Rapid preparation of polymersomes by a water addition/solvent evaporation method†
Hana Robson
Marsden
,
Luca
Gabrielli
and
Alexander
Kros
*
Department of Soft Matter Chemistry, Leiden Institute of Chemistry, Leiden University, P.O. Box 9502, 2300, RA, Leiden, The Netherlands. E-mail: a.kros@chem.leidenuniv.nl
First published on 10th August 2010
Abstract
In this article we demonstrate a rapid water addition/solvent evaporation method to produce polymersomes with controllable sizes. For this method a solution of an amphiphilic block copolymer in THF is quickly mixed with an aqueous solution, followed by organic solvent evaporation under reduced pressure using a rotary evaporator. The parameters that influence the formation, size, and stability of the polymersomes are easily controlled, and the entire process can take less than five minutes. The method was initially tested with a series of rod–rod peptidic block copolymers, where the hydrophilic block is a charged designed peptide, and the hydrophobic block is poly(γ-benzyl L-glutamate) with varying degrees of polymerization (35–250 monomers), and the polymersome formation was monitored and confirmed with dynamic light scattering, optical microscopy, and transmission electron microscopy. The widespread applicability of the technique was also proven with more traditional charged and non-charged coil–coil block copolymers of varying length. The method was found to be very robust with regards to salt concentration and initial mixing, and the polymersome size could be precisely adjusted over a wide range, with the same block copolymer forming polymersomes ranging from ∼200 nm to ∼2 µm in diameter. Given its simplicity, versatility, and speed, the water addition/solvent evaporation method described here is a very practical tool for polymersome preparation.
Introduction
Vesicles formed from lipids have been prepared since the early 1960s.1,2 During these decades many techniques have been developed to prepare lipid vesicles, such as film rehydration, bulk rehydration, electroformation, solvent injection, aqueous phase injection, emulsion based methods (solvent evaporation or dialysis), detergent removal techniques, and microfluidic formation.3 There are many variations on these methods, and also techniques to modify preformed vesicles such as vortexing, freeze-thawing, extrusion, or sonication.4 Each of these methods results in lipid vesicles with different properties, and as a result a remarkable range of properties are accessible: diameters from 25 nm to over 100 µm, unilamellar or multilamellar vesicles, with a relatively narrow or broad size distributions, and vesicles encapsulating varying amounts of compounds, either inside the aqueous interior or in the bilayer membrane. As a result, the physical properties required by a specific application often dictate which method of vesicle formation should be applied. For example, for tumor targeting and retention purposes 100 nm vesicles are favored,5 while for the study of cell mimics giant vesicles are preferred. However, there is no single method available that can be used to make this large range of vesicle sizes. In addition to the final vesicle characteristics, each technique has its associated advantages/disadvantages such as the use of high energy input or organic solvents, the necessity of specific equipment, or the time required for preparation. As a result, these aspects frequently direct the choice of vesicle preparation method. For instance, when proteins are involved a buffer of physiological ionic strength is often required, and sonication or organic solvents cannot be used, which narrows the possible preparation techniques.The formation of vesicles is not limited to the use of lipids, other amphiphiles can assemble into vesicles as well. Examples are block copolymers,6–12 modified cyclodextrins,13–15 and surfactants.16 Polymer vesicles are particularly interesting as there is a profusion of choice regarding block composition and length, which results in a large variety of potential vesicle properties. Research into polymer vesicles, referred to herein as ‘polymersomes’, has been carried out since 1995,8 and the remarkable vesicle properties arising from the choice of polymers have only been partially revealed. In order to allow the potential of polymersomes to be more fully exposed, the variety of preparation techniques that allows one to access different polymersomes needs to be extended. Most methods used to assemble lipids into vesicles cannot be transferred directly to polymers due to their different physical/chemical properties, primarily arising from the large size of the hydrophobic block.17,18 For example, many block copolymers cannot be directly hydrated as lipids can, due to the high glass-transition temperature of the hydrophobic block. Techniques used for lipid vesicles are therefore tailored to be applicable to polymers, or new procedures are developed. The original method of Eisenberg to prepare polymersomes, and currently the most commonly used, is to dissolve the polymer in an organic solvent followed by the gradual addition of water (typically over approx. hour), inducing the formation of aggregates. Next, the organic solvent is slowly removed by dialysis (typically days).8,19,20 Subsequent methods include other solvent exchange techniques which vary in the manner in which the organic solvent is replaced with an aqueous phase,21–26 hydration methods27,28 including electroformation7,29,30 and template methods,31 and a recently published detergent-aided method.32 The disadvantages of the current techniques are that it is generally impossible or not trivial to control the final polymersome sizes (with the exception of a microfluidic technique,26 and to a limited extent by extrusion29), for many only limited types of block copolymers can be used, and most polymersome formation procedures require at least one day to complete.
The water addition/solvent evaporation method described here is inspired by a rapid liposome preparation technique and is adapted to meet the requirements of block copolymers.33 We have recently developed a new class of peptides, polypeptide-b-peptides,25 that allows the combination of structure-inducing large hydrophobic blocks with the designed intermolecular interactions of sequence-specific peptides. Uniting these properties permits the highly controlled self-assembly of functional nanostructures. We have demonstrated this concept with a range of PBLG-b-coiled-coil-peptide polymers having different PBLG lengths. Utilizing the range of sizes of the PBLG blocks, and the coiled-coil folding of the designed peptide block,34 polymersomes with a range of diameters, membrane thicknesses, and surface functionality could be obtained. This modular control over polymersome properties will be very useful for future applications. However, for a given block copolymer it was not possible to control the polymersome diameters with the sonication method that was used. Additionally, not all of the block copolymers formed well-defined aggregates, and none of the other polymersome preparation techniques that were tested on this series of block copolymers were successful. To address these issues we developed the water addition/solvent evaporation method as demonstrated in this article. The method allows the adjustment of polymersome diameters over a wide range using the same block copolymer, and it is also applicable to the whole PBLG-b-coiled-coil-peptide series. Furthermore, the water addition/solvent evaporation method is also very generally applicable for diverse types of block copolymers, it requires only standard laboratory equipment, and can be completed within five minutes.
Experimental section
Materials
Peptides and peptide block copolymers were prepared and characterized as described previously.25,35 The synthetic coil–coil block copolymers PS106–PAA17 (P697–StAA), PEO80–PBD125 (P4753–BDEO), and PEO26–PBD46 (P9095–BdEO) were obtained from Polymer Source, Inc. The molecular characteristics of the peptides and block copolymers are given in Table 1. Phosphate buffered saline (PBS) contained 20 mM PO43− and 150 mM NaCl, and had a pH of 7.4. Rhodamine B was obtained from Fluka, and butylated hydroxytoluene stabilized tetrahydrofuran was obtained from Biosolve.| Name | Structure | M n/g mol−1 | PDIc |
|---|---|---|---|
| a Obtained from MALDI-TOF MS. b Based on 1H-NMR. c GPC calibrated with polystyrene standards. d As specified by Polymer Source, Inc. PEG: poly(ethylene glycol), PBLG: poly(γ-benzyl L-glutamate), amino acids in the designed peptides are represented by their one letter codes, Ac: acetyl, PS: polystyrene, PAA: poly(acrylic acid), PEO: poly(ethylene oxide) and PBD: polybutadiene. | |||
| E | Ac-G(E I A A L E K)3-NH2 | 2381a | |
| K | Ac-(K I A A L K E)3G-NH2 | 2378a | |
| K-PEG | Ac-(K I A A L K E)3G–PEG77 | 5832a,b | 1.05a |
| PBLG35-K | PBLG37-G(K I A A L K E)3-NH2 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 135b 135b |
1.3 |
| PBLG50-K | PBLG50-G(K I A A L K E)3-NH2 | 13279b |
1.5 |
| PBLG36-E | PBLG36-G(E I A A L E K)3-NH2 | 10230b |
1.1 |
| PBLG80-E | PBLG80-G(E I A A L E K)3-NH2 | 19877b |
1.4 |
| PBLG100-E | PBLG100-G(E I A A L E K)3-NH2 | 24262b |
1.4 |
| PBLG250-E | PBLG250-G(E I A A L E K)3-NH2 | 57148b |
1.7 |
| PS106–PAA17 | 12240d |
1.11d | |
| PEO26–PBD46 | 3800d | 1.04d | |
| PEO80–PBD125 | 10400d |
1.1d |
Preparation of polymersome suspensions
The polymersome suspensions were prepared as follows, unless stated otherwise in the text. The block copolymer (total concentration 0.02 µmol) was dissolved in 2 mL tetrahydrofuran (THF) in a 50 mL round bottomed flask. Next, 3 mL of PBS were quickly added and vortexed for 1 min at 200 rpm (see ESI†). The THF was evaporated using a rotary evaporator (40 rpm, water bath at 20 °C, 2 min), yielding the polymersome suspensions with a final concentration of 6.7 µM.For the encapsulation experiments, the samples were prepared as described above, with the addition of Rhodamine B (2 µM) to the PBS. For permeability experiments, the samples were prepared as described above, with the addition of 100 µL of Rhodamine B (20 µM) in PBS after the vortexing step.
For comparison with the water addition/solvent evaporation method described in this paper, samples were also prepared by the commonly used method developed by Eisenberg et al., in which the aqueous phase is added slowly to the organic polymer solution, and the organic solvent is then slowly removed.8,36 The block copolymer (0.02 µmol) was dissolved in 2 mL THF in a 50 mL round bottomed flask. 3 mL of water or PBS (330 mOsm kg−1 or 33 mOsm kg−1) were added dropwise at a rate of 0.2 wt% (4 µL) per min using a syringe pump (NE-300, just infusion, Prosense B.V.), while stirring at 700 rpm. The THF was then removed by dialysis. Dialysis tubing (Spectrum Laboratories, Inc.) with a molecular weight cut-off of 1000 g mol−1 was thoroughly rinsed with water or PBS, and the samples dialyzed against water or PBS at room temperature for at least 24 h with at least 5 changes of buffer.
Characterization of polymersome suspensions
Dynamic light scattering (DLS) was conducted using a Malvern Zetasizer Nano ZS equipped with a Peltier-controlled thermostatic cell holder. The laser wavelength was 633 nm and the scattering angle was 173°. Experimental diffusion coefficients, D, were measured at 25 °C. The Stokes–Einstein relationship D = kbT/3πηdh was used to estimate the hydrodynamic diameter, dh. Here kb is the Boltzmann constant and η is the solvent viscosity. To calculate the hydrodynamic diameter of objects in THF and mixtures of THF and water/PBS, viscosities and refractive indices were used from ref. 37.Transmission electron microscopy (TEM) was conducted on a JEOL 1010 instrument with an accelerating voltage of 60 kV. Samples for TEM were prepared by placing 5 µL of solution on carbon-coated copper grids. After ∼5 min, the droplet was removed from the edge of the grid. A drop of 2% phosphotungstic acid (PTA) or 2% osmium tetroxide (OsO4) stain was applied and removed after 2 min.
Differential interference contrast (DIC) and fluorescence optical micrographs were recorded with a Zeiss axiovert-200 inverted microscope equipped with a 63× objective long-range working lens. The images were recorded with a black and white CCD camera (AxioCam MRm) connected to image-recording and -processing software (Axiovision 4.4).
The amount of THF remaining in the samples prepared using the standard conditions was quantified with NMR analysis. 1H NMR spectra were recorded on a Bruker DPX300 spectrometer at room temperature. The proton resonance of water was used for ppm calibration (300 MHz, H2O, δ): 3.74 (m, 2H, CH2O), 1.88 (m, 2H, CH2). After solvent evaporation the residual THF peaks were integrated and compared with a series of water:THF standards. 97% of the THF was removed by evaporation for 2 min at 20 °C, with insignificant volumes of water evaporated.
Results and discussion
The new procedure for preparing polymersomes consists of three steps: (I) the amphiphilic block copolymer is dissolved in THF. (II) The aqueous phase is added all at once. THF and water are miscible, and the resulting decrease in solvent quality for the hydrophobic block leads to polymersome self-assembly. In step (III) the THF is removed by rotary evaporation under reduced pressure.Initially we focused on the application of the water addition/solvent evaporation method to the amphiphilic rod–rod block copolymer PBLG50-K, for which all previous efforts in assembling polymersomes had yielded undefined aggregates. Results are first shown for samples prepared using the standard conditions, and then the potential for adjusting the polymersome diameters is demonstrated. Finally, polymersomes are formed from a variety of different block copolymers, both peptidic and synthetic, to highlight the general applicability of this method.
Applying the standard conditions to PBLG50-K led to the formation of large structures with an average hydrodynamic diameter, dh, of ∼1200 nm, and a low polydispersity index (PDI ≈ 0.06) (Fig. 1A). The particles encapsulated the water-soluble dye Rhodamine B as observed by fluorescence microscopy (Fig. 1B), indicating the formation of polymersomes. Transmission electron microscopy confirmed the presence of polymersomes, with only a very minor quantity of spherical micelles (Fig. 1D). The average diameter of the polymersomes as measured by optical microscopy and dynamic light scattering was in agreement with one another, while the sizes of the dried polymersomes observed with electron microscopy were ∼50% smaller, which is most likely due to deflation upon drying. These polymersomes were stable for at least 4 months as determined by DLS.
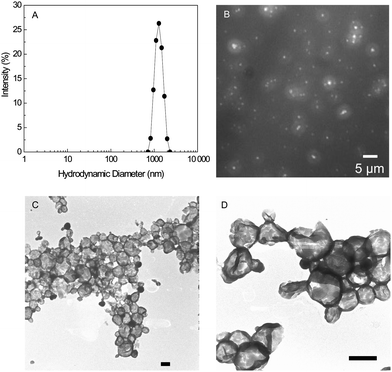 | ||
| Fig. 1 (A) DLS intensity distribution for PBLG50-K polymersomes prepared with the standard conditions. (B) Fluorescence microscope image of PBLG50-K polymersomes with encapsulated Rhodamine B. (C and D) Electron microscopy images of PTA stained PBLG50-K polymersomes. Scale bars = 500 nm. Conditions: 2 mL THF, 0.02 µmol PBLG50-K, 3 mL PBS, vortexing 1 min at 200 rpm and THF evaporated for 2 min at 20 °C. | ||
Adjusting polymersome diameters
 | ||
| Fig. 2 Particle characteristics in the mixed solvent system prior to THF evaporation. (A) DLS intensity distribution of PBLG50-K polymersomes measured ∼5 min after addition of PBS. (B) Optical bright field and (inset) fluorescence microscope images of PBLG50-K polymersomes with encapsulated Rhodamine B ∼2 min after addition of PBS. (C) The average dh of PBLG50-K assemblies as a function of the mixed solvent residence time. (D) Optical microscope image of PBLG50-K particles ∼30 min after addition of PBS. Scale bars = 5 µm. Conditions: 2 mL THF, 0.02 µmol PBLG50-K, 3 mL PBS. | ||
After THF removal the polymersome diameters in PBS are similar to the particle sizes in the mixed solvent phase. As the duration of the mixed solvent phase is extended and the particle sizes increase, the polymersome diameters after THF removal also increase (Fig. 3). These results show that there is continuity between the particles pre- and post-THF evaporation, i.e. there is not complete disassembly and reassembly of the structures during THF removal. In the final PBS solution the PBLG blocks are frozen due to the poor solvent quality, resulting in kinetically trapped polymersomes which are stable for many months.
 | ||
| Fig. 3 Average dh and the PDI of the particles in 2:3 v/v THF/PBS (○, +), and in PBS after removal of the THF at 20 °C (●, ×). | ||
 | ||
| Fig. 4 (A) TEM images of PTA-stained PBLG50-K polymersomes prepared at different temperatures. Scale bars = 500 nm. (B) The effect of the temperature at which the THF is evaporated on the average dh of PBLG50-K vesicles. (C) The average dh of PBLG50-K as a function of the time between PBS addition and THF evaporation at 20 °C (○) and 70 °C (●). | ||
When the THF was evaporated from the samples at elevated temperatures after extended periods of aging in the mixed solvent phase at room temperature, the growth in the mixed solvent phase was still correlated with larger polymersomes in PBS (Fig. 4C). However, the polymersome sizes in PBS were dominated by the temperature during solvent evaporation rather than the particle size in THF/PBS.
 | ||
| Fig. 5 Average dh of PBLG50-K polymersomes as a function of osmolality of the aqueous phase (physiological ionic strength PBS = 330 mOsm kg−1). | ||
Without the charge screening the corona repulsion ‘freezes’ the structures at ∼200 nm: the larger polymersomes that were accessible in physiological ionic strength PBS (i.e. by using low temperatures of THF evaporation, or by allowing for a period of growth prior to removing the THF) are not accessible due to the strong electrostatic repulsion between corona blocks. Also, the conditions which reduce the polymersome size in PBS (such as using an elevated temperature of solvent evaporation or omitting the mixing step) do not decrease the particle size in water because the repulsion within the corona overwhelms these effects. These experiments show that the effect of ionic strength (i.e. electrostatic repulsion) is the most important parameter controlling the polymersome diameters. This effect dominates the effect of temperature (presumably the physical size of the corona blocks), which in turn dominates the duration of the mixed solvent phase (i.e. reduction of core–solvent area).
Regarding the tolerance to different ionic strengths of the aqueous phase and to tuning the polymersome diameters, the water addition/solvent evaporation method is very flexible in comparison to the widely used method pioneered by Eisenberg and Zhang.8 In that method water is injected very slowly to a solution of the block copolymer in a water miscible organic solvent, typically at a rate of ∼0.2 wt% per minute. The water content is increased in this way until the polymersomes are in frozen equilibrium, and then the organic solvent is removed by dialysis, without significant changes to the frozen structures.44,45 This time consuming process (∼24 hours) results in polymersomes that are relatively close to thermodynamic equilibrium, with little scope for controlling polymersome sizes. Applying the slow water addition method to PBLG50-K with PBS as the aqueous phase (of physiological ionic strength or diluted ten-fold) resulted in insoluble disordered aggregates. This is presumably because the long period of stirring that this method requires combined with the reduced electrostatic repulsion between charged side chains led to the collapse of hydrated structures, i.e. the polymersomes are not thermodynamically stable. The slow water addition method could only be used to produce PBLG50-K polymersomes when using water as the aqueous phase (dh ≈ 450 nm, PDI 0.10, Fig. S4†). In contrast, the water addition/solvent evaporation method kinetically traps polymersomes by solvent evaporation, so it has greater resilience to destabilizing conditions. Additionally, because the structures are not in frozen equilibrium in the mixed solvent system, but are still mobile, a range of polymersome diameters can be accessed by aging or changing the temperature in the mixed solvent system. For PBLG50-K the polymersome diameters could be precisely defined over an order of magnitude, from 200–2000 nm, with a low polydispersity. Due to the slow dynamics of block copolymers, lengthy procedures are often applied for supramolecular structure formation, however, using this rapid procedure the polymersomes have reproducible diameters and low PDIs, which means that the limited time available for mobility of the hydrophobic block is nevertheless sufficient for well-defined ordering of PBLG50-K.
Scope of the WASE method
In order to test the scope of this simple and fast technique it was also applied to range of different peptide-based and traditional block copolymers, all of which yielded polymersomes (Table 2).| Samplea | d h/nm | PDI |
|---|---|---|
| a Prepared using the standard conditions, unless stated otherwise. 2 mL THF, 0.02 µmol polymer, 3 mL PBS, 1 min vortexing at 200 rpm, 2 min rotary evaporation at 20 °C. b The range of dh and PDI values correspond to the different polymersomes characteristics that are accessible by altering the stated parameters (0–10 min aging in mixed solvent system, rotary evaporation at 5–70 °C, osmolality of the aqueous phase 0–330 mOsm kg−1). | ||
| PBLG50-K | 1200 | 0.06 |
| Aging in mixed solvent systemb | 650–2100 | 0.01–0.15 |
| Temperature of solvent evaporationb | 450–2250 | 0.06–0.12 |
| Ionic strength of aqueous phaseb | 200–1200 | 0.05–0.16 |
| PBLG35-K | 1170 | 0.05 |
| PBLG35-K/E | 400 | 0.16 |
| PBLG36-E | 210 | 0.18 |
| PBLG36-E/K | 180 | 0.15 |
| PBLG36-E/K-PEG | 170 | 0.12 |
| PBLG80-E | 130 | 0.18 |
| PBLG100-E | 150 | 0.12 |
| PBLG250-E | 230 | 0.19 |
| PS106–PAA17 | 650 | 0.27 |
| PEO26–PBD46 | 200 | 0.16 |
| PEO80–PBD125 | 180 | 0.20 |
Using the standard conditions, PBLG35-K self-assembled into polymersomes of similar sizes and PDI to those composed of PBLG50-K. The PBLG-K block copolymers were designed such that in aqueous solution the hydrophilic block, the designed peptide K, forms a specific noncovalent complex with peptide E, which has a complementary amino acid sequence (Table 1).46 E and K form a parallel dimer (denoted E/K), hence the binding of PBLG-K with E can be exploited to alter the properties of the corona.35 In THF the peptide blocks K and E do not fold into the coiled-coil conformation because the polarity is too low.25 When the water addition/solvent evaporation procedure was applied to an equimolar mixture of PBLG35-K and E the resulting polymersomes were markedly smaller than PBLG-K polymersomes (dh ≈ 400 nm, PDI 0.09). The smaller polymersome diameters are expected because the larger area of the corona block in the polymeric complex increases the curvature of the corona–core interface.
The method was also applied to a related series of block copolymers, PBLGn-E. Whereas peptide K has a net charge of 3+ in PBS, peptide E has a net charge of 3−. Several PBLGn-E block copolymers were used, with n varying between 36 and 250, and it was shown that all samples formed polymersomes, which had not been achieved using any other method. The membrane core thickness ranged from 33 ± 7 nm for the copolymer with the shortest PBLG block to 140 ± 50 nm for the largest copolymer (Fig. S5†). For PBLG36-E the pathway-dependence of the kinetically trapped polymersomes was investigated as detailed above for PBLG50-K, and all trends were the same as those observed for PBLG50-K. The major difference between the self-assembly of the PBLG-K and PBLG-E block copolymers was that the sizes of the polymersomes formed from the PBLG-E series were significantly smaller than for the PBLG-K block copolymers. The electrostatic interactions within the corona therefore have a strong influence on which polymersome size is energetically favorable, as was described in detail for PBLG50-K. Thus far polymersomes have been prepared with negatively charged (E), positively charged (K), or neutral peptidic coronas (E/K, E/K-PEG). The ability of E and K to form a coiled coil during the solvent evaporation method allows the possibility of preparing polymersomes with different surface functionalities, tailored to particular applications.
To further investigate its scope the technique was also applied to traditional synthetic coil–coil block copolymers. Polystyrene-b-poly(acrylic acid) (PS106–PAA17) is a charged polymer with block proportions typical of those that have been demonstrated to pack into ‘crew cut’ polymersomes by the gradual water addition/dialysis method.39 Using the water addition/solvent evaporation technique PS106–PAA17 formed polymersomes with an average dh of ∼150 nm in water. In physiological strength PBS the polymersome sizes were larger, ∼650 nm, which is due to reduced repulsion between the ionic corona chains, as was also observed for the PBLG-K and PBLG-E series. The method was also tested with non-charged poly(ethylene oxide)-b-polybutadiene block copolymers of two different sizes. PEO26–PBD46 and PEO80–PBD125 formed unilamellar polymersomes with observed membrane thicknesses of 13.5 ± 3.4 nm and 16.9 ± 2.5 nm respectively (Fig. 6).
 | ||
| Fig. 6 OsO4 stained TEM images of polymersomes in PBS prepared from (A) PS106–PAA17 (inset, optical microscope image), (B) PEO26–PBD46, (C) PEO80–PBD125. Scale bars = 200 nm. | ||
These results show that the WASE method can be used to kinetically trap block copolymers into vesicle assemblies, which are not stable using polymersome-forming methods in which the structures are closer to thermodynamic equilibrium, and it is also applicable to block copolymers with a wide range of hydrophobic and hydrophilic block properties. Although the scope of the WASE method is very broad with regards to the possible polymersome components and diameters, it does require the use of THF, which may limit its scope for the preparation of polymersomes with encapsulated biomacromolecules.
Conclusions
In this paper we demonstrate a rapid, simple, and robust process for preparing polymersomes with controllable diameters. Block copolymers that are molecularly dispersed in the organic solvent THF assemble into dynamic polymersomes upon quick addition of an aqueous phase. The structures are frozen during evaporation of the THF under reduced pressure. The method requires only standard laboratory equipment and is fast compared with other polymersome preparation methods. The method also introduces the ability to adjust the polymersome sizes during water addition, aging in the mixed solvent phase, or solvent evaporation, depending on the characteristics of the block copolymer in question. The potential of this method to tune the polymersome sizes is demonstrated with PBLG50-K. Because PBLG50-K has a charged and structured corona the temperature at which the THF is removed and the ionic strength of the aqueous phase are strong determinants of the size of the polymersomes. By using different preparation parameters the same block copolymer assembles into polymersomes with the average size precisely specified within the 200 nm to 2 µm range. As well as producing a variety of polymersome sizes, there is more potential for varying other polymersome properties than with many established methods, as polymersomes can be prepared from numerous types of block copolymers: peptide based rod–rod block copolymers having a range of hydrophobic block lengths, and with positively or negatively charged coronas, noncovalent polymer complexes, and synthetic charged and non-charged coil–coil polymers. The practicality of this WASE method, in terms of time, equipment, the applicability to different block copolymers, and the ability to adjust the polymersome sizes, mean that it will be a suitable choice for the preparation of polymersomes with a wide variety of properties.Acknowledgements
This work was supported by an ERC-starting grant (240394) from the European Research Council.References
- L. Saunders, D. Gammack and J. Perrin, J. Pharm. Pharmacol., 1962, 14, 567–572 CAS.
- A. D. Bangham and R. W. Horne, J. Mol. Biol., 1964, 8, 660–668 CrossRef CAS.
- S. Ota, S. Yoshizawa and S. Takeuchi, Angew. Chem., Int. Ed., 2009, 48, 6533–6537 CrossRef CAS.
- K. Kita-Tokarczyk, J. Grumelard, T. Haefele and W. Meier, Polymer, 2005, 46, 3540–3563 CrossRef CAS.
- A. Nagayasu, K. Uchiyama and H. Kiwada, Adv. Drug Delivery Rev., 1999, 40, 75–87 CrossRef CAS.
- D. Lensen, D. M. Vriezema and J. C. M. van Hest, Macromol. Biosci., 2008, 8, 991–1005 CrossRef CAS.
- B. M. Discher, Y. Y. Won, D. S. Ege, J. C. M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef.
- L. F. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CrossRef CAS.
- A. Kros, W. Jesse, G. A. Metselaar and J. Cornelissen, Angew. Chem., Int. Ed., 2005, 44, 4349–4352 CrossRef CAS.
- J. A. Opsteen, R. P. Brinkhuis, R. L. M. Teeuwen, D. Lowik and J. C. M. van Hest, Chem. Commun., 2007, 3136–3138 RSC.
- S. F. M. van Dongen, H. P. M. de Hoog, R. Peters, M. Nallani, R. J. M. Nolte and J. C. M. van Hest, Chem. Rev., 2009, 109, 6212–6274 CrossRef CAS.
- E. G. Bellomo, M. D. Wyrsta, L. Pakstis, D. J. Pochan and T. J. Deming, Nat. Mater., 2004, 3, 244–248 CrossRef CAS.
- F. Versluis, I. Tomatsu, S. Kehr, C. Fregonese, A. W. J. W. Tepper, M. C. A. Stuart, B. J. Ravoo, R. I. Koning and A. Kros, J. Am. Chem. Soc., 2009, 131, 13186–13187 CrossRef CAS.
- B. J. Ravoo and R. Darcy, Angew. Chem., Int. Ed., 2000, 39, 4324–4326 CrossRef CAS.
- P. Falvey, C. W. Lim, R. Darcy, T. Revermann, U. Karst, M. Giesbers, A. T. M. Marcelis, A. Lazar, A. W. Coleman, D. N. Reinhoudt and B. J. Ravoo, Chem.–Eur. J., 2005, 11, 1171–1180 CrossRef CAS.
- E. W. Kaler, K. L. Herrington, A. K. Murthy and J. A. N. Zasadzinski, J. Phys. Chem., 1992, 96, 6698–6707 CrossRef CAS.
- R. C. Hayward and D. J. Pochan, Macromolecules, 2010, 43, 3577–3584 Search PubMed.
- D. E. Discher and F. Ahmed, Annu. Rev. Biomed. Eng., 2006, 8, 323–341 CrossRef CAS.
- H. W. Shen, L. F. Zhang and A. Eisenberg, J. Am. Chem. Soc., 1999, 121, 2728–2740 CrossRef CAS.
- A. A. Choucair, A. H. Kycia and A. Eisenberg, Langmuir, 2003, 19, 1001–1008 CrossRef CAS.
- M. E. Yildiz, R. K. Prud'homme, I. Robb and D. H. Adamson, Polym. Adv. Technol., 2007, 18, 427–432 CrossRef CAS.
- W. Mueller, K. Koynov, K. Fischer, S. Hartmann, S. Pierrat, T. Basché and M. Maskos, Macromolecules, 2009, 42, 357–361 CrossRef CAS.
- K. T. Kim, J. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Adv. Mater., 2009, 21, 2787–2791 CrossRef CAS.
- H. C. Shum, J. W. Kim and D. A. Weitz, J. Am. Chem. Soc., 2008, 130, 9543–9549 CrossRef.
- H. R. Marsden, J.-W. Handgraaf, F. Nudelman, N. A. J. M. Sommerdijk and A. Kros, J. Am. Chem. Soc., 2010, 132, 2370–2377 CrossRef CAS.
- J. Thiele, D. Steinhauser, T. Pfohl and S. Förster, Langmuir, 2010, 26, 6860–6863 CrossRef CAS.
- C. P. O'Neil, T. Suzuki, D. Demurtas, A. Finka and J. A. Hubbell, Langmuir, 2009, 25, 9025–9029 CrossRef CAS.
- S. Rameez, I. Bamba and A. F. Palmer, Langmuir, 2010, 26, 5279–5285 CrossRef CAS.
- J. C. M. Lee, H. Bermudez, B. M. Discher, M. A. Sheehan, Y. Y. Won, F. S. Bates and D. E. Discher, Biotechnol. Bioeng., 2001, 73, 135–145 CrossRef CAS.
- D. M. Vriezema, A. Kros, R. de Gelder, J. Cornelissen, A. E. Rowan and R. J. M. Nolte, Macromolecules, 2004, 37, 4736–4739 CrossRef CAS.
- J. R. Howse, R. A. L. Jones, G. Battaglia, R. E. Ducker, G. J. Leggett and A. J. Ryan, Nat. Mater., 2009, 8, 507–511 CrossRef CAS.
- H. R. Marsden, C. B. Quer, E. Y. Sanchez, L. Gabrielli, W. Jiskoot and A. Kros, Biomacromolecules, 2010, 11, 833–838 CrossRef CAS.
- A. Moscho, O. Orwar, D. T. Chiu, B. P. Modi and R. N. Zare, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 11443–11447 CrossRef CAS.
- H. R. Marsden and A. Kros, Angew. Chem., Int. Ed., 2010, 49, 2988–3005 CrossRef.
- H. R. Marsden, A. V. Korobko, E. N. M. van Leeuwen, E. M. Pouget, S. J. Veen, N. Sommerdijk and A. Kros, J. Am. Chem. Soc., 2008, 130, 9386–9393 CrossRef CAS.
- H. W. Shen and A. Eisenberg, J. Phys. Chem. B, 1999, 103, 9473–9487 CrossRef CAS.
- M. A. Saleh, S. Akhtar, M. S. Ahmed and M. H. Uddin, Phys. Chem. Liq., 2001, 39, 551–563 CrossRef CAS.
- PBLG50-K is α-helical in THF, as determined by circular dichroism. The length of a PBLG50-K cylinder with complete α-helical secondary structure is 11 nm. The effective ‘spherical’ diameter of rods can be obtained by: L/ln(L/D), where L is the length of the rod, and D is the diameter. For PBLG50-K this gives ∼6.5 nm. For small objects the hydrodynamic diameter can be considerably larger than the actual object, hence a hydrodynamic diameter of 8 nm is in the expected range for an object with an effective diameter of 6.5 nm.
- P. L. Soo and A. Eisenberg, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 923–938 CrossRef CAS.
- D. J. Adams, C. Kitchen, S. Adams, S. Furzeland, D. Atkins, P. Schuetz, C. M. Fernyhough, N. Tzokova, A. J. Ryan and M. F. Butler, Soft Matter, 2009, 5, 3086–3096 RSC.
- K. Bryskhe, S. Bulut and U. Olsson, J. Phys. Chem. B, 2005, 109, 9265–9274 CrossRef CAS.
- L. Zhang and A. Eisenberg, Macromolecules, 1996, 29, 8805–8815 CrossRef CAS.
- A. V. Korobko, W. Jesse, A. Lapp, S. U. Egelhaaf and J. R. C. van der Maarel, J. Chem. Phys., 2005, 122, 024902 CrossRef CAS.
- L. Chen, H. W. Shen and A. Eisenberg, J. Phys. Chem. B, 1999, 103, 9488–9497 CrossRef CAS.
- L. Zhang, K. Yu and A. Eisenberg, Science, 1996, 272, 1777–1779 CAS.
- J. R. Litowski and R. S. Hodges, J. Biol. Chem., 2002, 277, 37272–37279 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional data and discussion of the addition of the aqueous phase and structure formation in the mixed solvent phase, temperature dependent CD spectra of peptide K and additional TEM images. See DOI: 10.1039/c0py00172d |
| This journal is © The Royal Society of Chemistry 2010 |