One-pot synthesis of polymeric nanomaterials via RAFT dispersion polymerization induced self-assembly and re-organization
Wen-Ming
Wan
and
Cai-Yuan
Pan
*
Department of Polymer Science and Engineering, CAS Key Laboratory of Soft Matter Chemistry, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China. E-mail: pcy@ustc.edu.cn
First published on 3rd August 2010
Abstract
A novel strategy for preparation of multiple nanostructural materials, which is the creation of such materials directly from controlled radical dispersion polymerization in one pot, has been developed. In the formation of polymeric nanomaterials, the chain length ratio of the hydrophobic to hydrophilic blocks is altered continuously, which induces two phase transitions, phase separation to form spherical micelles and re-organization of the resulting spheres to yield multiple morphologies including nanorods, nanotubes, vesicles and doughnuts. This is quite different from self-assembly of the block copolymer in a selective solvent, where nature and solubility parameter of the solvents are changed. In the reversible addition-fragmentation chain transfer (RAFT) polymerization of styrene in methanol using poly(4-vinylpyridine) as macro RAFT agent, the resultant polymeric nanomaterials with various morphologies coexisted generally, however, uniform nanowires and vesicles could be prepared by appropriately selecting concentration of monomer and feed ratio, as well as by strict control of reaction conditions. One advantage of this strategy is that the nanomaterials with a concentration as high as 0.5 g mL−1 can be achieved, this provides possibility for studying extensive applications of the various nanomaterials.
1. Introduction
Nanoscience and nanotechnology have gained great attention in the past few decades due to their potential applications in various fields.1 Compared to the widely investigated inorganic and organic nanomaterials,2 studies of the properties and applications of the polymeric nanomaterials are relatively limited. One probable reason is the limitation of the present preparation methods because studies of properties and applications need a certain amount of nanomaterials. So finding a simple and feasible method for production of the polymeric nanomaterials in a relatively large scale would be highly desired.Generally, two main strategies for preparation of the polymeric nanomaterials, which are heterogeneous polymerization (such as: emulsion polymerization and dispersion polymerization, etc) and self-assembly of the block copolymer in a selective solvent, have been extensively investigated.3 The emulsion polymerization yielded limited morphologies, and their formation mechanism was complicated and not clear.4 The self-assembling strategy is very important and has been utilized in the preparation of a broad range of intricate, biomimetic nanomaterials, including spherical micelles, nanowires, vesicles, tubes and doughnuts, etc.5 But formation of the fine nanostructural materials by self-assembling method is difficult to control, requires multiple-steps, and is time-consuming, while the capacity of every preparation is relatively low. Thus, great efforts have been made to find a new strategy for solving these problems.
As we know, dispersion polymerization is a useful technique for preparation of the polymeric materials with narrow size distribution.6 All reaction ingredients are dissolved in the polymerization medium, and the polymerization proceeds homogeneously at initial polymerization. With evolution of the polymerization, the polymeric materials in the form of spheres (micron and sub-micron in size) are formed and are dispersed in the reaction medium heterogeneously, and then further polymerization takes place mainly in the monomer-swollen particles.7 In the past decade, controlled/living dispersion polymerization has been studied.8 And the results showed that only a polymerization-induced self-assembly process occurred when the macro initiator was used as both initiator and stabilizer. In these polymerization systems, the core-shell micelles at the nanoscale were always formed. Therefore, we have two questions: why were the formed nanomaterials always spherical micelles? Could the multiple nanostructural materials, which are generally formed by self-assembly of the block copolymers in a selective solvent, be prepared via controlled radical polymerizations?
By analysis of the results reported,8 we can find that in the self-assembly of the amphiphilic block copolymers in a selective solvent, one of the determining factors for formation of the morphologies is the polymer composition, and the spherical micelles are formed at the lowest chain length ratio of the hydrophobic to the hydrophilic blocks in the formation of various morphologies.9 In these controlled/living polymerization systems, the chain length of the macro initiator were fixed, the second blocks grew gradually, when their chain length increased to a critical value, the polymerization-induced phase separation always produced spherical micelles because the chain length ratio at phase separation met firstly the requirements for the formation of spherical micelles. This is why the spherical micelles were always obtained in the previous investigations. A feasible strategy for achieving multiple polymeric materials through controlled radical polymerization is to transfer the formed spherical micelles into other different nanostructures. According to the theory of self-assembly, the morphologies of aggregates are mainly determined by a force balance involving the stretching of the core chains, the surface tension between the core and outside solvent, and repulsion among the corona chains.9 So, changing the force balance would result in the transformation of morphologies, and altering the composition of block copolymers might be one efficient method for change of the force balance. In addition, the morphology formed by phase separation was generally locked because the polymerization temperature was below the glass transition temperature (Tg) of the second block.9 Therefore, to achieve transformation of the spherical micelles to other morphologies, continuous propagation and lowering Tg of the core chains after phase separation are two key points. In our previous investigations, we found that the propagation of the core polymer chains almost stopped, but continuous polymerization of the core chains was achieved by swelling of the core because this not only increased the diffusion rate of the monomer from the solution into the cores, but also decreased the Tg of the core polymer.6 So, it is reasonable to presume that if the cores in the spherical micelles are swollen, propagation of the core chains will continue at reasonable rate after phase separation, and transition of the morphology may occur. This idea was confirmed by our previous reports.10,11
In this article, more data are presented to demonstrate the transition of the spherical micelles not only into vesicles,10 but also into other morphologies. Since reversible addition-fragmentation chain transfer (RAFT) polymerization has been extensively applied in the preparation of block copolymers including functional block copolymers12 and also in dispersion polymerization.13 Therefore, we studied preparation of various nanomaterials directly from RAFT dispersion polymerization of styrene (St) in methanol using trithiocarbonate-terminated poly(4-vinylpyridine) (P4VP-TC) and 2,2′-azobisisobutyronitrile (AIBN) as macro RAFT agent and initiator, respectively. With evolution of the polymerization, the phase separation and the re-organization of the formed spherical micelles were observed.
2. Experimental
2.1 Materials
St (Shanghai Chem. Co.) was washed with an aqueous solution of sodium hydroxide (5 wt%) three times and then washed with water until neutralization. After dried with anhydrous magnesium sulfate, St was distilled under reduced pressure. 4VP (Acros, 96%) was dried over CaH2 and then was distilled under reduced pressure prior to use. The AIBN was purified by recrystallization from ethanol. All other reagents with analytical grade were used as received.2.2 Synthesis of S-1-dodecyl-S-(α,α′-dimethyl-α′′-acetic acid) trithiocarbonate (TC)
TC was synthesized in yield of 78.4% according to the method described in ref. 14. 1H NMR, δ( TMS, ppm): 0.90 (t, 3H, –CH3), 1.37–1.47 (m, 20H, –(CH2)10–), 1.75 (s, 6H, 2–CH3), 3.42 (t, 2H, –CH2S), 13.05 (s, 1H, –COOH).2.3 Preparation of TC-terminated poly(4VP) (P4VP-TC)
The general procedure is as follows. The 4VP (2.12 g, 20 mmol), TC (36 mg, 0.1 mmol), isopropanol (2 mL) and AIBN (2 mg, 0.01 mmol) were added into a 10 mL polymerization tube with a magnetic bar. After three freeze-evacuate-thaw cycles, the tube was sealed under high vacuum, and then the sealed tube was placed in an oil bath at 80 °C while stirring. After 6 h, the tube was cooled to room temperature with ice-water, and then the tube was opened. The polymer was precipitated by pouring a polymer solution in isopropanol into excess diethyl ether while stirring. The precipitate obtained by filtration was dried in a vacuum oven at room temperature overnight, 1.08 g of the product was obtained, and the conversion measured by 1H NMR method was 50%. Its 1H NMR spectrum is shown in Fig. 7Aa′.2.4 RAFT polymerization of St using P4VP-TC as the macro chain transfer agent
A typical procedure is as follows. The P4VP-TC (0.106 g, 0.01 mmol), St (1.05 g, 10 mmol), AIBN (0.2 mg, 1 μmol) and methanol (1 mL) were successively added to a 5 mL glass tube with a magnetic bar, and then the system was degassed by three freeze-pump-thaw cycles. The tube was sealed under vacuum, and then the sealed tube was placed in an oil bath at 80 °C while stirring the contents. After polymerization was carried out for 6 h, the reaction mixture was cooled to room temperature with ice-water, and then the tube was opened. The polymer was precipitated by pouring a polymer solution in methanol into excess petroleum ether while stirring. The precipitate was collected by filtration, and then dried in a vacuum oven at room temperature overnight, 0.65 g of the product was obtained, and the conversion was 52%.For preparation of nanowires, St (5.20 g, 50 mmol), P4VP (106 mg, 1 × 10−2 mmol) and AIBN (0.20 mg, 1 × 10−3 mmol) were dissolved in methanol (3.5 g), the resultant solution was equally divided into 5 portions and every portion was placed in a 5 mL polymerization tube. The polymerizations were performed at 80 °C for 2 h, 4 h, 12 h, 24 h and 48 h, respectively, and the other procedure was the same with that described above.
For studying the kinetics of RAFT dispersion polymerization, the P4VP-TC, St and AIBN with the feed molar ratio = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50000:1 were added into methanol in eight 5 mL glass tubes, respectively. The polymerizations were carried out at the same conditions mentioned above, and then stopped at 0.5, 1, 2, 4, 6, 8, 12 and 24 h respectively. After the polymerization, the solution was treated with the same procedure mentioned above; the white powder products were obtained.
:50000:1 were added into methanol in eight 5 mL glass tubes, respectively. The polymerizations were carried out at the same conditions mentioned above, and then stopped at 0.5, 1, 2, 4, 6, 8, 12 and 24 h respectively. After the polymerization, the solution was treated with the same procedure mentioned above; the white powder products were obtained.
For preparation of vesicles, the same procedure and conditions with preparation of nanowires were performed. Every polymerization tube contained St (2.08 g, 20 mmol), P4VP (21.2 mg, 2 × 10−3 mmol), AIBN (0.033 mg, 2 × 10−4 mmol) and methanol (1.1 g), and the polymerizations were performed at 80 °C for 2, 4, 12 and 24 h, respectively.
The kinetics study was performed according to the same procedure described in the preparation of nanowires except the molar ratio of P4VP-TC/St/AIBN = 10:100000:1.
2.5 Characterization
3. Results and discussion
In the RAFT dispersion polymerization, St, P4VP-TC, AIBN and methanol are used as monomer, macro RAFT agent, initiator and solvent, respectively. The St, P4VP-TC and AIBN are soluble in methanol, the initial polymerization proceeds homogeneously, and the block copolymers, P4VP-b-PSs are formed as shown in Scheme 1A. Since PS is insoluble in methanol, when the PS blocks grow to a critical value, phase separation occurs to form spherical micelles (Scheme 1B), which is called polymerization-induced self-assembling.6–8 Generally, the polymerization almost stopped after the spherical micelles were formed.8 If the polymerization media can swell easily the cores of micelles the propagation of core PS chains will continue at reasonable rate. Increasing of the PS chain length resulted in increase of the packing parameter, leading to curvature decreasing of the polymeric assemblies according to critical packing parameter of amphiphilic molecules, a general concept in determining the shape of molecular assemblies.15 As a result, the spherical micelles are transferred to other morphologies along with decrease of curvature as shown in Scheme 1C, which is called polymerization-induced re-organization. Thus, for preparation of the multiple morphologies, two processes, polymerization-induced self-assembling and re-organization (PISR) should occur in the polymerization. In this article, we focus mainly on re-organization of the formed spheric micelles, which is different from the previous articles.6–8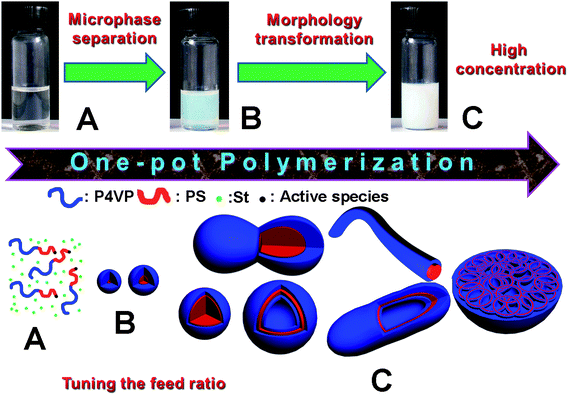 | ||
| Scheme 1 The formation mechanism of the multiple morphologies in the RAFT polymerization of St in methanol using P4VP-TC as macro RAFT agent, (A) formation of the soluble block copolymer, PS-b-P4VP; (B) phase separation to form spherical micelles; (C) re-organization of the resulting spheres to yield multiple morphologies. | ||
PISR in the RAFT polymerization
We selected methanol as solvent because it can dissolve P4VP and St, but it is a nonsolvent of the PS. The macro RAFT agent, P4VP-TC with Mn = 10400 g mol−1 and Mw/Mn = 1.08 was used in this study except where specially mentioned, and it acts also as stabilizer of the nanomaterials formed. When the polymerization with a feed molar ratio of P4VP/St (1 g)/AIBN =10:5000:1 was carried out in methanol (1 g) at 80 °C, and we saw a transition of the polymerization solution from transparent to blue opalescence (Fig. 1Aa) where aggregates formed. The aggregates were spherical micelles with PS core and P4VP corona (Fig. 1Ab) because the P4VP is soluble, but the PS is insoluble in methanol. When DLS was used to follow this polymerization, as shown in Fig. 1A, we can see that the diameter of the aggregates increased from 16 nm at 0.5 h of polymerization to 98 nm at 4 h rapidly, and then the increasing rate of the aggregates' diameter slowed down, after 24 h of polymerization, the spherical micelles with a diameter of 120 nm were formed. Since the solubility parameter (δ) of methanol (δ = 29.7) is much higher than that of the PS (δ = 16.6–20.3),16 the PS cores were tightly shrunk, leading to very slow diffusion rate of the monomer molecules from the solution into the cores. The propagation of the core PS chains almost ceased; as a result, transition of the spherical micelles to other morphologies was not achieved, which was observed in the previous reports.8
 | ||
| Fig. 1 The relationship between Dh and reaction time, (A) for the RAFT polymerization with a feed ratio of P4VP/St/AIBN = 10:5000:1 at 80 °C, St: 1 g; methanol: 1 g; (B) for RAFT polymerization with a feed molar ratio of P4VP-TC/St/AIBN =10:50000:1 at 80 °C, St: 1 g; methanol: 1 g. | ||
To achieve the re-organization of the formed spherical micelles to yield other morphologies, it is very important to maintain the same propagation rate of the core-forming chains with that before the phase separation. This can be realized by improving compatibility of the reaction media with the core PS chains. Consider the δ value of St is 19.0,16 which is similar to the δ value of PS. So, St is a good solvent for PS. For simplifying the recipe and easy discussion, we did not adopt the mixing solvents; high concentration of St in methanol was used in the following RAFT polymerization because St can act as monomer, also as solvent before its polymerization. In addition, a high feed molar ratio of monomer to macro RAFT agent used can enhance polymerization rate, while increase of the chain length ratio of PS to P4VP in the resultant diblock copolymers is achieved. Fig. 1B is the results obtained from the RAFT polymerization with a feed molar ratio of P4VP/St (1 g)/AIBN =10:50000:1 in methanol (1 g) at 80 °C. We saw that the polymerization system at 0.1 h was transparent (Fig. 2a), it became blue opalescence after 2 h polymerization (Fig. 2b), which indicates the phase separation occurred, and spherical micelles were formed. The DLS was utilized to measure the size of spherical micelles formed at 2 h of polymerization, and the average diameter (Dh) was approximately 80 nm. With evolution of the polymerization, the aggregates continuously increased to Dh = 595 nm, and then the size increase was levelled off (Fig. 1B), and the polymerization system became milky solution (Fig. 2c). Compare to the result in Fig. 1A, this system displayed a big different polymerization behavior. In order to know what happened in this system, TEM was used to observe the aggregates formed at 24 h of polymerization. As shown in Fig. 2A, various nanostructures, such as spherical micelles (B), vesicles (C), doughnuts (D) and lamellae (E), and rod-like aggregates were obtained. This demonstrates that the re-organization of the formed spherical micelles might occur.
 | ||
| Fig. 2 Optical digital photos of the RAFT polymerization with a feed molar ratio of P4VP/St (1 g)/AIBN = 10:50000:1 in methanol (1 g) at 80 °C for 0.1 h (a), 2 h (b) and 24 h (c); A: TEM image of the multiple morphologies formed at 24 h with scale bar of 500 nm; B–E: magnified images of B–E in A. | ||
In order to find out whether more other morphologies could be prepared via the PISR process, we tried to tune the reaction conditions and the feed molar ratios in the RAFT dispersion polymerization, and the resultant aggregates were observed by TEM. Every RAFT polymerization for 24 h created multiple morphologies, generally, nanorods or vesicles coexisting with various morphologies. We selected two or three morphologies from every TEM image, and these select morphologies are shown in Fig. 3. We can see that the multiple morphologies including vesicles, nanotubes, large compound vesicles and other complex aggregates were created depending upon the polymerization conditions and the feed molar ratios. So, multiple morphologies could be prepared by controlled radical polymerization. However, are these nanostructural materials really formed via the PISR process? And can uniform morphologies be prepared? We will focus on these questions in the next sections.
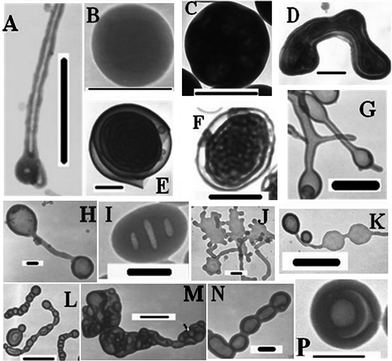 | ||
| Fig. 3 TEM images of the multiple morphologies were prepared by RAFT polymerization induced self-assembly and re-organization at 80 °C for 24 h, A–D: with the feed molar ratio of P4VP-TC/St/AIBN= 10:200000:1 in methanol (P4VP-TC: 5 mg; methanol: A, D, 1 g; B, 0.8 g; C, 0.5 g); E–H: with feed molar ratio of P4VP-TC/St/AIBN = 10:100000–200000:1 in methanol (P4VP: 10 mg; methanol: H, 1 g; E, 0.8 g; F and G, 0.5 g); I–P: P4VP-TC/St/AIBN = 10:50000–150000:1 in methanol (P4VP-TC: 20 mg; methanol: I and L, 0.5 g; P, 0.8 g; J, K, M and N, 1 g). The scale bar is 1000 nm, except for B, H, J, L, N and P where it is 200 nm. | ||
Preparation of uniform nanowires
One-dimensional nanostructures, particularly nanowires, gained a great attention due to their potential utilizations as active components or interconnect in the construction of nanoscale electronic or electromechanical devices.17 Thus, we firstly studied preparation of the uniform nanowires.In order to clearly understand the formation mechanism of the morphologies, DLS, TEM and FESEM were used to trace the transition of the morphologies. Since the ratio of gyration radius (Rg) to hydrodynamic radius (Rh) is very sensitive to the morphologies of aggregates,18 and we can speculate the resultant morphologies based on the Rg/Rh values, generally, the values for spherical micelles, vesicles with thin-layer, vesicles with wall thickness about 1/3 radius of the vesicle and rods are 0.77, 1.0, 0.86 and >1.0 (determined by the ratio of length to diameter) respectively.19 Thus, DLS was applied to follow the RAFT polymerization in methanol with a feed molar ratio of P4VP/St/AIBN = 10:50000:1 (St: 1.04 g; methanol: 0.7 g) at 80 °C, and the results are shown in Fig. 4. The relationship of Rh and Rg/Rh with reaction time in Fig. 4A exhibited only one peak at 1.3 nm, which demonstrated that the block copolymer P4VP-b-PS chains formed at 1 h of polymerization were molecularly dissolved in the reaction media. After 2 h of polymerization, two peaks were observed, one small peak is ascribed to the block copolymer, and the other big peak at Rh = 12.4 nm belongs to the spherical micelles because their Rg/Rh is 0.78 (Fig. 4B). This must result from phase separation of the formed P4VP-b-PS chains. When the polymerization lasted 3 h the size increased from Rh = 12.4 nm to 32 nm, but their Rg/Rh did not vary obviously, so, the spherical micelles remained. This can be further confirmed by their FESEM and TEM images shown in Fig. 5A and Fig. 6A. These two images displayed spherical micelles with a diameter of 36 nm and 34 nm, respectively. As the polymerization continued to 4 h, the resulting aggregates exhibited two peaks (Fig. 4A), a large one at Rh = 87 nm may result from re-organization of the spherical micelles, and a small one belongs to the spherical micelles. The Rg/Rh of the new and big aggregates was 1.2 (Fig. 4B), so they might be the rod-like micelles, which can be clearly seen in FESEM image in Fig. 5B. We can see that the rod-like micelles coexisted with a small amount of the spherical micelles. Their D is approximately 70 nm with a narrow distribution. It looks as though the rod-like micelles are the result of fusion of the spherical micelles (inset of Fig. 5B). Very a few collapsed spheres, which are the vesicles, which is verified by the TEM image in Fig. 6B, were observed as shown in Fig. 5B. In order to identify what morphology was formed, the TEM images of the aggregates formed at 3.5 h and 4 h of polymerization were recorded. As shown in Fig. 6B and 6C, several vesicles can be clearly seen. The exact reason needs further investigation, we presumed that it must be related to the molecular weight distribution of the block copolymers formed. But we did not find such vesicles on the TEM image of the aggregates formed at 5 h of polymerization in Fig. 6D. It is probable that the exchange between the block copolymer chains in the vesicles and that in the spherical micelles occurred, leading to instability of the vesicles and then formation of the rod-like micelles. The exact reason is unclear. When the polymerization continued for 12 h and 24 h, the Rh and Rg/Rh increased as shown in Fig. 4B. This may be due to the ratio increase of the length to the diameter of the rod-like micelles. For 12 h of polymerization, the lower size peak disappeared completely, and a symmetrical curve appeared at Rh = 107 nm (Fig. 4A). The Rg/Rh of the aggregates was 1.8, which indicates that they were nano-wires.
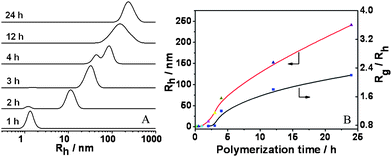 | ||
| Fig. 4 A: The DLS results of the reaction solutions obtained at different polymerization times. B: The relationship of hydrodynamic radius (Rh) and the ratio of gyration radius to hydrodynamic radius (Rg/Rh) with the polymerization times at a feed molar ratio of P4VP/St/AIBN= 10:50000:1 in methanol (St: 1.04 g; methanol: 0.7 g) at 80 °C. | ||
 | ||
| Fig. 5 FESEM images of the polymer nanowires prepared by RAFT polymerization with a feed molar ratio of P4VP/St/AIBN = 10:50000:1 at 80 °C, methanol: 0.7 g; St: 1.04 g for A: 3 h; B: 4 h; C: 12 h: D: 24 h, respectively. The scale bars are 500 nm, 2000 nm, 1000 nm and 1000 nm, respectively. | ||
 | ||
| Fig. 6 TEM images of the nanowires prepared by RAFT polymerization with a feed molar ratio of P4VP/St/AIBN = 10:50000:1 at 80 °C (St: 1.04 g; methanol: 0.7 g) for different polymerization times, A: 3 h; B: 3.5 h; C: 4 h; D: 5 h; E: 12 h and F: 24 h, respectively. The scale bars are 200 nm except for A, where the scale bar is 100 nm. | ||
FESEM and TEM images of the aggregates formed at 12 and 24 h of polymerization can further support the results obtained by DLS measurements. Fig. 5C shows that the aggregate formed at 12 h of polymerization is the long nanowires with hemispherical caps and there is no spherical micelle in this aggregate. The average D of the nanowires is 70 nm, which is similar to that formed at 4 h of polymerization (Fig. 5B). This might result from continuous fusion of the spherical micelles and the short nanorods. After 24 h of polymerization, the resulting long nanowires had a D of approximately 93 nm, and the most of them were over 1 μm long (Fig. 5D). Their TEM images in Fig. 6E and 6F demonstrated the similar results observed by FESEM.
As we know, the multiple morphologies can be prepared either by self-assembling of the block copolymers in a selective solvent or through the PISR process, but their variable parameter is quite different.11 For the morphologies prepared by the self-assembling strategy, the block copolymers with a definite chain length ratio are previously prepared, the morphologies are formed by adding poor solvent into the solution of block copolymers, and the δ of the solvents changed continuously in the process, so the variable is the solubility of solvents. However, in the PISR process, when the recipe and the reaction conditions are fixed, the variable is the composition of block copolymer chains because the chain length of PS block gradually increased with evolution of the polymerization, which alters the force balance in the spherical micelles, leading to transformation of the morphology,11 and in this process, variation of the solubility parameter is relatively small. Thus the variable is the chain length ratio of the PS to P4VP blocks. In order to understand the effects of variable in PISR process and the polymerization conditions on the morphologies formed, the GPC and 1H NMR were used to trace the RAFT polymerization. Fig. 7A is 1H NMR spectra of P4VP-TC and PS-b-P4VPs formed at 1 h and 4 h polymerization. In Fig. 7Ab′ and 7Ac′, we can see clearly the characteristic proton signals of the pyridine and phenyl rings in the P4VP-b-PS appeared at 8.29, 6.57 ppm and 7.06, 6.46 ppm, respectively, and the molecular weights (Mn(NMR)) of P4VP were calculated based on the integral values of the signals at δ = 3.4 (f) and 8.31 ppm (e) in Fig. 7Aa′. Similarly, the Mn(NMR)s of PS-b-P4VP and the degrees of polymerization (DPs) of the P4VP and PS blocks formed at different polymerization times were also calculated based on the DP of P4VP and integration ratio of the signals at 8.31 to that at 7.07 ppm. From the GPC results shown in Fig. 7B, we can see unimodal and symmetric curves. Fig. 7C shows relationship of Mn(NMR)s and conversions with polymerization time. The difference between Mn(NMR) and Mn(GPC) for the same polymer is due to calculation of Mn(GPC)s based on the narrow polystyrene standards. We can see a turning point at 2 h of polymerization on the curve of conversions against polymerization time (Fig. 7C), at which the phase separation occurred, forming the spherical micelles. After this point, the PS chains propagated at reasonable rate still. The critical degree of polymerization (CDP = 375) of the PS block at phase separation was estimated based on the plot of the molecular weight against polymerization time, and the composition of the block copolymer was P4VP99-b-PS375 (the subscripts refer to the number-averaged DP). By calculating the change of chain length and the variation of δ, we can see big change of the PS chain length (from DP = 0 to 375) and very small δ variation of the reaction media (from 23.74 to 23.96) in comparison with starting polymerization. Therefore, the morphology formed in PISR process is determined mainly by the increase of PS chain length. After phase separation, the polymerization took place mainly in the cores of the spherical micelles. Like in the emulsion polymerization of St, the PS particles were highly swollen by St monomer,20 so, the PS chain length increased continuously with relatively high rate (Fig. 7C). With evolution of polymerization, the polymerization media became progressively worse to PS block due to decrease of the St in the solution. These altered the stretching of the PS chains in the core, the surface tension between the PS core and the outside solvents, and repulsion among the corona P4VP chains, and all these made the spherical micelles unstable. When the DP of the PS blocks increased to approximately 770 at 4 h of polymerization, the new force balance drove the spherical micelles to coagulate each other forming rod-like micelles.9 When the polymerization continued to 12 h, the PS blocks grew progressively to DP = 1150, forming a block copolymer, P4VP99-b-PS1150. In this process, the formed spherical micelles and short nanowires were continuously fused to produce the long nanowires without obviously altering the diameter (∼70 nm). After the short nanorods disappeared completely, continuous growth of the PS chains mainly increased the diameter of nanowires, so the DP of the PS chains increased from 1150 to 1246, the diameter of nanowires enhanced from 70 nm to 93 nm (Fig. 5D).
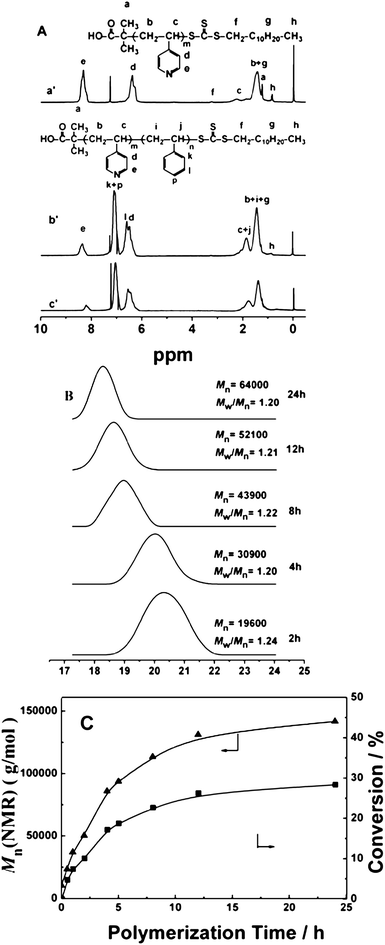 | ||
| Fig. 7 A: 1H NMR spectra of:P4VP-TC (a′) and the block copolymers, P4VP-b-PS formed at 1 h (b′) and 4 h (c′); B: GPC traces of the block copolymers obtained at different polymerization times, and C: the relationship between Mn(NMR) and polymerization time, and conversion and polymerization time for RAFT polymerization in methanol at 80 °C. Feed molar ratio of P4VP-TC/St/AIBN= 10:50000:1; St: 1.04 g; methanol: 0.7 g. | ||
Preparation of uniform vesicles
To further illustrate the formation mechanism of the uniform morphology via the PISR process, creation of the uniform vesicles was selected as an example because the vesicles have been prepared by self-assembling strategy9 and displayed interesting properties and potential applications in various fields such as the catalysis, biomedical, cosmetic and food industries.21 Study of their preparation via PISR can increase understanding of the PISR process.According to a previous study on self-assembling of amphiphilic block copolymers,9 increasing the chain length of PS blocks forming rod-like micelles will create vesicular aggregates. To achieve this purpose, one convenient method is to increase the concentration of St in methanol because St has the solubility parameter similar to that of the PS, and the propagation rate of the PS chains should be fast in the high concentration of St, which will yield the block copolymers with high chain length ratio of PS to P4VP blocks. Therefore, the RAFT dispersion polymerization with a feed molar ratio of P4VP/St/AIBN = 10:100000:1 and the concentration of St (2 g) in methanol (1.1 g) was carried out, the morphology of aggregates obtained at 24 h of polymerization is shown in Fig. 8D. We can see clearly that all aggregates are vesicles, thus preparation of the uniform vesicles is possible.
 | ||
| Fig. 8 TEM images of the vesicles prepared by RAFT polymerization with a feed molar ratio of P4VP/St/AIBN = 10:100000:1 at 80 °C in methanol (St: 2.08 g; methanol: 1.1 g) for different polymerization times, A: 2 h; B: 3 h; C: 4 h and D: 24 h, respectively. FESEM image of the vesicles obtained at 24 h (E). The scale bar is 100 nm for A and B, 200 nm for C and D, and 500 nm for E. | ||
To understand the formation mechanism of the vesicles, the DLS, TEM and FESEM were used to trace the polymerization. Fig. 9A shows DLS results of the polymerization system formed at different times. The polymerization solution at 1 h of polymerization displayed only one peak at Rh = 1.9 nm, which is ascribed to the molecularly dissolving of the formed block copolymer, P4VP-b-PS in the polymerization media, and no aggregation occurred. Compare to the polymerization for preparation of the nanowires (Fig. 4), the larger Rh of the block copolymer formed at the same time might be due to that the high concentration of St in methanol resulted in high polymerization rate of St. Owing to better solubility of the polymerization media to PS, the phase separation was prolonged leading to higher molecular weight of the block copolymer at phase separation. This was supported by GPC results of the copolymers, for example, Mn(GPC)s of the copolymers obtained at 2 h of polymerization were 39800 g mol−1 for preparation of the vesicles (Fig. 10A) and 19600 g mol−1 for preparation of nanowires (Fig. 7B), respectively. When the polymerization proceeded for 2 h, the peak at 1.9 nm decreased greatly, a new peak appeared at Rh = 14.8 nm (Fig. 9A), and its Rg/Rh = 0.77 indicates the formation of the spherical micelles, which are the result of phase separation. The TEM image in Fig. 8A supports the spherical micelles formed at 2 h of polymerization, and their average diameter is about 25 nm. When the polymerization continued to 3 h, only a small amount of the molecularly dissolved block copolymer remained, and the aggregates became large (Rh = 48 nm) as shown in Fig. 9A. But the Rg/Rh was 0.78, so the spherical micelles remained. The TEM image of the aggregates shows spheres morphology with an average diameter of 86 nm (Fig. 8B). The molecularly dissolved P4VP-b-PS was completely disappeared when the polymerization went on further to 4 h, but we can see two peaks, a big peak at Rh = 56 nm and a small peak at 138 nm as shown in Fig. 9A. The big one is the spherical micelles and the small one may be the vesicles, which was identified based on the Rg/Rh = 0.83. To make sure the morphologies formed, TEM observation of this sample was performed, and the result is shown in Fig. 8C. Most of the aggregates are spherical micelles; a few vesicles were observed. This indicates that some of the spherical micelles transformed to the vesicles. Coexistence of the vesicles and rod-like micelles was not observed. So, we presumed that the spherical micelles tended to be fused, and then transformed directly to the vesicles. After 12 h of polymerization, the peak of the spherical micelles disappeared completely, and only one peak appeared at Rh = 216 nm. Their Rg/Rh was 1.04, which demonstrated that the transformation of spherical micelles to the vesicles was achieved. Further polymerization up to 24 h didn't change the vesicle morphology, which was identified based on the Rg/Rh = 1.08. TEM image of this sample evidenced the formation of vesicles. As shown in Fig. 8D, all aggregates are vesicles. Thus, by appropriately selecting the recipe and strict control of the polymerization conditions, preparation of the uniform vesicles is feasible.
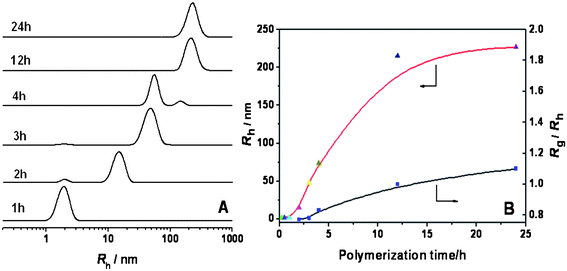 | ||
| Fig. 9 A: The DLS results of the polymerization system obtained at different times. B: The relationship between the hydrodynamic radius (Rh) and polymerization time, and the ratio of gyration radius to hydrodynamic radius (Rg/Rh) and polymerization time for RAFT polymerization with a feed molar ratio of P4VP/St/AIBN = 10:100000:1 (St: 2.08 g; methanol: 1.1 g) at 80 °C for different polymerization times. | ||
 | ||
| Fig. 10 A: GPC traces of the block copolymers obtained at different polymerization times, and B: The relationship of molecular weights and conversion with the polymerization time for RAFT polymerization with feed molar ratio of P4VP/St/AIBN = 10:100000:1 in methanol (St: 2.08 g; methanol: 1.1 g) at 80 °C for different polymerization times. | ||
To further understand the formation mechanism of vesicles in the polymerization, 1H NMR and GPC were used to trace the polymerization, and the results are shown in Fig. 10. All GPC curves of the block copolymers obtained at different polymerization times are unimodal and symmetric, and their molecular weights increased with evolution of polymerization as shown in Fig. 10A. Similar to the polymerization for preparation of the nanowires, the molecular weights, Mn(NMR)s and compositions of the copolymers obtained at different polymerization times were calculated based on the 1H NMR data, relationship of the Mn(NMR) and the conversion with polymerization time is shown in Fig. 10B. The compositions of copolymers formed at phase separation and re-organization were P4VP99-b-PS797 and P4VP99-b-PS1803, respectively. These CDP values of the PS blocks are higher than the relevant CDPs (375 and 770) for preparation of nanowires. This should be due to high solubility of the reaction media to PS chains, leading to the phase separation and re-organization occurred at high chain length ratios of PS to P4VP blocks. Compared to the data in Fig. 7B and 7C, we can find that propagation rate of the PS blocks in the case of the vesicles' preparation was much higher than that in the case of the nanowires' preparation. This is ascribed to the high concentration of St. From the above discussion, we can further understand that the phase separation and re-organization are mainly determined by the growth of PS block, which is the same as for the formation of nanowires. In addition, one point we noticed is that all factors influencing the propagation rate of PS block, such as temperature and agitation, will affect the formation of morphologies in addition to the feed molar ratio and the concentration of St.
It is worth mentioning that multi-morphologies obtained via the PISR process can be conducted in concentrations as high as 500 mg mL−1. This is unimaginable for self-assembly of the block copolymers in a selective solvent, in which 1 mg mL−1 of the copolymers is generally used. This will be possible to provide enough amounts of the polymeric nanomaterials for extensive investigation of their applications. At the same time, the solution of these nanomaterials is very stable, and no precipitation was observed for at least 10 months. After the polymer nanomaterials were dried, they are easily re-dispersed. For example, we dried a sample, whose morphology is the long nanowires with average diameter of 93 nm, and then the solid was put into methanol. After shaking strongly, a well-dispersed solution was obtained as shown in Fig. 11A. The morphology after re-dispersion was measured by FETEM, and the Fig. 11B displays almost the same morphology with the original one.
 | ||
| Fig. 11 A: Dryness-redispersion tests of the resulting nanowires prepared by RAFT polymerization with a feed molar ratio of P4VP/St (1 g)/AIBN= 10:50000:1 in methanol (0.7 g) at 80 °C for 24 h; B: FETEM image of re-dispersed nanowires. The scale bar is 1000 nm. | ||
4. Conclusions
We developed a facile and feasible strategy for preparation of the polymeric nanomaterials with multi-morphologies via one pot RAFT dispersion polymerization. The formation of various morphologies is through two phase transitions, the phase separation to form spherical micelles and re-organization of the spherical micelles to form other morphologies. The two phase transitions are mainly induced by continuous composition change of the block copolymers. This is different from the self-assembling strategy, which is induced by continuous variation of the solubility parameter. By tuning the feed molar ratios and strict control of polymerization conditions, polymeric nanomaterials with multiple morphologies can be prepared. The nanomaterials obtained by this strategy are very stable and are easily to dry and to re-disperse in a selective solvent. This method makes the study of the extensive applications of polymeric nanomaterials possible.5. Acknowledgements
We thank the National Natural Science Foundation of China for financial support under contract No. 50673086 and 50633010.Notes and references
-
(a) M. Salerno, P. Landoni and R. Verganti, Technol. Forecasting Soc. Change, 2008, 75, 1202–1223 Search PubMed
; (b) R. N. Kostoff, R. G. Koytcheff and C. G. Y. Lau, J. Technol. Transfer, 2008, 33, 472–484 CrossRef
-
(a) A. P. Alivisatos, ACS Nano, 2008, 2, 1514–1516 CrossRef CAS
-
(a) P. B. Zetterlund, Y. Kagawa and M. Okubo, Chem. Rev., 2008, 108, 3747–3794 CrossRef CAS
-
(a) G. Delaittre, C. Dire, J. Rieger, J.-L. Putaux and B. Charleux, Chem. Commun., 2009, 2887–2889 RSC
-
(a) L. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CrossRef CAS
-
(a)
K. E. J. Barrett. Dispersion Polymerization in Organic Media, Wiley, New York, 1975 Search PubMed
- S. E. Shim, H. Jung, H. Lee, J. Biswas and S. Choe, Polymer, 2003, 44(19), 5563–5572 CrossRef CAS
-
(a) G. H. Zheng and C. Y. Pan, Macromolecules, 2006, 39, 95–102 CrossRef CAS
- L. Zhang and A. Eisenberg, Polym. Adv. Technol., 1998, 9, 677–699 CrossRef CAS
- W. M. Wan, X. L. Sun and C. Y. Pan, Macromolecules, 2009, 42, 4950–4952 CrossRef CAS
-
(a) W. M. Wan, C. Y. Hong and C. Y. Pan, Chem. Commun., 2009, 5883–5885 RSC
-
(a) C. L. McCormick, B. S. Sumerlin, B. S. Lokitz and J. E. Stempka, Soft Matter, 2008, 4, 1760–1773 RSC
-
(a) L. F. Yan and W. Tao, Polymer, 2010, 51, 2161–2167 CrossRef CAS
-
(a) J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS
-
(a) J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525–1568 RSC
-
J. Brandrup, E. H. Immergut, E. A. Grulke, Polymer Handbook, New York, Wiley-Interscience, 4th edn, 1999, vol. 3, pp. VII699–VII701, VII711 Search PubMed
- S. Y. Song, Y. Zhang, J. Feng, X. Ge, D. P. Liu, W. Q. Fan, Y. Q. Lei, Y. Xing and H. J. Zhang, CrystEngComm, 2009, 11, 1987–1993 RSC
- W. Burchard and W. Richtering, Prog. Colloid Polym. Sci., 1989, 80, 151–163 CAS
- H. J. Dou, M. Jiang, H. S. Peng, D. Y. Chen and Y. Hong, Angew. Chem., Int. Ed., 2003, 42, 1516 CrossRef CAS
-
G. Odian, Principles of Polymerization, John Wiley & Sons, Inc, 3rd edn, 1991, p. 341 Search PubMed
-
(a) J. M. Dean, N. E. Verghese, H. Q. Pham and F. S. Bates, Macromolecules, 2003, 36, 9267–9270 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2010 |