Ring-opening polymerization of L-lactide using N-heterocyclic molecules: mechanistic, kinetics and DFT studies†
Vimal
Katiyar
and
Hemant
Nanavati
*
Department of Chemical Engineering, Indian Institute of Technology Bombay, Powai, Mumbai, 400 076, India. E-mail: vimk@risoe.dtu.dk; hnanavati@che.iitb.ac.in; Fax: +91 22 2572 6895; Tel: +91 22 2576 7215
First published on 10th August 2010
Abstract
We report here our investigations of the mechanism and kinetics of N-heterocyclic molecules as initiators for PLA synthesis. This is a step towards employing life saving drugs based on N-heterocyclic functional groups, to generate carrier-drug complexes in situ, via ring opening polymerization (ROP) of L-lactide. During administration, the drug may be released with well-defined release rate kinetics. With this motivation, five and six membered N-heterocyclic molecules such as 4-pyrrolidino-pyridine (PDP), 2-methyl-pyridine, pyridine, pyrrole and imidazole, are examined as initiators of bulk ROP of L-lactide, to yield metal-free poly(L-lactide) (PLA). NMR, IR and MALDI-TOF MS analyses, establish the reacting species in the polymerization. Side reactions such as formation of macrocycles, occur when permitted by lack of steric hindrance, as when polymerization is initiated by PDP + benzyl alcohol or by imidazole. Accordingly, we propose a PDP-based anionic polymerization mechanism, and then examine the effect of reaction parameters. The kinetics studies indicate an overall first order reaction with lactide, and low activation energy. Density functional theory (DFT) calculations of the initiators' proton affinities, provide a fundamental understanding, to enable selection of more efficient various N-heterocyclic drug molecules, which can be utilized as initiators, to generate PLA chains as carriers for drug administration. We find that PDP has the highest proton affinity, and that the order of the calculated proton affinities correlates broadly with the resulting PLA molecular weights. We also find that PDP appears to the most effective initiator, yielding Mw ∼28,000 Da in 90 min, which is in line with the correlation between initiator effectiveness and proton affinities, of the investigated five and six member N-heterocyclic molecules.
Introduction
Poly (L-Lactide) (PLA) belongs to the family of aliphatic polyesters. It is a thermoplastic, high strength biodegradable polymer, and can be made from annually renewable sources such as corn-starch.1–4 Advances in PLA synthesis, along with improved material properties, have resulted in its extensive biomedical applications.5 PLA synthesis is generally carried out in solution6,7 or in bulk.8–10 The basic advantages of solution polymerization over bulk, are minimization of side reactions (inter and intra-chain transesterification reactions) and the ability to execute living polymerization. However, the rate of solution polymerization, is slower than that of bulk polymerization, and is not favorable for large-scale production of PLA.11 Hence, our study focuses on bulk polymerization of L-lactide. Various bulk synthesis methods, have been extensively reported for PLA, such as direct polycondensation,12 azeotropic-dehydrative condensation,13 melt/solid polycondensation14 and ring opening polymerization (ROP).7,10,15,16 However, only bulk ROP of lactide, yields high molecular weight (MW) PLA, in a short polymerization time.To date, several metallic and organometallic catalysts, have been examined for PLA synthesis.17,18 These include compounds of tin, aluminium, zinc, calcium, mixed ligand lithium aggregates and rare earth metals. The most extensively used catalyst in the synthesis of high molecular weight PLA by ROP, is stannous octoate (tin(II)-2-ethylhexanoate). It is highly active, with typical bulk reaction times ranging from a minute to a few hours, is capable of producing stereo-controlled PLA, and the American Food and Drug Administration (FDA) has approved this compound as a food additive. However, the toxicity19 and downstream polymer degradation, associated with compounds based on most metals including tin, is a major concern for biomedical applications.
Although quantitative toxicity analyses of PLA-catalyst complexes are rare, considering its tremendous potential for biomedical applications, it is of interest to consider purely organic initiators for PLA synthesis. In particular, the motivation for investigating ROP of L-lactide using N-heterocyclic molecules as initiators, is that there are various drugs (antiviral,20 anti-HIV,21 anti-inflammatory22 drugs, etc.) which contain functional groups with similar heterocyclic nitrogen atoms, which can effectively connect to the PLA molecules. Therefore, an in situ generated drug carrier (PLA) along with the drug molecule, could yield well-defined release rate kinetics, as drug release becomes a chemically driven process.
Along these lines, organic molecules, as optimized initiators for PLA synthesis, make PLA useful for a range of biomedical applications, including drug delivery carriers,23–25 orthopedic fixation devices (i.e., bone nails, screws, plates, rods and pins),26–30 wound closure (resorbable sutures, suture anchors and surgical staples),27 stents,31 artificial tissues (cartilage, bone, liver and intestine),32,33 scaffolds for cell transplantation,34,35 adhesion barriers, dental implants and vascular grafts.36
In order to synthesize metal-free PLA, phosphine derived compounds such as P(Bu)3, PhP(Me)2, Ph2P(Me), PPh3, have been discovered for controlled ROP of lactide.7 Purely organic catalytic approaches, have also been explored using N-heterocyclic carbenes.37,38 Thiourea-amine initiators are also extraordinarily selective catalysts for controlled ROP of lactide, to generate well-defined homopolymers and block copolymers.39 Hassner and Alexanian,40 have proposed 4-(N, N-dialkylamino) pyridine based compounds such as 4-(N,N-dimethylamino) pyridene (DMAP) and 4-pyrrolidino-pyridine (PDP) as initiators, in the presence of dicyclohexyldicarbodiimide (DCC), for the synthesis of ester from carboxylic acid and alcohol. Further, DMAP initiated solution polymerization of lactide, has been reported to yield moderate MW PLAs with a number average MW, Mn ∼17,300 D, after 20 h.41 However, to be useful, an organically initiated PLA, should be synthesized under the more rapid (reaction time ∼ a few hours) bulk (i.e., solvent-free) conditions.
As a first step towards employing drugs containing heterocyclic nitrogen based functional groups, as initiators for lactide ROP, we have examined a group of N-heterocyclic molecules (Fig. 1) comprising PDP, 2-methyl-pyridine (MP), pyridine (Py), pyrrole (Pyl) and imidazole (Imd), for PLA synthesis. We have synthesized PLA of weight-averaged molecular weights (Mw), ranging from a few hundred to ∼28,000 D, within 2 h, under solvent-free, melt conditions. We find that the PDP-initiated ROP yields the highest MW PLA, and therefore, we propose here its reaction mechanism, and provide our estimates of the kinetics parameters. We also find that the initiator activity of these N-heterocyclic molecules, is correlated to their cationic affinities (obtained via DFT studies), and that the initiator with the highest proton affinity, PDP, yields the highest MW PLA. Our investigations also indicate that the steric hindrance by the initiator, i.e., its molecular geometry, affects the occurrence of backbiting and macrocyclic forming side reactions.
 | ||
| Fig. 1 Structures of N-heterocyclic molecules, investigated as initiators for ROP of lactide. | ||
Results and discussion
The data from our polymerization experiments, are reported in Table 1 (an extended data table is provided in the ESI, Appendix A†). These experiments examine the behavior of the initiator during bulk ROP of L-lactide, both, in absence and in presence, of a co-initiator such as benzyl alcohol (BA).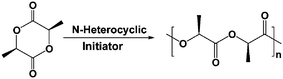
| Entry | Initiator:/L/[I] | T/°C | t/min | M n | DP n | M w | M w /Mn | Conv. (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | PDP-50 | 120 | 90 | 18,000 | 126 | 28,000 | 1.52 | 98 |
| 2 | PDP-100 | 120 | 90 | 14,000 | 98 | 20,000 | 1.43 | 95 |
| 3 | PDP-250 | 120 | 90 | 7,000 | 52 | 8,900 | 1.19 | 51 |
| 4 | PDP-500 | 120 | 90 | 6,200 | 43 | 7,700 | 1.24 | 49 |
| 5 | PDP-50 | 100 | 90 | 15,000 | 107 | 18,000 | 1.18 | 70.5 |
| 6 | PDP-50 | 140 | 90 | 15,000 | 107 | 24,000 | 1.54 | 90.5 |
| 7 | PDP-50 | 160 | 90 | 10,400 | 73 | 14,500 | 1.39 | 97.5 |
| 8 | PDP-50 | 180 | 90 | 9,000 | 62 | 12,000 | 1.3 | 98 |
| 9 | PDP/BA 50 | 120 | 90 | 8,800 | 61 | 11,000 | 1.28 | 99.7 |
| 10 | PDP/BA −100 | 120 | 90 | 9,900 | 69 | 11,000 | 1.13 | 96.2 |
| 11 | PDP/BA −250 | 120 | 90 | 9,300 | 65 | 11,000 | 1.15 | 76.3 |
| 12 | PDP/BA −500 | 120 | 90 | 10,000 | 70 | 12,000 | 1.19 | 67.4 |
| 13 | PDP/BA 50 | 100 | 90 | 12,000 | 84 | 14,000 | 1.18 | 92 |
| 14 | PDP/BA −50 | 140 | 90 | 6,700 | 47 | 9,700 | 1.45 | 98 |
| 15 | PDP/BA −50 | 160 | 90 | 6,000 | 42 | 9,300 | 1.56 | 99 |
| 16 | PDP/BA −50 | 180 | 90 | 5,900 | 41 | 7,700 | 1.30 | 99 |
| 17 | pyridine −50 | 120 | 90 | 280 | 2 | 300 | 1.05 | 30 |
| 18 | pyrrole −50 | 120 | 90 | 460 | 3 | 490 | 1.07 | 12 |
| 19 | imidazole −50 | 120 | 90 | 6,700 | 47 | 8,200 | 1.22 | 66.7 |
| 20 | 2-methyl-pyridine −50 | 120 | 90 | 880 | 6 | 940 | 1.05 | 16.2 |
| 21 | pyridine/BA −50 | 120 | 90 | 780 | 6 | 800 | 1.02 | 9.9 |
| 22 | pyrrole/BA −50 | 120 | 90 | 460 | 3 | 460 | 1.00 | 5.9 |
| 23 | 2-methyl-pyridine/BA −50 | 120 | 90 | 760 | 6 | 810 | 1.01 | 21.3 |
| 24 | Imidazole/BA −50 | 120 | 90 | 4,600 | 32 | 5,400 | 1.16 | 94.8 |
Reacting species in bulk polymerization of L-lactide
| M1c = 72x + MPDP (148) + MH+ (1) | (1) |
| M2c = 72x + MBA(108) + MNa+ (23) | (2) |
| M3c = 72x + MImd(68) + MH+ (1) | (3) |
| Mr = 72x | (4) |

PLA synthesized by PDP + BA (Fig. 3), contains three different types of PLA chains, located at masses, consistent with eqn (1), (2) and (4). These correspond to four distinct population envelopes, A, B, C and D. Envelope A confirms the growth of a PLA chain with one PDP terminal group, similar to that in Fig. 2, with masses at intervals of 144 D, representing insertion of lactide molecules, without influence of side reactions. BA substituted PLA chains are represented by both, envelope B (x is an odd number of lactyl repeat units) and envelope C (x is an even number of lactyl repeat units). These envelopes of even or odd x, appear due to backbiting of the propagating anions, which yields initiator-free macrocyclics (eqn (4)), designated by envelope D. Of these, BA terminated chains are more intense, and hence, BA is recognized as a chain transfer agent.
![MALDI TOF MS analysis of PLA synthesized using PDP + BA as initiator. Conditions: [L]/[I] = 50 at 120 °C for 90 min. Envelopes A, B, C and D, correspond to species satisfying eqn (1), (2) (with odd values for x), (2) (with even values for x) and (4), respectively.](/image/article/2010/PY/c0py00125b/c0py00125b-f3.gif) | ||
| Fig. 3 MALDI TOF MS analysis of PLA synthesized using PDP + BA as initiator. Conditions: [L]/[I] = 50 at 120 °C for 90 min. Envelopes A, B, C and D, correspond to species satisfying eqn (1), (2) (with odd values for x), (2) (with even values for x) and (4), respectively. | ||
The series of polymerizations using Imd as the initiator, yields PLA chains with Imd as a terminal group (eqn (3)), along with macrocyclic compounds (eqn (4)). Cyclization occurs when Imd is used as the initiator, as in cases of chains with BA as a terminal group (obtained when PDP is used with BA as co-initiator). This indicates that the lower steric hindrance by Imd of the terminal carbonyl carbon of the PLA chain permits the backbiting reaction through the terminal anion.
MALDI TOF MS spectra (Fig. 4) of PLA synthesized by imidazole, also contain four different envelopes of chain populations. Envelope A represents the insertion of lactide molecules into the Imd initiated PLA chains, corresponding to even values of x in eqn (3). Envelope B corresponds to odd values of x in eqn (3), possibly due to side reactions, such as formation of macrocyclic compounds. These macrocyclic compounds (eqn (4)) are evidenced by population envelopes C and D, corresponding to even and odd values of x, respectively, at 144 D intervals.
![MALDI TOF MS analysis of PLA synthesized using imidazole as initiator. Conditions: [L]/[I] = 50 at 120 °C for 90 min. Envelopes A, B, C and D, correspond to species satisfying eqn (3) (with even values for x), 3 (with odd values for x), (4) (with even values for x) and (4) (with odd values for x), respectively.](/image/article/2010/PY/c0py00125b/c0py00125b-f4.gif) | ||
| Fig. 4 MALDI TOF MS analysis of PLA synthesized using imidazole as initiator. Conditions: [L]/[I] = 50 at 120 °C for 90 min. Envelopes A, B, C and D, correspond to species satisfying eqn (3) (with even values for x), 3 (with odd values for x), (4) (with even values for x) and (4) (with odd values for x), respectively. | ||
Hence, we infer that PLA synthesized by PDP, is free from unwanted side reactions such as cyclization, since the bulky PDP group attached to the terminal carbonyl carbon, prevents PDP substitution, and thus prevents the macrocycle-forming backbiting reactions of the propagating anion.
![13C NMR spectrum (400 MHz.) in CDCl3 of PLA via PDP initiated ROP. Conditions: [L]/[I] = 25 at 120 °C, ROP time = 90 min. Species 1b as per Scheme 1.](/image/article/2010/PY/c0py00125b/c0py00125b-f5.gif) | ||
| Fig. 5 13C NMR spectrum (400 MHz.) in CDCl3 of PLA via PDP initiated ROP. Conditions: [L]/[I] = 25 at 120 °C, ROP time = 90 min. Species 1b as per Scheme 1. | ||
13C NMR spectra of PLA synthesized by PDP with BA (Fig. 6), exhibit five additional peaks. The peaks corresponding to benzyl ring carbons, are at around 128.5, 128.6 and 128.7 ppm, with a very small peak at 140.4 ppm, for ipso-carbon. There is also a peak at ∼67 ppm, corresponding to the –CH2 of BA attached to the PLA chain. 1H NMR spectra of the PLA synthesized by PDP with BA (ESI, Appendix B, Fig. B1 and B2†), also exhibit additional benzenoid proton peaks at ∼7.3 ppm, along with a quartet at 4.35 ppm, assigned to PLA's terminal –CH–OH group. Hence, benzyl alcohol partly substitutes the PDP on the PLA chain. This observation is also confirmed by the MALDI TOF MS spectra (Fig. 3).
![13C NMR spectrum (400 MHz) in CDCl3 of PLA by PDP + BA (molar ratio = 1) initiated ROP. Conditions: [L]/[I] = 50 at 120 °C, ROP time = 90 min. Species 1b and 1c, as per Scheme 1.](/image/article/2010/PY/c0py00125b/c0py00125b-f6.gif) | ||
| Fig. 6 13C NMR spectrum (400 MHz) in CDCl3 of PLA by PDP + BA (molar ratio = 1) initiated ROP. Conditions: [L]/[I] = 50 at 120 °C, ROP time = 90 min. Species 1b and 1c, as per Scheme 1. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), νas (CH3), νs (CH3), ν (COC), and ν (C–COO), are at 1758, 2997, 2948, 1274 and 865 cm−1, respectively. Bending frequencies for δs (CH3), δas (CH3) and δ (CO), have been identified at 1385, 1458 and 760 cm−1, respectively. The sharp peak corresponding to ν (CO–N) at ∼1645 cm−1, highlights the presence of an amide group. The two distinct sharp bands appearing at 576 and 642 cm−1, are due to the in-plane bending of the OC–N group, formed by the PDP fragment, which lies at the end of the PLA chain, as confirmed by MALDI-TOF-MS analysis. The intensities of these peaks, are significantly reduced in case of PDP + BA as initiator, because of the partial substitution of PDP by benzyl oxide groups. This is in good agreement with its MALDI-TOF-MS spectra (Fig. 3), confirming the presence of both species, satisfying eqn (1) and (2).
O), νas (CH3), νs (CH3), ν (COC), and ν (C–COO), are at 1758, 2997, 2948, 1274 and 865 cm−1, respectively. Bending frequencies for δs (CH3), δas (CH3) and δ (CO), have been identified at 1385, 1458 and 760 cm−1, respectively. The sharp peak corresponding to ν (CO–N) at ∼1645 cm−1, highlights the presence of an amide group. The two distinct sharp bands appearing at 576 and 642 cm−1, are due to the in-plane bending of the OC–N group, formed by the PDP fragment, which lies at the end of the PLA chain, as confirmed by MALDI-TOF-MS analysis. The intensities of these peaks, are significantly reduced in case of PDP + BA as initiator, because of the partial substitution of PDP by benzyl oxide groups. This is in good agreement with its MALDI-TOF-MS spectra (Fig. 3), confirming the presence of both species, satisfying eqn (1) and (2).
 | ||
| Fig. 7 Comparative FTIR spectra of the PLA synthesized by PDP alone (upper spectrum) and with BA (lower spectrum). | ||
Furthermore, a broad peak appears at 3446 cm−1, which may possibly correspond to terminal ν (C–O⋯H). However, the same peak is shifted to 3506 cm−1, in the region of free –OH, when polymerization is carried out using PDP with BA as a co-initiator, in which case, the hydroxyl group also lies at the end of the PLA chain. Additionally, in combination with a broad peak around 3100 cm−1, corresponding to aromatic C–H stretching of the benzyl group, combination bands appear around 2000 cm−1 (C–O stretching) and at 707 cm−1 (ring CC), which indicate the presence of benzyl alcohol terminated PLA chains (Fig. 7, lower spectrum). These also corroborate the MALDI-TOF-MS spectra (Fig. 3) conclusions, in terms of species satisfying eqn (2). Spectra of PLA synthesized, both, with and without co-initiator (BA), exhibit peaks at 760 cm−1 and 865 cm−1, which correspond to the crystalline and amorphous phases, respectively, indicating that PDP synthesized PLA, possesses a semicrystalline morphology.
We propose a reaction pathway as depicted in Scheme 1. The polymerization first initiates with PDP, and forms an active anionic adduct (1a), which is responsible for sequential addition of the L-lactide molecules, to form a linear PLA chain with a PDP terminus (1b), as confirmed by the eqn (1) in MALDI TOF MS analysis (Fig. 2). However, if lactide ROP is initiated by PDP in presence of BA acting as a co-initiator, a lower MW PLA is obtained, when compared with PLA via ROP, initiated by PDP alone, although higher conversions are achieved when co-initiator is present, under otherwise identical conditions. This improvement in the conversion, may be because the benzyl oxide anion of BA, participates in transfer reactions with some of the growing PLA (1b) chains, and follows path 1 (Scheme 1), to yield isolated benzyl oxide terminated 1c, while releasing PDP. The released PDP could be further utilized by the residual L-lactide. This notion is corroborated by Fig. 6, which indicates the absence of L-lactide. Additionally, comparing entries 1 and 9 in Table 1, indicates that lactide conversion increases, and approaches 100%, possibly because the released PDP, reacts with the residual lactide.
 | ||
| Scheme 1 The proposed scheme for ROP of L-lactide by PDP alone, yields 1a and 1b. ROP by PDP with BA, yields 1c and 1d along with 1b. | ||
We have also observed the formation of macrocyclics (1d), when BA used as co-initiator with PDP, as indicated in MALDI TOF MS spectra (Fig. 3), which contains peaks corresponding to eqn (4). This may also be one of the reasons, for the lower MW than for PLA by ROP, initiated by PDP alone. Additionally, in case of Imd as initiator, the PLA chains' MW's, are consistent with eqn (3) and (4). This suggests that the terminal anionic oxygen replaces imdazole from the carbonyl carbon, to form macrocyclics, because the smaller size of Imd, creates a less hindered occupancy, near the terminal carbonyl carbon.
Therefore, we infer that anionic chain-growth ROP of L-lactide, occurs while using N-heterocyclic molecules, along with partial transesterification side-reactions The latter occur, when a small co-initiator is employed, or when there is no steric hindrance, as evidenced in case of imidazole, which is the smaller molecule (Fig. 1). Such side reactions are common, particularly, when ROP is carried out in the melt state.
Effect of reaction parameters on melt polymerization of L-lactide
![GPC determined Mn as function of PDP-initiated L-lactide polymerization time, [L]/[I] = 50. Trendlines are added as a visual aid.](/image/article/2010/PY/c0py00125b/c0py00125b-f8.gif) | ||
| Fig. 8 GPC determined Mn as function of PDP-initiated L-lactide polymerization time, [L]/[I] = 50. Trendlines are added as a visual aid. | ||
Fig. 9 indicates that the maximum Mn values, are obtained at 120 °C for [L]/[I] molar ratios of 50 and 100, for PLA of PDP initiated ROP. In contrast, as explained above, PDP + BA yields a monotonically decreasing MW above 100 °C. This system yields PLA of a comparatively lower MW, since BA behaves as a chain transfer agent, and forms partly isolated PLA (1c) with macrocyclic compounds, whose peak locations satisfy eqn (4). Therefore, to achieve high Mn PLA, polymerization should preferentially be carried out at a low temperature (120 °C) in absence of benzyl alcohol. Reduction in molecular weight at [L]/[I] = 50, is more rapid than at [L]/[I] = 100 at higher temperatures, because of the availability (at [L]/[I] = 50) of more active sites for side reactions. This effect dominates, especially when most of the monomer is consumed, and conversion approaches 100%.
![M
n
as function of ROP temperature for [L]/[I] = 50 and 100, ROP time = 90 min. Trendlines are added as a visual aid.](/image/article/2010/PY/c0py00125b/c0py00125b-f9.gif) | ||
| Fig. 9 M n as function of ROP temperature for [L]/[I] = 50 and 100, ROP time = 90 min. Trendlines are added as a visual aid. | ||
![GPC determined Mn as function of [L]/[I], L-lactide polymerization initiated by PDP at 120 °C and 140 °C, ROP time = 90 min. Trendlines are added as a visual aid.](/image/article/2010/PY/c0py00125b/c0py00125b-f10.gif) | ||
| Fig. 10 GPC determined Mn as function of [L]/[I], L-lactide polymerization initiated by PDP at 120 °C and 140 °C, ROP time = 90 min. Trendlines are added as a visual aid. | ||
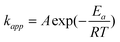 , employed for kapp values from Fig. 11 (0.0394 min−1, 0.0536 min−1 and 0.0651 min−1, at 120 °C, 140 °C and 160 °C, respectively) yields an activation energy value of 5.6 Kcal mol−1. This activation energy is lower than that for coordination insertion polymerization of L-lactide (19.55 Kcal mol−1).51 In spite of a lower activation energy, melt polymerization of L-lactide is not possible below its melting point (97 °C). At 100 °C (close to the melting point of L-lactide), diffusion limitation is an obstacle for the growing polymer chain, since viscosity increases with polymerization. Hence, only low molecular weight polymer, is obtained at 100 °C for both, with and without benzyl alcohol co-initiator.
, employed for kapp values from Fig. 11 (0.0394 min−1, 0.0536 min−1 and 0.0651 min−1, at 120 °C, 140 °C and 160 °C, respectively) yields an activation energy value of 5.6 Kcal mol−1. This activation energy is lower than that for coordination insertion polymerization of L-lactide (19.55 Kcal mol−1).51 In spite of a lower activation energy, melt polymerization of L-lactide is not possible below its melting point (97 °C). At 100 °C (close to the melting point of L-lactide), diffusion limitation is an obstacle for the growing polymer chain, since viscosity increases with polymerization. Hence, only low molecular weight polymer, is obtained at 100 °C for both, with and without benzyl alcohol co-initiator.
![L-lactide monomer conversion (−Ln([L]/[L0])) vs. time during PDP initiated ROP, at 120 °C, 140 °C, 160 °C for [L]/[I] (molar ratio) = 50.](/image/article/2010/PY/c0py00125b/c0py00125b-f11.gif) | ||
| Fig. 11 L-lactide monomer conversion (−Ln([L]/[L0])) vs. time during PDP initiated ROP, at 120 °C, 140 °C, 160 °C for [L]/[I] (molar ratio) = 50. | ||
PDP initiated anionic polymerization is also feasible at low temperature in solution, as reported by Nederberg et al.,41 although this process yields only low DPn (32 lactide units or Mn ≈ 4,600 D) PLA, even after long polymerization times (20 h).
| −d[L]/dt = {kabs[I]0.2} × [L]0.8 | (5) |
![L-lactide monomer conversion (−Ln([L]/[L0])) vs. time during PDP initiated ROP, for [L]/[I] (molar ratio) = 50 and 100 at 120 °C.](/image/article/2010/PY/c0py00125b/c0py00125b-f12.gif) | ||
| Fig. 12 L-lactide monomer conversion (−Ln([L]/[L0])) vs. time during PDP initiated ROP, for [L]/[I] (molar ratio) = 50 and 100 at 120 °C. | ||
The initiator exponent in the rate equation indicates that some molecules of the initiator may not be available for complexation with monomer. This could be due to poor dispersion of initiator within the polymerization system, since the reaction is carried out in the melt state without mixing, and because some of the initiator, may be deactivated in the presence of trace quantity of water, possibly present with L-lactide.
Correlation of initiator activities with their structure using DFT studies
As shown in Scheme 1, lactide polymerization is initiated by the interaction (affinity) of the lone pair electrons of the N-heterocyclic nitrogen atom, with the carbonyl carbon atom of the lactide molecule. We report the results of our DFT calculations (details in ESI, Appendix C†) for both, 5-membered ring N-heterocyclic molecules, Imd and Pyl, and 6-membered ring N-heterocyclic molecules, PDP, MP and Py. This analysis provides a quantitative explanation for the selection of initiator from such N-heterocyclic molecules, on the basis of their electron donation tendencies and their effects on the final PLA molecular weight.Our findings suggest a correlation between initiation activities of these molecules, with their corresponding proton affinity values, for a given geometry (defined in this case, by the number of members in the ring). The calculated proton affinities for the five and six member N-heterocyclic molecules that we have examined, are found in the order: Imd > Pyl and PDP > MP > Py, respectively. PDP possesses the highest proton affinity value (235.33 kcal mol−1), which corresponds well with PDP-initiated ROP, yielding PLA of the highest molecular weight (Mn ∼19,000 D). The proton affinities of MP and Py are lower, 219.31 and 215.03 kcal mol−1, respectively, and their corresponding PLA molecular weights are 880 D and 280 D. Similar trends of proton affinities have been observed for Imd (217.92 kcal mol−1) and Pyl (181.15 kcal mol−1), with corresponding molecular weights, 6,700 D and 460 D, respectively, under identical polymerization conditions. Hence, in continuation with these predictions, one can utilize DFT studies to determine the optimal nucleophilic molecules, in order to discover superior metal-free initiators, to achieve high MW PLA.
Experimental section
Materials
L-Lactide, 4-pyrrolidino-pyridine, 2-methyl-pyridine, imidazole and CHCl3-d were purchased from Sigma Aldrich. Chloroform, benzyl alcohol, hexane and methanol were obtained from Merck-India.Polymerization procedure
Bulk polymerization reactions have been carried out in vacuum-sealed glass ampoules. First, the glass ampoule is charged with monomer (L-Lactide), and dried for two hours under high vacuum at 50 °C. Subsequently, the initiator (PDP, MP, Py, Pyl or Imd) is added, with L-lactide to initiator molar ratio ([L]/[I]), lying in the range from 50 to 500. In polymerization runs carried out with initiator and co-initiator such as benzyl alcohol, the initiator: co-initiator composition is always kept equimolar. The ampoule is sealed under high vacuum, and immersed in a temperature controlled oil bath, for polymerization reactions in the temperature range 100–180 °C. At predetermined times (10–120 min), a glass ampoule is removed, the molten reactive polymer mixture is cooled by immersing the sealed ampoule into liquid nitrogen to arrest the polymerization, and the contents are analyzed. The polymer is precipitated in hexane, to remove unreacted lactide and residual initiator, to confirm the reacting species in the polymerization and to determine the end groups in the PLA chains. However, GPC analyses have been performed directly on the crude reaction mixture, without precipitation, to avoid fractionation of the sample, which would incorrectly influence the analyses.Characterization
The conversion for kinetics studies, is determined vialH NMR analysis (400 MHz) of crude reaction mixtures. Spectra have been obtained for 1% (w/v) solutions in CDC13. The concentration of residual L-lactide, has been determined at various ROP conditions from the relative integral ratio of the lactide (Iq,LLA) and polymer (Iq,PLLA) methine quartets, located at 5.04 and 5.2 ppm, respectively (eqn (6)).
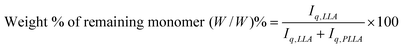 | (6) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 v/v CHC: acetonitrile), is added into the analyte. The differences between the measured and the calculated masses of peaks, are within 0.5 D. We examine whether the peaks correspond to PLA chains, bearing the initiator as a terminal group.
:50 v/v CHC: acetonitrile), is added into the analyte. The differences between the measured and the calculated masses of peaks, are within 0.5 D. We examine whether the peaks correspond to PLA chains, bearing the initiator as a terminal group.
Conclusions
The present work describes successful ROP of L-lactide in the presence of N-heterocyclic molecules. Understanding of these N-heterocylic molecules as initiators for PLA synthesis, provides an immense opportunity for utilizing similar N-functionalized drug molecules to generate an in situ PLA-drug complex, as a drug-carrier system. Accordingly, a mechanistic understanding of the chemical association of these molecules with the PLA chain, could provide a well defined release rate. We summarize here our preliminary results in this direction.Of the initiators examined here, ROP initiated by 4-pyrrolidino-pyridine, produces PLA chains of the highest molecular weight. The other initiator molecules examined, are 2-methypyridine, imidazol, pyridine and pyrole.
In combination with NMR spectroscopy, MALDI TOF MS measurement and IR analyses, we have determined that PLA chains propagate during ROP, via the N-functionalized initiation from these initiators; e.g., analyses of representative products of PDP-initiated ROP, reveal the existence of a PDP terminus on each chain. Our investigation also reveals the possibility of side reactions during ROP of L-lactide, which can also yield macrocyclic molecules, and such effects depend on the geometry of the initiator molecules; i.e., the reactions occur when there are no steric hindrances on the initiator, to prevent them.
On the basis of our analyses, we have proposed a lactide anion-based ROP mechanism, with or without presence of co-initiator. Kinetics studies of this system suggest that the rate expression is first order in monomer concentration. The rate constant of the reaction, determined as a function of temperature, yields the activation energy of the PDP initiated ROP, which is lower than that for the conventional, tin octoate initiated ROP.
In order to correlate the experimental initiation activity with electron releasing capabilities of these N-heterocyclic molecules, the proton affinities have been calculated by density functional theory. PDP has the highest gas phase proton affinity in the series of N-heterocyclics. The orders of the calculated proton affinity are Imd > Pyl and PDP > MP > Py, for 5 and 6-membered rings, respectively. These correlate with the resultant MW trends for ROPs carried out with these initiators.
Acknowledgements
The authors are grateful to the NMITLI program (Ref. 5/258/25/2003), Council of Scientific and Industrial Research (CSIR), India, for providing financial support to carry out this investigation. The authors gratefully acknowledge Dr R. P. Sunoj and Mr Dipankar Roy in Department of Chemistry at IIT Bombay, for their assistance with the DFT calculations.References
- Q. Fang and M. A. Hanna, Ind. Crops Prod., 1999, 10, 47 CrossRef CAS.
- J. Guan, K. M. Eskridge and M. A. Hanna, Ind. Crops Prod., 2005, 22, 109 CrossRef CAS.
- J. Guan and M. A. Hanna, Ind. Eng. Chem. Res., 2005, 44, 3106 CrossRef CAS.
- J. Lunt, Polym. Degrad. Stab., 1998, 59, 145 CrossRef CAS.
- A.-C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466 CrossRef CAS.
- Ph. Dubois, N. Ropson, R. Jérôme and Ph. Teyssié, Macromolecules, 1996, 29, 1965 CrossRef CAS.
- M. Myers, E. F. Connor, T. Glauser, A. Möck, G. Nyce and J. L. Hedrick, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 844 CrossRef CAS.
- X. Wang, K. Liao, D. Quan and Q. Wu, Macromolecules, 2005, 38, 4611 CrossRef CAS.
- A. J. Nijenhuis, D. W. Grijpma and A. J. Pennings, Macromolecules, 1992, 25, 6419 CrossRef CAS.
- K. Shinno, M. Miyamoto, Y. Kimura, Y. Hirai and H. Yoshitome, Macromolecules, 1997, 30, 6438 CrossRef CAS.
- D. Garlotta, J. Polym. Environ., 2001, 9(2), 63 CrossRef CAS.
- S.-H. Hyon, K. Jamshidi and Y. Ikada, Biomaterials, 1997, 18, 1503 CrossRef CAS.
- K. Enomoto, M. Ajioka and A. Yamaguchi, US Pat., 5 310 865, 1994.
- S.-I. Moon, C.-W. Lee, I. Taniguchi, M. Miyamoto and Y. Kimura, Polymer, 2001, 42, 5059 CrossRef CAS.
- P. R. Gruber, E. S. Hall, J. J. Kolstad, M. L. Iwen, R. D. Benson and R. L. Borchardt, US Pat., 5 142 023, 1992.
- N. Nomura, R. Ishii, M. Akakura and K. Aoi, J. Am. Chem. Soc., 2002, 124, 5938 CrossRef CAS.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147 CrossRef.
- B. J. O'Keefe, M. A. Hillmyer and W. B. Tolman, J. Chem. Soc., Dalton Trans., 2001, 2215 RSC.
- K. S. Stridsberg, R. Maria and A.-C. Albertsson, Adv. Polym. Sci., 2002, 157, 41 CAS.
- G. Zoidis, C. Fytas, C. Pannecouque, G. B. Foscolos, G. Fytas, A. Kolocouris, E. De-Clercq, E. Padalko, L. Naesens, J. Neyts and N. Kolocouris, Bioorg. Med. Chem., 2006, 14(10), 3341–48 CrossRef CAS.
- G. Stamatiou, G. B. Foscolos, G. Fytas, A. Kolocouris, N. Kolocouris, C. Pannecouque, M. Witvrouw, E. Padalko, J. Neyts and E. De-Clercq, Bioorg. Med. Chem., 2003, 11(24), 5485–92 CrossRef CAS.
- V. Kh. Syundyukova, E. G. Neganova, B. K. Beznosko and E. N. Tsvetkov, Pharm. Chem. J., 1992, 26, 543–655 Search PubMed.
- G. Ruan and Si-S. Feng, Biomaterials, 2003, 24, 5037 CrossRef CAS.
- T. Riley, T. Govender, S. Stolnik, C. D. Xiong, M. C. Garnett, L. Illum and S. S. Davis, Colloids Surf., B, 1999, 16, 147 CrossRef CAS.
- Y. Ikarashi, T. Tsuchiya, M. Kaniwa and A. Nakamura, Biol. Pharm. Bull., 2000, 23(12), 1470 CAS.
- D. Bendix, Polym. Degrad. Stab., 1998, 59, 129 CrossRef CAS.
- E. S. DeJong, T. M. DeBerardino, D. E. Brooks and K. Judson, Arthroscopy, 2004, 20(5), 511 CrossRef.
- D. K. Gilding and A. M. Reed, Polymer, 1979, 20, 1459 CrossRef CAS.
- T. Hayashi, Prog. Polym. Sci., 1994, 19, 663 CrossRef CAS.
- T. Saito, A. Iguchi, M. Sakurai and K. Tabayashi, Ann. Thorac. Surg., 2004, 77, 684 CrossRef.
- Y. Saito, K. Minami, M. Kobayashi, Y. Nakao, H. Omiya, H. Imamura, N. Sakaida and A. Okamura, J.Thorac. Cardiovasc. Surg., 2002, 123, 161 CrossRef.
- F. Yang, R. Murugan, S. Ramakrishna, X. Wang, Y.-X. Ma and S. Wang, Biomaterials, 2004, 25, 1891 CrossRef CAS.
- A. G. Mikos and J. S. Temenoff, Electron. J. Biotechnol., 2000, 3(2), 114 Search PubMed.
- P. A. Gunatillake and R. Adhikari, Eur. Cells Mater., 2003, 5, 1 CAS.
- E. Sachlos and J. T. Czernuszka, Eur. Cells Mater., 2003, 5, 29 CAS.
- U. Izhar, H. Schwalb, J. B. Borman, G. R. Hellener, A. H. Salomon, G. Marom, T. Stern and D. Cohn, J. Surg. Res., 2001, 95, 152 CrossRef CAS.
- J. C. Garrison and W. J. Younqs, Chem. Rev., 2005, 105, 3978 CrossRef CAS.
- E. F. Connor, G. W. Nyce, M. Myers, A. Möck and J. L. Hedrick, J. Am. Chem. Soc., 2002, 124(6), 914 CrossRef CAS.
- R. C. Pratt, Bas G. G. Lohmeijer, D. A. Long, P. N. P. Lundberg, A. P. Dove, H. B. Li, C. G. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39(23), 7863–7871 CrossRef CAS.
- A. Hassner and V. Alexanian, Tetrahedron Lett., 1978, 19, 4475 CrossRef.
- F. Nederberg, E. F. Connor, M. Möller, T. Glauser and J. L. Hedrick, Angew. Chem., Int. Ed., 2001, 40(14), 2712 CrossRef CAS.
- (a) M. J. Frisch et al., Gaussian 98, revision A.11, Gaussian, Inc., Pittsburgh, PA, 1998 Search PubMed; (b) M. J. Frisch et al. , Gaussian 03, revision C.02, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- (a) A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (c) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785 CrossRef CAS.
- E. Tajkhorshid, B. Paizs and S. Suhai, J. Phys. Chem. B, 1999, 103, 4518 CrossRef CAS.
- (a) Energy is scaled by 0.97; (b) P. S. Zuev and R. S. Sheridan, J. Am. Chem. Soc., 2004, 126, 12220 CrossRef CAS.
- J. Sun, W. Shi, D. Chen and C. Liang, J. Appl. Polym. Sci., 2002, 86, 3312 CrossRef CAS.
- S. Junquan, C. Liqing and W. Langtine, Chin. J. Funct. Polym., 1996, 2, 252 Search PubMed.
- M. K. Samantaray, V. Katiyar, K. Pang, D. Roy, H. Nanavati, R. Stephen, R. B. Sunoj and P. Ghosh, Eur. J. Inorg. Chem., 2006, 2975 CrossRef CAS.
- L. Ray, V. Katiyar, M. J. Raihan, M. M. Shaikh, H. Nanavati and P. Ghosh, Eur. J. Inorg. Chem., 2006, 3724 CrossRef CAS.
- K. Stridsberg, M. Ryner and A.-C. Albertsson, Macromolecules, 2000, 33, 2862 CrossRef CAS.
- C. Yu, L. Zhang, K. Tu and Z. Shen, Polym. Bull., 2004, 52(5), 329 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details for the ROP of L-lactide, 1H NMR of organically initiated ROP of L-lactide and DFT calculations. See DOI: 10.1039/c0py00125b |
| This journal is © The Royal Society of Chemistry 2010 |