yu, P. Hu, C. Ye, Y. Liu, J. Qin and Z. Li, Macromolecules, 2009, 42, 1589–1596 CrossRef CAS.
- Z. A. Li, G. Yu, Y. Liu, C. Ye, J. Qin and Z. Li, Macromolecules, 2009, 42, 6463–6472 CrossRef CAS.
- X. Xue, J. Zhu, W. Zhang, Z. Zhang and X. Zhu, Polymer, 2009, 50, 4512–4519 CrossRef CAS.
- X. Xue, J. Zhu, Z. Zhang, N. Zhou, Y. Tu and X. Zhu, Macromolecules, 2010, 43, 2704–2712 CrossRef CAS.
- C. Saravanan, S. Senthil and P. Kannan, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7843–7860 CrossRef CAS.
- J. del Barrio, L. Oriol, R. Alcalá and C. Sánchez, Macromolecules, 2009, 42, 5752–5760 CrossRef CAS.
- J. del Barrio, L. Oriol, R. Alcalá and C. Sánchez, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1538–1550 CrossRef CAS.
- M. Li, E. Zhou, J. Xu, C. Yang and X. Tang, Polym. Bull., 1995, 35, 65–72 CrossRef CAS.
- D. M. Haddleton, M. C. Crossman, B. H. Dana, D. J. Duncalf, A. M. Heming, D. Kukulj and A. J. Shooter, Macromolecules, 1999, 32, 2110–2119 CrossRef CAS.
- M. Munteanu, S. Choi and H. Ritter, Macromolecules, 2008, 41, 9619–9623 CrossRef CAS.
- B. S. Sumerlin, N. V. Tsarevsky, G. Louche, R. Y. Lee and K. Matyjaszewski, Macromolecules, 2005, 38, 7540–7545 CrossRef CAS.
- H. Zhang, B. Klumperman, W. Ming, H. Fischer and R. Van der Linde, Macromolecules, 2001, 34, 6169–6173 CrossRef CAS.
- J. Geng, J. Lindqvist, G. Mantovani and D. M. Haddleton, Angew. Chem., Int. Ed., 2008, 47, 4180–4183 CrossRef CAS.
- M. Niemann and H. Ritter, Makromol. Chem., 1993, 194, 1169–1181 CrossRef CAS.
- A. M. Donald and A. H. Windle, Liquid crystalline polymers. Cambridge University Press, Cambridge, U.K., 1992 Search PubMed.
- S. Hvilsted, F. Andruzzi, C. Kulinna, H. W. Siesler and P. S. Ramanujam, Macromolecules, 1995, 28, 2172–2183 CrossRef CAS.
- J. L. Zhou, X. F. Chen, X. H. Fan, C. P. Chai, C. X. Lu, X. D. Zhao, Q. W. Pan, H. Y. Tang, L. C. Gao and Q. F. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4532–4545 CrossRef CAS.
- H. Finkelmann, H. Ringsdorf and J. H. Wendorff, Makromol. Chem., 1978, 179, 273–276 CrossRef CAS.
- S. Montserrat and R. Colomer, Polym. Bull., 1984, 12, 173–180 CAS.
- X. F. Chen, K. K. Tenneti, C. Y. Li, Y. Bai, X. Wan, X. Fan, Q. F. Zhou, L. Rong and B. S. Hsiao, Macromolecules, 2007, 40, 840–848 CrossRef CAS.
- G. Wang, X. Tong and Y. Zhao, Macromolecules, 2004, 37, 8911–8917 CrossRef CAS.
- H. Akiyama and N. Tamaoki, Macromolecules, 2007, 40, 5129–5132 CrossRef CAS.
- Our primary results revealed that the high molecular weight azo polymers showed better mechanical properties in comparison with their low molecular weight counterparts. For instance, the solid film of P6-F proved to be much more brickle than that of P6-C.
Footnote |
| † Electronic supplementary information (ESI) available: SEC chromatograms of poly(PgMA), P4-C, and P6-C as well as the dependence of the UV absorbance of the solutions of P6-F and P6-C in DMF on their concentrations. See DOI: 10.1039/c0py00138d |
| This journal is © The Royal Society of Chemistry 2010 |