Polymer micelles decorated by gadolinium complexes as MRI blood contrast agents: design, synthesis and properties
Mathurin
Grogna
a,
Rudi
Cloots
b,
André
Luxen
c,
Christine
Jérôme
a,
Catherine
Passirani
d,
Nolwenn
Lautram
d,
Jean-F.
Desreux
e and
Christophe
Detrembleur
*a
aCenter for Education and Research on Macromolecules (CERM), University of Liège, B6 Sart-Tilman, B-4000, Liège, Belgium. E-mail: christophe.detrembleur@ulg.ac.be; Fax: +32 4-3663497; Tel: +32 4-3663465
bLaboratoire de chimie inorganique structurale, University of Liège, B6 Sart Tilman, B-4000, Liège, Belgium
cCyclotron Research Centre, University of Liège, B6 Sart Tilman, B-4000, Liège, Belgium
dINSERM, U 646, 10 Rue A Boquel, 49000, Angers, France
eCoordination and Radiochemistry, University of Liège, B6 Sart Tilman, B-4000, Liège, Belgium
First published on 6th August 2010
Abstract
New micellar macrocontrast agents with improved contrast at high frequencies were designed by grafting a gadolinium based contrast agent onto functional stealth micelles formed by poly(ethylene oxide)-b-poly(ε-caprolactone) (PEO-b-PCL) in water. As evidenced by relaxometry measurements and the hemolytic CH50 test, the new contrast agents are of interest as MRI blood pool agents.
Introduction
Nuclear Magnetic Resonance Imaging (MRI) is presently the leading imaging modality in diagnostic medicine.1 MRI is noninvasive and allows imaging biological objects (soft tissues for instance) and processes at the molecular level. The image contrast in MRI depends essentially on differences in relaxation times and proton density between different tissues. To reach the full potential of MRI for advanced imaging applications, the inherently low sensitivity of MRI has to be enhanced with contrast agents (CAs) containing paramagnetic metal ions such as Gd(III).2The contrast agents currently approved for clinical uses are based on low molecular weight Gd(III) chelates, such as Magnevist® and Dotarem® (Scheme 1). However, modern MRI instruments are operating at high magnetic fields3–5 (i.e. frequencies) where contrast agents have a low relaxivity (the longitudinal relaxation rates per second and per mmol of metal ion that governs the contrasting efficiency). Consequently, relatively large amounts of these potentially toxic contrast agents are needed per injection and further improvements are required to alleviate this problem. Three main directions can be considered for that purpose: (1) enhancing the relaxivity of Gd(III) based complexes, (2) improving the circulation lifetime in the case of blood pool agents, and (3) targeting them to the desired tissue. Firstly, the relaxivity of Gd(III) based complexes can be much improved by reducing their tumbling rate in solution. Slowing down rotational motions was successfully achieved by immobilizing gadolinium complexes onto macromolecules of different sizes and shapes (proteins,6–10 polylysine,11 dendrimers,12–14 polysaccharides (i.e. dextran15,16) and micelles (or liposomes)4,17–21). Although those strategies have allowed to push rotational correlation times to their theoretical maximum, they are all too often compromised either by nonoptimal water residency times, poor solubility in water, reduced target affinity and selectivity, or lack of realistic usefulness in the actual application. The pharmacokinetic behavior of these agents is also a concern and has to be determined case-by-case.21 Secondly, the decoration of high molecular weight metal complexes by stealth molecules (such as polyethylene oxide, PEO) is a way to increase their blood residence time that allows ill tissues to be reached.22 Finally, the efficacy of the contrast agents can be improved by functionalizing them with targeting peptides20,23 and/or by grafting them to macromolecules that passively target tumor tissues through a combination of reduced renal clearance and exploitation of the enhanced permeation and retention (EPR) effect, which prevails for fast-growing tumors.24–26
 | ||
| Scheme 1 Clinically used contrast agents for MRI (left: Magnevist® and right: Dotarem®). | ||
In the following studies, we aim at designing novel gadolinium based blood pool contrast agents3 with improved relaxivity at high frequencies resulting from the grafting in mild conditions of the gadolinium complex (S-2-(4-aminobenzyl)-diethylenetriamine pentaacetic acid, p-NH2-Bn-DTPA(Gd3+), Scheme 2) at the surface of biocompatible micelles formed by an amphiphilic poly(ethylene oxide)-b-poly(ε-caprolactone) block copolymer (PEO-b-PCL) in water. The relaxivity should be improved by decreasing the tumbling rate of the gadolinium complex while the PEO chains should ensure the repulsion of opsonins allowing for a prolonged blood circulation. These improvements are intensively searched for decreasing the doses of gadolinium required per injection while maintaining satisfactory image acquisition times.
![General procedure for the synthesis of the macromolecular contrast agent [(a) (1) Napht-K, EO (THF, 40 °C); and (2) isopropanol; and (b) Sn(oct)2, ε-CL (toluene, 120 °C)].](/image/article/2010/PY/c0py00117a/c0py00117a-s2.gif) | ||
| Scheme 2 General procedure for the synthesis of the macromolecular contrast agent [(a) (1) Napht-K, EO (THF, 40 °C); and (2) isopropanol; and (b) Sn(oct)2, ε-CL (toluene, 120 °C)]. | ||
Experimental section
General procedures
Commercial tetrahydrofuran (THF), 3,3-diethoxy-1-propanol, naphthalene, toluene, heptane, isopropanol, tin(II) 2-ethylhexanoate [Sn(oct)2, tin octoate, 95%], 6-aminofluorescein and caprolactone (ε-CL) were purchased from Sigma-Aldrich. S-2-(4-Aminobenzyl)-diethylenetriamine pentaacetic acid (p-NH2-Bn-DTPA) was purchased from Macrocyclics. Ethylene oxide (EO, 99.9%) was purchased from Chemogas and was used as received. ε-Caprolactone (ε-CL) was dried over calcium hydride at room temperature for 48 h and distilled under reduced pressure just before use. THF and toluene were dried with a sodium/benzophenone system and distilled just before use.Synthesis
1H NMR (CDCl3, TMS, 250 MHz): δ = 1.19 ppm (t, 6H, CH3–CH2–), δ = 1.88 ppm (q, 2H, –CH–CH2–CH2–), δ = 3.65 ppm (m, 364H, –CH2–CH2–O–), δ = 4.63 ppm (t, 1H, –O–CH–CH2–). Mn,NMR = 4000 g mol−1, DPn,NMR = 91, Mn,SEC = 3900 g mol−1, Mw/Mn = 1.11.
α-Acetal-PEO1600-OH and α-acetal-PEO2500-OH were synthesized using the same experimental procedure, except that the monomer to initiator ratio was adapted accordingly.
1H NMR (CDCl3, TMS, 250 MHz): δ = 1.19 (t, 6H, CH3–CH2–), δ = 1.37 (m, 46H, –CH2–CH2–CH2–CH2–CH2–O–), δ = 1.64 (m, 92H, –CH2–CH2–CH2–CH2–CH2–O–), δ = 2.30 (t, 46H, –CH2–CH2–CH2–CH2–CH2–O–), δ = 3.65 (m, 364H, –CH2–CH2–O–), δ = 4.05 (t, 46H, –CH2–CH2–CH2–CH2–CH2–O–), δ = 4.63 (t, 1H, –O–CH–CH2–). Mn,NMR = 6600 g mol−1, DPn,NMR = 114, Mn,SEC = 6100 g mol−1, Mw/Mn = 1.16.
1H NMR (CDCl3, TMS, 250 MHz): δ = 1.37 (m, 46H), δ = 1.64 (m, 92H), δ = 2.30 (t, 46H), δ = 3.65 (m, 364H), δ = 4.05 (t, 46H), δ = 9.35 (s, 0.6 H). Mn,NMR = 6600 g mol−1, DPn,NMR = 114, Mn,SEC = 6100 g mol−1, Mw/Mn = 1.16.
Characterizations
1H NMR spectra of the different polymers were recorded at 298 K with a Bruker spectrometer (250 MHz) in CDCl3 ((D1) 2 s, 16 scans, 5 wt% of polymer). Size exclusion chromatography (SEC) of the polymers was carried out in dimethylformamide containing 25 mM LiBr (flow rate: 1 ml min−1) at 55 °C with a Waters 600 liquid chromatrograph equipped with a 410 refractive index detector and four Waters Styragel columns [HR 1 (100–5000), HR 3 (500–30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), HR 4 (5000–500000), and HR 5 (2000–4000000) (7.8 × 300 mm)]. Poly(ethylene glycol) standards were used for calibration. Copolymers were analyzed for their size and charge distribution using a Malvern Zetasizer® Nano Series DTS 1060 (Malvern Instruments S.A., Worcestershire, UK). Copolymers concentrations were adjusted to 1 mg ml−1 in a solution composed of 750 µl of deionised water and 250 µl of Veronal-buffered saline containing 0.15 mM Ca2+ and 0.5 mM Mg2+ in order to ensure a convenient scattered intensity on the detector.
000), HR 4 (5000–500000), and HR 5 (2000–4000000) (7.8 × 300 mm)]. Poly(ethylene glycol) standards were used for calibration. Copolymers were analyzed for their size and charge distribution using a Malvern Zetasizer® Nano Series DTS 1060 (Malvern Instruments S.A., Worcestershire, UK). Copolymers concentrations were adjusted to 1 mg ml−1 in a solution composed of 750 µl of deionised water and 250 µl of Veronal-buffered saline containing 0.15 mM Ca2+ and 0.5 mM Mg2+ in order to ensure a convenient scattered intensity on the detector.
Complement consumption testing (CH50 test)
Complement consumption was assessed in normal human serum (NHS) (provided by the Etablissement Français du Sang, CHU, Angers, France) by measuring the residual haemolytic capacity of the complement system after contact with particles. The technique consisted in determining the amount of serum able to lyse 50% of a fixed number of sensitized sheep erythrocytes with rabbit antisheep erythrocyte antibodies (CH50), according to the procedure described elsewhere.30,31 Complement activation was expressed as a function of the surface in order to compare particles with different mean diameters. Nanoparticle surface areas were calculated as described elsewhere,32 using the equation: S = n4πr2 and V = n(4/3)(πr3) leading to S = 3m/rρ where S is the surface area (cm2) and V the volume (cm3) of n spherical beads of average radius r (cm), weight m (µg) and volumetric mass ρ (µg cm−3).Results and discussion
Synthesis of the macrocontrast agent (DTPA(Gd3+)-PEO-b-PCL)
The general procedure for the synthesis of the macrocontrast agent consists in first preparing an aldehyde functionalized poly(ethylene oxide)-b-poly(ε-caprolactone) (ald-PEO-b-PCL) diblock copolymer, followed by imination with S-2-(4-aminobenzyl)-diethylenetriamine pentaacetic acid (NH2-Bn-DTPA(Gd3+)) and reduction with sodium cyanoborohydride (Scheme 2).The procedure described by Scholz et al.28 for the synthesis of aldehyde-ended poly(ethylene glycol)-b-poly(lactide) block copolymer was adapted for the preparation of the aldehyde functionalized poly(ethylene glycol)-b-poly(ε-caprolactone) (Scheme 2).
We first synthesized poly(ethylene oxide) of different molecular weights (Mn = 1300–2500–4000 g mol−1) bearing an acetal group at the α position (α-acetal-PEO-OH) by ring-opening polymerization of EO using potassium 3,3-diethoxy-1-propanolate as initiator, as reported elsewhere.27 Next, the terminal hydroxyl group of α-acetal-PEO-OH was used to initiate the ring-opening polymerization of ε-caprolactone with Sn(oct)2 to produce the corresponding block copolymer α-acetal-PEO-b-PCL. The composition and polydispersity of the prepared copolymers have been determined by 1H NMR and SEC, respectively, and are summarized in Table 1 (columns 2 and 3). After precipitation and drying, the copolymer was dissolved in THF and added dropwise to water under vigorous sonication to form the block copolymer micelles with a hydrophobic PCL core and a hydrophilic PEO corona bearing the acetal groups. Deprotection of the acetal groups into aldehyde was performed by decreasing the pH of the solution to 2. Finally, the pH was increased to 7 and the product was dialyzed against water. Lyophilization led to the corresponding aldehyde functional copolymer (α-aldehyde-PEO4000-b-PCL2600). 1H NMR analysis (Fig. 1) of the block copolymer before and after deprotection allowed us to demonstrate that the reaction was quantitative thanks to the complete disappearance of the typical peaks of the acetal groups at 1.19 ppm (–O–CH–CH2–CH3) and 4.63 ppm (–O–CH–CH2–CH3). The appearance of a singlet at 9.35 ppm is characteristic of the aldehyde function that represents however only 60% of the initial acetal groups. This discrepancy is the result of the aldehyde/hydrate equilibrium that establishes itself easily in water.33
| Entry | NMR and SEC data of acetal-PEO-b-PCL (in CDCL3 and DMF respectively) | DLS data of aldehyde-PEO-b-PCL micelles in water | DLS data of DTPA(Gd3+)-PEO-b-PCL micelles in water | |||
|---|---|---|---|---|---|---|
| M n/g mol−1 (DP)a | M w/Mnb | Diameterc/nm | PDId | Diameterc/nm | PDId | |
| a Degree of polymerization [DP] determined by NMR. b Molecular weight distribution determined by size exclusion chromatography. c Micelles diameter. d Polydispersity determined by dynamic light scattering. | ||||||
| 1 | 1300-b-1500 (30–13) | 1.16 | 15 | 0.24 | 20 | 0.22 |
| 2 | 1300-b-3000 (30–26) | 1.19 | 23 | 0.2 | 30 | 0.18 |
| 3 | 2500-b-1200 (57–11) | 1.17 | 14 | 0.21 | 20 | 0.28 |
| 4 | 2500-b-2000 (57–18) | 1.18 | 26 | 0.25 | 35 | 0.2 |
| 5 | 2500-b-5000 (57–44) | 1.15 | 40 | 0.28 | 49 | 0.3 |
| 6 | 4000-b-1600 (91–14) | 1.16 | 30 | 0.3 | 40 | 0.27 |
| 7 | 4000-b-2600 (91–23) | 1.18 | 37 | 0.25 | 50 | 0.27 |
 | ||
| Fig. 1 1H NMR spectra of α-acetal-PEO4000-OH (top), α-acetal-PEO4000-b-PCL2600 (middle) and α-aldehyde-PEO4000-b-PCL2600 (bottom). | ||
Size exclusion chromatography analysis of the block copolymer before (B curve, Fig. 2) and after (C curve, Fig. 2) deprotection clearly shows that no degradation of the PCL block occurred in these mild experimental conditions.
 | ||
| Fig. 2 Gel permeation chromatograms in DMF of acetal-PEO4000-OH (A, line), acetal-PEO4000-b-PCL2600 (B, double lines) and aldehyde-PEO4000-b-PCL2600 (C, dashed line). | ||
The aldehyde functional PEO-b-PCL micelles in water (0.5 mg ml−1) with a hydrophobic PCL core and a hydrophilic PEO corona bearing the aldehyde groups have been characterized by dynamic light scattering measurements (Table 1, columns 4 and 5). Depending on the size and composition of the block copolymers, micelles have a diameter between 14 and 40 nm with a size distribution between 0.2 and 0.3. As expected, for a same PEO block, increasing the size of the hydrophobic block increases the size of the micelles.
To these micellar solutions was added an equimolar (relative to the aldehyde functions) aqueous solution of the contrast agent (NH2-Bn-DTPA(Gd3+)) prepared by chelation of gadolinium cations by the commercially available NH2-Bn-DTPA. After reaction of the amino group of NH2-Bn-DTPA(Gd3+) with the aldehyde moieties at the micelle surface, an excess of sodium cyanoborohydride (NaBH3CN) was added to reduce the so-formed imine into the corresponding stable amine moieties. For each copolymer, the reaction yield was equal to 60% as determined by ICP analysis of the gadolinium content after removing the ungrafted NH2-Bn-DTPA(Gd3+) by micelles dialysis. The average diameter and size distribution of the DTPA(Gd3+)-PEO-b-PCL were measured by DLS (Table 1) and compared with the average diameter of the aldehyde-copolymers. Micelles have a diameter between 20 and 50 nm with a size distribution between 0.18 and 0.3. As expected, grafting of NH2-Bn-DTPA(Gd3+) on aldehyde-PEO-b-PCL increases the size of the micelles.
Relaxivities of the DTPA(Gd3+)-PEO-b-PCL micelles
The contrasting efficiencies of the synthesized macrocontrast agents (DTPA(Gd3+)-PEO-b-PCL) in micellar form are compared in Table 2. The relaxivity (r1) of the different contrast agents was calculated from formula 1 after determining the longitudinal relaxation time (T1) of water protons.34,35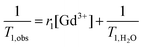 | (1) |
| Entry | Copolymer PEO-b-PCL | Relaxation time/ms (20 MHz, 25 °C) | Relaxivity/mM−1 s−1 |
|---|---|---|---|
| 1 | 1300-b-1500 | 84.3 | 11.9 (±0.2) |
| 2 | 1300-b-3000 | 107.2 | 9.3 (±0.2) |
| 3 | 2500-b-1200 | 85.1 | 11.8 (±0.1) |
| 4 | 2500-b-2000 | 84.5 | 11.8 (±0.2) |
| 5 | 2500-b-5000 | 106 | 9.4 (±0.1) |
| 6 | 4000-b-1600 | 97.2 | 10.3 (±0.2) |
| 7 | 4000-b-2600 | 99.2 | 10.1 (±0.1) |
The relaxivities of the different macrocontrast agents at 20 MHz were found to vary between 9 and 12 mmol−1 s−1 and are significantly higher than that of the free and low molecular weight contrast agent DTPA(Gd3+) (r1 = 4.3 mmol−1 s−1) (Table 2). The relaxivity enhancements noted for the macrocontrast agents result from an increase of their rotational correlation lifetime due to their bulkiness. The observed enhancements are only slightly lower than other micellar systems based on gadolinium (r1 ≈ 18 mmol−1 s−1 at 20 MHz) because of the high flexibility of the PEO chains of our PEO-b-PCL micelles. Comparing the characteristics of the micelles prepared using the different PEO-b-PCL block copolymers (Table 1) and the relaxivities of the corresponding DTPA(Gd3+)-PEO-b-PCL micelles at 20 MHz (Table 2) clearly shows that there is no simple relationship between the size of the micelles and the relaxivities. Most of the relaxivities are in the same range (10 ≤ r1 ≤ 12 mmol−1 s−1), except for the micelles formed from the block copolymers with the PCL sequence longer than the PEO one (r1 ≈ 9.5 mmol−1 s−1, Table 2 entries 2 and 5).
Full relaxometric data were measured for two DTPA(Gd3+)-PEO-b-PCL based micelles over a large magnetic field range (from 0.01 to 100 MHz) and were compared to DTPA(Gd3+) (Fig. 3). At low frequency (0.01 to 5 MHz), the relaxivity of the macrocontrast agent is about twice higher than that of free DTPA(Gd3+). Importantly, the effect of the immobilization of gadolinium on micelles has an even more pronounced effect on relaxivity at high frequencies (10 to 80 MHz). Indeed, the micelle relaxivities remain strongly enhanced when compared to DTPA(Gd3+) whose relaxivity drops drastically to 4 mM−1 s−1. The maximum relaxivity for the best macrocontrast agent we have developed, i.e. DTPA(Gd3+)-PEO1300-b-PCL1500, was obtained at 40 MHz (r1 = 12.6 mM−1 s−1) with a 300% relaxivity increase upon attachment of DTPA(Gd3+) onto the micelles by comparison with free DTPA(Gd3+). Once again, when the PCL sequence of the PEO-b-PCL block copolymer is longer than the PEO one, the relaxivities are lower. Symmetrical PEO-b-PCL or block copolymers with a shorter PCL sequence are thus preferred for optimized relaxivities.
 | ||
| Fig. 3 Comparison of the 1H NMRD profiles of DTPA(Gd3+)PEO1300-b-PCL1500 (full triangles), DTPA(Gd3+)PEO2500-b-PCL5000 (full circles) and NH2-Bn-DTPA(Gd3+) (empty circles) at 298 K. | ||
Complement activation test36
The improvement of the relaxivity of a contrast agent by immobilizing it onto polymeric micelles is the first step towards the reduction of the dose needed for satisfactory image acquisition. The second step consists in preventing the recognition of the macrocontrast agent by the immune system that is responsible for its rapid elimination from the blood circulation, restricting timing for studies. For that purpose, poly(ethylene oxide) is the most popular polymer used for imparting stealthiness to molecules or particles injected in the body.37–41The PEO corona at the macrocontrast agent surface should therefore improve its blood circulation time. One of the macrocontrast agents (DTPA(Gd3+)-PEO2500-b-PCL5000) was therefore evaluated by the hemolytic CH50 test and compared to a PEO4900-b-PCL3900 block copolymer known for stealthiness.42 Both copolymers are very poor activators of the complement system in comparison with a P(MMA-co-MA) copolymer chosen as positive control43 (Fig. 4). Interestingly, Fig. 4 clearly shows that the grafting of the gadolinium complex onto PEO-b-PCL micelles does not activate the complement. This very low activation means that the macrocontrast agent is expected to have a long blood circulation time and could be ready to be evaluated by in vivo test like plasma clearance test.
 | ||
| Fig. 4 Consumption of CH50 unit's vs. surface area of DTPA(Gd3+)PEO2500-b-PCL5000 (full circles), P(MMA-co-MA) (full triangles) and PEO4900-b-PCL3900 (empty circles). | ||
Conclusion
Aldehyde functionalized micelles based on a hydrophilic corona of poly(ethylene oxide) and a hydrophobic core of poly(ε-caprolactone) were prepared in water and grafted with a gadolinium based contrast agent. Relaxometry measurements of these novel micellar macrocontrast agents have evidenced the strong increase (up to 300%) of relaxivity at high frequencies compared to low molecular weight contrast agents due to the decrease of their tumbling rate in solution. Moreover, the hemolytic CH50 test has demonstrated that the PEO chains at the surface of the macrocontrast agents prevent their recognition by the immune system, imparting long-circulating properties to molecules. The improved relaxivity and the potential long circulation time of the macrocontrast agent make them good potential candidates for MRI blood pool contrast agent.Acknowledgements
C.D., M.G. and C.J. are much indebted to the “Politique Scientifique Fédérale” for financial support in the frame of the « Interuniversity Attraction Pôles Programme (IAP VI/27): Supramolecular Chemistry and Supramolecular Catalysis » and to the National Funds for Scientific Research (F.R.S.-FNRS). The authors also thank Dr Jérôme Bejaud [Ingénierie de la Vectorisation Particulaire (Inserm)—Angers, France] for skilful assistance in CH50 tests. J.F.D. gratefully thanks the FNRS and the IISN of Belgium for financial support.References
- M. Rudin and R. Weissleder, Nat. Rev. Drug Discovery, 2003, 2, 123–131 CrossRef CAS.
- P. Caravan, Chem. Soc. Rev., 2006, 35, 512–523 RSC.
- R. B. Clarkson, Top. Curr. Chem., 2002, 221, 201–235 CAS.
- P. Hermann, J. Kotek, V. Kubicek and I. Lukes, Dalton Trans., 2008, 3027–3047 RSC.
- E. Toth, L. Helm and A. E. Merbach, Top. Curr. Chem., 2002, 221, 61–101 CAS.
- R. B. Lauffer, T. J. Brady, R. D. Brown, III, C. Baglin and S. H. Koenig, Magn. Reson. Med., 1986, 3, 541–548 CrossRef CAS.
- P. Niemi, T. Reisto, I. Hemmila and M. Kormano, Invest. Radiol., 1991, 26, 820–824 CrossRef CAS.
- H. Paajanen, T. Reisto, I. Hemmila, M. Komu, P. Niemi and M. Kormano, Magn. Reson. Med., 1990, 13, 38–43 CrossRef CAS.
- U. Schmiedl, M. E. Moseley, M. D. Ogan, W. M. Chew and R. C. Brasch, J. Comput.-Assisted Tomogr., 1987, 11, 306–313 Search PubMed.
- M. Spanoghe, D. Lanens, R. Dommisse, A. Van der Linden and F. Alder Weireldt, Magn. Reson. Imaging, 1992, 10, 913–917 CrossRef CAS.
- G. Schuhmann-Giampieri, H. Schmitt-Willich, T. Frenzel, W. R. Press and H. J. Weinmann, Invest. Radiol., 1991, 26, 969–974 CAS.
- L. H. Bryant, Jr, M. W. Brechbiel, C. Wu, J. W. Bulte, V. Herynek and J. A. Frank, J. Magn. Reson. Imaging, 1999, 9, 348–352 CrossRef CAS.
- Z. Jaszberenyi, L. Moriggi, P. Schmidt, C. Weidensteiner, R. Kneuer, A. E. Merbach, L. Helm and E. Toth, JBIC, J. Biol. Inorg. Chem., 2007, 12, 406–420 CrossRef CAS.
- J. Rudovsky, M. Botta, P. Hermann, K. I. Hardcastle, I. Lukes and S. Aime, Bioconjugate Chem., 2006, 17, 975–987 CrossRef CAS.
- R. Rebizak, M. Schaefer and E. Dellacherie, Bioconjugate Chem., 1997, 8, 605–610 CrossRef CAS.
- R. Rebizak, M. Schaefer and E. Dellacherie, Bioconjugate Chem., 1998, 9, 94–99 CrossRef CAS.
- J. P. Andre, E. Toth, H. Fischer, A. Seelig, H. R. Macke and A. E. Merbach, Chem.–Eur. J., 1999, 5, 2977–2983 CrossRef CAS.
- E. Nakamura, K. Makino, T. Okano, T. Yamamoto and M. Yokoyama, J. Controlled Release, 2006, 114, 325–333 CrossRef CAS.
- G. Zhang, R. Zhang, X. Wen, L. Li and C. Li, Biomacromolecules, 2008, 9, 36–42 CrossRef CAS.
- A. Accardo, D. Tesauro, G. Morelli, E. Gianolio, S. Aime, M. Vaccaro, G. Mangiapia, L. Paduano and K. Schillen, JBIC, J. Biol. Inorg. Chem., 2007, 12, 267–276 CrossRef CAS.
- A. J. L. Villaraza, A. Bumb and M. W. Brechbiel, Chem. Rev., 2010, 110, 2921–2959 CrossRef CAS.
- A. L. Ayyagari, X. Zhang, K. B. Ghaghada, A. Annapragada, X. Hu and R. V. Bellamkonda, Magn. Reson. Med., 2006, 55, 1023–1029 CrossRef CAS.
- W. J. M. Mulder, G. J. Strijkers, J. W. Habets, E. J. W. Bleeker, D. W. J. van der Schaft, G. Storm, G. A. Koning, A. W. Griffioen and K. Nicolay, FASEB J., 2005, 19, 2008–2010 CAS.
- M. E. Fox, F. C. Szoka and J. M. J. Frechet, Acc. Chem. Res., 2009, 42, 1141–1151 CrossRef CAS.
- H. Maeda, G. Y. Bharate and J. Daruwalla, Eur. J. Pharm. Biopharm., 2009, 71, 409–419 CrossRef CAS.
- T. Seki and H. Maeda, Kekkan Igaku, 2008, 9, 397–409 Search PubMed.
- Y. Nagasaki, T. Okada, C. Scholz, M. Iijima, M. Kato and K. Kataoka, Macromolecules, 1998, 31, 1473–1479 CrossRef CAS.
- C. Scholz, M. Iijima, Y. Nagasaki and K. Kataoka, Macromolecules, 1995, 28, 7295–7297 CrossRef CAS.
- X. Shuai, H. Ai, N. Nasongkla, S. Kim and J. Gao, J. Controlled Release, 2004, 98, 415–426 CrossRef CAS.
- A. Aqil, S. Vasseur, E. Duguet, C. Passirani, J. P. Benoit, A. Roch, R. Muller, R. Jerome and C. Jerome, Eur. Polym. J., 2008, 44, 3191–3199 CrossRef CAS.
- A. Vonarbourg, C. Passirani, P. Saulnier, P. Simard, J. C. Leroux and J. P. Benoit, J. Biomed. Mater. Res., Part A, 2006, 78a, 620–628 CrossRef CAS.
- C. Passirani, G. Barratt, J.-P. Devissaguet and D. Labarre, Life Sci., 1998, 62, 775–785 CrossRef CAS.
- Y. Zou, D. E. Brooks and J. N. Kizhakkedathu, Macromolecules, 2008, 41, 5393–5405 CrossRef CAS.
- R. B. Lauffer, Chem. Rev., 1987, 87, 901–927 CrossRef CAS.
- C. Vanasschen, PhD Thesis, University of Liège, 2009 Search PubMed.
- C. Passirani and J.-P. Benoit, Biomaterials for Delivery and Targeting of Proteins and Nucleic Acids, ed. Ram I. Mahato, 2005, pp. 187–230 Search PubMed.
- A. Vonarbourg, C. Passirani, P. Saulnier and J.-P. Benoit, Biomaterials, 2006, 27, 4356–4373 CrossRef CAS.
- K. Shiraishi, K. Kawano, T. Minowa, Y. Maitani and M. Yokoyama, J. Controlled Release, 2009, 136, 14–20 CrossRef CAS.
- V. P. Torchilin, Adv. Drug Delivery Rev., 2002, 54, 235–252 CrossRef CAS.
- R. Gref, M. Luck, P. Quellec, M. Marchand, E. Dellacherie, S. Harnisch, T. Blunk and R. H. Muller, Colloids Surf., B, 2000, 18, 301–313 CrossRef CAS.
- E. Owens Donald, 3rd and A. Peppas Nicholas, Int. J. Pharm., 2006, 307, 93–102 CrossRef CAS.
- K. Van Butsele, P. Sibret, C. A. Fustin, J. F. Gohy, C. Passirani, J. P. Benoit, R. Jerome and C. Jerome, J. Colloid Interface Sci., 2009, 329, 235–243 CrossRef CAS.
- J. Rieger, C. Passirani, J.-P. Benoit, K. Van Butsele, R. Jerome and C. Jerome, Adv. Funct. Mater., 2006, 16, 1506–1514 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |