Facile and selective synthesis of aldehyde end-functionalized polymers using a combination of catalytic chain transfer and rhodium catalyzed hydroformylation†
Niels M. B.
Smeets‡
,
Jan
Meuldijk
,
Johan P. A.
Heuts
and
Ard C. J.
Koeken§
*
Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, P.O. Box 513, Eindhoven, The Netherlands. E-mail: a.c.j.koeken@uu.nl
First published on 28th May 2010
Abstract
A novel synthetic pathway towards aldehyde end-functionalized polymers is presented from a combination of catalytic chain transfer polymerization (CCTP) and rhodium catalyzed hydroformylation in supercritical carbon dioxide. CCTP allows for the synthesis of well-defined macromonomers in terms of the average molecular weight and the terminal unit carrying the unsaturated bond. The rhodium catalyzed hydroformylation allows for a high selectivity towards aldehyde end-group functionalized polymers. The introduction of the synthetically versatile aldehyde end-group opens up a broad range of possible applications.
Introduction
There is a continuing interest in controlled radical polymerization (CRP) as these techniques provide a means to control the molecular architecture of the polymer, such as star, comb/brush, network/crosslinked or dendritic/hyperbranched morphologies.1–6 CRP also offers the possibility of end-functionalization of the polymer by post-polymerization modifications, even further expanding the array of possible molecular architectures, e.g. by click chemistry.4,7–11Herein, we demonstrate a novel route for the selective synthesis of functionalized polymeric building blocks for the preparation of complex molecular architectures. We report the synthesis of aldehyde-end-functionalized polymers by a combination of catalytic chain transfer polymerization (CCTP) and subsequent hydroformylation using a very active homogeneous rhodium catalyst in supercritical carbon dioxide (scCO2), see Fig. 1.
 | ||
| Fig. 1 Synthetic approach to aldehyde end-functionalized polymers. Conditions: (A) methyl methacrylate, 0.15 w/w% AIBN, 23 ppm COBF, toluene 50 w/w%, 80 °C; (B) styrene, 0.13 w/w% AIBN, 63 ppm COBF, toluene 50 w/w%, 80 °C; (C) styrene : α-methyl styrene = 18 : 1 (weight basis), 0.25 w/w% AIBN, 7.4 ppm COBF, toluene 50/50 w/w%, 80 °C; (D, E, and F) 3–25 g macromonomer, 52 µmol Rh, Rh : L1 (tris(2,4-di-tert-butylphenyl) phosphite) = 1 : 10, 1.3 mol CO2, 0.8–1 mol CO and H2, 90 °C. | ||
In CCTP low-spin Co(II) complexes mediate the polymerization, which catalyze the chain transfer to monomer reaction and hence provide a means for the control of the average molecular weight in free radical polymerization.12–18 CCTP yields polymer chains with a vinyl end-group functionality, commonly referred to as macromonomers. Due to the vinyl ω-end functionality, the formed macromonomers can be used as starting material for more complex architectures, e.g. methacrylate macromonomers can be copolymerized with a broad variety of monomers resulting in the formation of block,19,20 comb and star like architectures21,22 or self-reinforcing hydrogels.23,24
The ω-end-group of a macromonomer can be controlled by direct CCTP, where a monomer with a low rate coefficient of propagation (kp) and a high rate coefficient of transfer (ktr) is copolymerized with a monomer with a high kp and a low ktr.25–27 An example is the CCTP of styrene (S) and α-methyl styrene (AMS) where macromonomers with nearly 100% AMS end-groups can be obtained.23,28
Control over the ω-end-group functionality can be achieved in direct CCTP via the incorporation of monomers carrying a specific functional group27 or by isomerizational CCTP of monomers containing e.g. an α-hydroxymethyl group.29–31 The disadvantage of using direct CCTP of functional monomers is that the desired functionality is present throughout the whole polymer chain, which does not allow for site-specific chain extension or click-chemistry reactions. Isomerizational CCTP does not suffer from this phenomenon, however, the number of monomers containing e.g. α-hydroxymethyl groups is rather limited.13 Post-functionalization of the vinyl ω-end-group of the macromonomer could provide a more versatile synthetic route towards end-group functionalized macromonomers.
Hydroformylation is an important example of homogeneous catalysis32 and provides a very effective means to extend an alkene with an aldehyde group through the addition of carbon monoxide and hydrogen to the carbon–carbon double bond, giving access to a broad range of further functional groups and more complex molecular structures by chemical modification.33 Next to the research into the improvement of catalyst selectivity and activity,34,35 the use of alternative reaction media, like supercritical carbon dioxide (scCO2), is receiving considerable interest.36–39 The specific advantages of the application of CO2 as an alternative solvent include the possibility to create a one-phase supercritical reaction mixture in which phase boundaries are absent, high diffusivity of the different species, and high solubility of CO and H2.40–43 It is not expected that the macromonomers investigated in this study will dissolve extensively in CO2. However, it is expected that CO2 can expand the liquid macromonomer phase and will allow for a significant reduction in the viscosity.44,45 Lucien and co-workers have demonstrated for CCTP of methyl methacrylate in a single phase CO2-rich46 or CO2 expanded system47,48 that the resulting oligomers do not precipitate under chosen conditions. In these cases methyl methacrylate acted as a co-solvent. The distribution of styrene and methyl methacrylate oligomers of different chain lengths between the supercritical and the polymer phase has been reported by Morbidelli and co-workers49 and Beckman.43
Very recently, the use of a rhodium catalyst modified with the bulky ligand tris(2,4-di-tert-butylphenyl) phosphite (L1) and CO2 as a solvent were combined, which resulted in a catalytic system displaying high rates at relatively mild reaction conditions.50 In what follows we show that this new catalytic system can be used for the effective hydroformylation of macromonomers. The CO2 allows for an enhancement in mass transfer as a result of a lower viscosity of the CO2 expanded macromonomer phase and an increase in CO and H2 concentrations near the rhodium catalyst, which results in the very efficient conversion of sterically hindered vinyl bonds in macromonomers.
Experimental
Materials
Carbon dioxide (CO2), carbon monoxide (CO), and hydrogen (H2), grade 5.0, 4.7, and 5.0, respectively, were obtained from Hoekloos (The Netherlands). Prior to use CO2 was passed over a Messer Oxisorb filter to remove oxygen and moisture. The rhodium precursor rhodium(I) dicarbonyl acetylacetonate ([Rh(CO)2acac], Fluka) and ligand tris(2,4-di-tert-butylphenyl) phosphite (L1, Aldrich, 98%) were stored under argon prior to use. The catalytic chain transfer agent, bis[(difluoroboryl)dimethylglyoximato]-cobalt(II) (COBF), was prepared according to the method of Bakač et al.51 The chain transfer activity of the complex was determined in methyl methacrylate (MMA) bulk polymerization and found to be equal to 30 × 103. For all experiments, a single batch of catalyst was used. The monomers, MMA (Aldrich, 99%), styrene (S, Aldrich, ≥99%) and α-methyl styrene (AMS, Aldrich, 99%), were purified by passing over a column of activated basic aluminium oxide (Aldrich) or inhibitor remover (Aldrich) to remove the inhibitor and stored at −24 °C prior to usage. The initiator, 2,2-azobis(2-methylpropionitrile) (AIBN, Aldrich, 98%), was re-crystallized from methanol (Biosolve, HPLC supra gradient) and stored at −24 °C prior to usage. Toluene (Tol, Merck, analytical grade) and tetrahydrofuran (THF, Merck, analytical grade) were used as received.Macromonomer synthesis
The required amounts of COBF and AIBN were placed inside a three-necked round bottom flask and deoxygenized by a repeated vacuum–argon cycle, see Table 1. The toluene monomer mixture (50 : 50 w/w%, 100 g) was purged for 1 h with argon and subsequently added to the three-necked round bottom flask whilst maintaining an oxygen-free environment. The three-necked round bottom flask was connected to a reflux condenser and subsequently placed inside a pre-heated oil bath at 80 °C. The polymerization was allowed to continue for 4 h, after which the majority of the AIBN initiator has decomposed. The reaction mixture was passed over a basic aluminium oxide column to remove the cobalt complex, the solvent and residual monomer were removed by rotary evaporation and the remaining macromonomer analyzed by SEC, MALDI-TOF-MS and 1H-NMR analysis.Macromonomer hydroformylation in CO2
The details of the high-pressure batch reactor setup are described elsewhere.52 The general procedure for a hydroformylation experiment was started by charging the desired amounts of macromonomer (pMMA and pS), [Rh(CO)2acac] (0.013 g, 52 µmol) and the phosphite ligand (0.335 g, 0.52 mmol) into the empty reactor, which is closed, flushed with argon and subsequently evacuated for three times. The desired amount of carbon monoxide and hydrogen gas (1 : 1) was fed to the reactor at room temperature usually up to a pressure of 4.9 MPa. The reactor contents were heated to 90 °C and simultaneously the reactor was charged with carbon dioxide and the stirrer was switch on with a rate of 700 rpm. After a total reaction time of 24 h, the reactor was cooled to room temperature and carefully depressurized. The reactor was opened and 50 to 70% of the initial weight of macromonomer was collected as a yellow to orange coloured liquid (containing some white coloured solid material). The product solids were allowed to dissolve in the viscous product liquid. For pS a large part of the hydroformylation product was dissolved in THF as the polymer proved to be too viscous to be removed from the reactor conveniently. The resulting yellow to orange coloured viscous liquid was further analyzed by SEC, MALDI-TOF-MS and 1H-NMR. For the hydroformylation of the p(S-co-AMS) the procedure was similar, except that after charging the reactor with CO and H2, the reactor contents were initially heated to a temperature of 70 °C, the stirring was switched on and consecutively CO2 was charged into the reactor at a constant flow up to a total pressure of about 28 MPa. After 20 min at 70 °C the reactor contents were heated to the desired reaction temperature of 90 °C. The experimental conditions for the hydroformylations are collected in Table 2.| Rh/µmol | L1/mmol | Macromonomer/g | ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) b/mmol b/mmol |
CO/mmol | H2/mmol | CO2/mol | P max/MPa | |
|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: T = 90 °C; V = 0.104 dm3. b The amount of unsaturated end-groups was calculated based on the number-average molecular weight of the prepared macromonomer. c The macromonomer contained 2.7 g toluene as concluded from 1H-NMR analysis. | ||||||||
| pMMAc | 49 | 0.52 | 15.5 | 22 | 78 | 81 | 1.4 | 39.6 |
| pS | 52 | 0.52 | 25.5 | 11 | 75 | 73 | 1.3 | 40.1 |
| p(S-co-AMS) | 49 | 0.50 | 2.91 | 1.4 | 101 | 108 | 1.3 | 34 |
Chemical analysis
Size exclusion chromatography (SEC) was performed using a Waters GPC equipped with a Waters model 510 pump and a model 410 differential refractometer. A set of two mixed bed columns (Mixed-C, Polymer Laboratories, 30 cm) were used in series at 40 °C. THF, stabilized with 3,5-di-tert-butyl-4-hydroxytoluene, was used as the eluent at a flow rate of 1 mL min−1, and the system was calibrated using narrow molecular weight polystyrene standards ranging from 600 to 7 × 106 g mol−1. Matrix-assisted laser desorption/ionization time-of-flight mass spectroscopy (MALDI-TOF-MS) analysis was carried out on a Voyager DE-STR from Applied Biosystems using trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]-malononitril (DCTB) as matrix material and potassium trifluoroacetate in the case of pMMA and silver trifluoroacetate in the case of pS and p(S-co-AMS) as ionization agent. All spectra were recorded in the reflector mode. Samples were prepared by dissolving approximately 3 mg mL−1 in THF. The ratio of ionization agent to matrix to polymer sample was 1 : 4 : 4 (w/w). Proton nuclear magnetic resonance (1H-NMR) spectroscopy was performed using a VARIAN 400 NMR at 20 °C. Samples were diluted in CDCl3 to 30–50 mg mL−1. Data were processed using VNMR-software.Results and discussion
Synthesis of the macromonomers
Poly(methyl methacrylate) (pMMA), polystyrene (pS) and poly(styrene-co-α-methyl styrene) (p(S-co-AMS)) macromonomers were synthesized by CCTP in toluene up to low conversion to prevent any possible side reactions, see Fig. 1A, B and C. The reaction time of 4 h was chosen to significantly reduce the amount of remaining initiator (half-life time of AIBN at 80 °C is 1.2 h, resulting in 10% residual initiator) to minimize complications during the consecutive hydroformylation step. The final properties of the prepared macromonomers are collected in Table 3.| COBFa (ppm) | x (—) | SEC | MALDI-TOF-MS | ||
|---|---|---|---|---|---|
| M n /103 g mol−1 | PDI (—) | M n/103 g mol−1 | |||
| a The amount of COBF is defined as moles of COBF per 106 moles of monomer. b The reported conversion is the conversion obtained after 4 h of polymerization. c Number-average molecular weight determined by size exclusion chromatography. d Could not be determined accurately by SEC as the molecular weight exceeds the calibration range of the column set. | |||||
| pMMA | 23 | 0.34 | ∼0.5 to 0.7d | N/Ad | 0.8 |
| pS | 63 | 0.32 | 2.4 | 2.24 | 2.6 |
| p(S-co-AMS) | 7.4 | 0.33 | 2.1 | 1.92 | 1.8 |
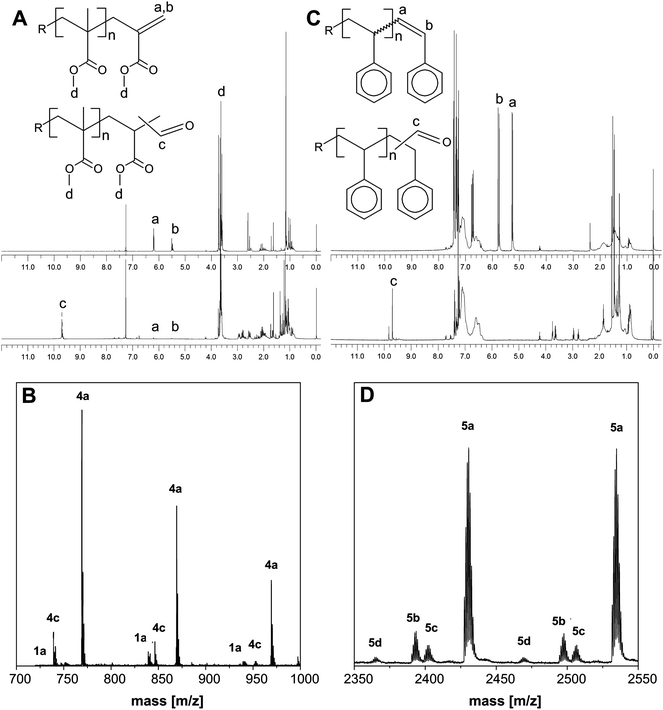
CCTP with monomers containing α-methyl groups relative to the radical centre is known to be highly efficient. CCTP of MMA yields macromonomers with a number-average molecular weight (Mn) of 700 g mol−1 as determined from 1H-NMR analysis, where the intensity of the vinyl ω-end-group (CH2, δ = 6.2 ppm, 2H, Fig. 2A, peak a) is compared to the intensity of the methoxy groups of the repeating units and terminal unit in the polymer backbone (–OCH3δ = 3.6 ppm, 3H, Fig. 2A, peak d). SEC analysis was not reliable as the molecular weight of the macromonomers exceeded the calibration range of the low molecular weight column set.
 | ||
| Fig. 2
1H-NMR and MALDI-ToF-MS spectra for the pMMA and pS aldehyde end-functionalized macromonomers. (A) 1H-NMR spectra of the pMMA macromonomer (1a) and the hydroformylated product (4a). (B) MALDI-ToF-MS spectrum of the aldehyde end-functionalized pMMA polymer, where (4a) H–(MMA)n–CHO; (4c) H–(MMA)n–H2 and (1a) H–(MMA)n–CH2–C(CO–O–CH3)CH2. (C) 1H-NMR spectra of the pS macromonomer (2a) and the hydroformylated product (5a). (D) MALDI-ToF-MS spectrum of the aldehyde end-functionalized pS polymer, where (5a) H–(STY)n–CHO; (5b) NC(CH3)2–(STY)n–CHO; (5c) H–(STY)n–H2 and (5d) NC(CH3)2–(STY)n–H2. | ||
CCTP of styrenics is less efficient, as styryl radicals are able to form reversible cobalt–carbon bonds, which decreases the concentration of the active Co(II) species.53,54 Poly(styrene) macromonomers can be functionalized with an AMS end-group as shown by Kukulj et al.25 The functionalized p(S-co-AMS) macromonomers in this study were prepared by the CCT copolymerization of AMS and S. The CCT homopolymerization of S and the CCT copolymerization of S and AMS yielded pS and p(S-co-AMS) macromonomers with a Mn of 2100 and 2400 g mol−1, respectively, with a polydispersity index of approximately 2 as determined by SEC analysis, see Table 3. The SEC and MALDI-TOF-MS analysis for the different macromonomers are presented in Fig. S1 to S5 of the ESI†.
MALDI-TOF-MS analysis revealed that for the used weight fraction of AMS, 70% of the formed macromonomers carried an AMS end-group. Evidently, this amount can be optimized by increasing the AMS weight fraction. Due to the extremely low kp value of AMS (AMS hardly homopolymerizes), 88% of the p(S-co-AMS) macromonomers contained only a single terminal AMS monomer unit. Note that the ionization efficiency in MALDI-TOF-MS can differ significantly depending on the end-group.55–57 However, due to the structural similarity between the p(S-co-AMS) and pS macromonomers, comparable ionization efficiencies were assumed for the calculation of the fraction of macromonomers carrying an AMS end-group.
The pMMA and pS macromonomers represent a range of macromonomers containing vinyl bonds with different structural orientation (i.e. terminal or internal, respectively) which will likely display differences in the hydroformylation behaviour,34 see Fig. 1. The p(S-co-AMS) macromonomer is an alternative example that illustrates that macromonomers can easily be functionalized with an ω-end-group carrying an external vinyl bond, see Fig. 1.
Macromonomer hydroformylation
The 1H-NMR and MALDI-TOF-MS spectra of the hydroformylated pMMA and pS macromonomers are presented in Fig. 2. The CCTP of MMA resulted in macromonomers exclusively carrying a hydrogen initiating group and, consequently, no macromonomers carrying cyanoisopropyl initiating group (from the AIBN initiator) are observed in the MALDI-TOF-MS spectrum, see Fig. 2B. This is commonly observed for monomers which display efficient CCTP, e.g. methacrylates.14 The formation of the aldehyde end-functionalized polymers is confirmed by 1H-NMR, where the disappearance of the signals of the vinyl end-group (CH2, δ = 6.2 ppm, δ = 5.5 ppm, 2H, Fig. 2A, peaks a and b, respectively) and the appearance of the signal of the proton of the aldehyde functional group (–CHO, δ = 9.7 ppm, 1H, Fig. 2A, peak c) were observed. Moreover, based on the intensity of the signals of the vinyl protons it can be concluded that after 20 h of hydroformylation near complete conversion of the pMMA macromonomers is achieved.
The CCTP conditions for synthesis of the pS macromonomers were less efficient and consequently a significant fraction of the macromonomers contained a cyanoisopropyl initiating fragment (Fig. 2D, peaks 5b and 5d). As can be seen from Fig. 2D, this results in a situation where the hydroformylation products of macromonomers with different chemical structures are obtained. Moreover, the Rh catalyzed hydroformylation of pS macromonomers results in a situation where the aldehyde functionality can be present on either carbon atom of the vinyl group.34 Therefore, the two aldehyde peaks in the 1H-NMR spectrum of pS (CHO, δ = 9.7 and 9.9 ppm, 1H, Fig. 2C, peak c) probably originate from these two most likely aldehyde group positions. The conversion of the pS macromonomers after 20 h was nearly complete, which was concluded from MALDI-TOF-MS and 1H-NMR analysis, see Fig. 2C and D.
Hydrogenation of carbon–carbon double bonds is a common side reaction under hydroformylation conditions.34 Hydrogenation products are observed in the hydroformylation of both pMMA and pS macromonomers, see Fig. 2. The selectivity of the hydroformylation towards aldehyde end-functionalization cannot be quantified from 1H-NMR or MALDI-TOF-MS analysis. The proton signals of the hydrogenated products overlap with signals of the CH2 and CH3 groups present in the pMMA and pS polymer backbone in 1H-NMR analysis. In MALDI-TOF-MS, due to the significant structural differences between the hydroformylated (CHO end-group) and hydrogenated (H-end-group) products, differences in the ionization efficiencies can be expected.55–57 This was illustrated, e.g., for a series of end-functionalized polystyrenes where the ionization efficiency differed for hydrogen, hydroxyl, tertiary amine and quaternary ammonium end-groups.58
From the MALDI-TOF-MS spectrum, however, it can be concluded that the pMMA macromonomers (Fig. 2B, peak 1a) are converted with a high selectivity towards the aldehyde functionality (Fig. 2B, peak 4a). High selectivity towards the aldehyde functionality is also observed for the hydrogen (Fig. 2D, peak 5a) and cyanoisopropyl initiated (Fig. 2D, peak 5b) pS macromonomers. The selectivity towards the aldehyde functionality can be increased by further optimization of the reaction conditions, for example, by varying the concentration of carbon monoxide and hydrogen.59
The 1H-NMR and MALDI-TOF-MS spectra of the p(S-co-AMS) macromonomer hydroformylation product are presented in Fig. 3. The synthesis of the p(S-co-AMS) macromonomers resulted in the formation of hydrogen and cyanoisopropyl initiated macromonomers. Moreover, under the given copolymerization conditions approximately 32% of the macromonomers carry a S end-group and 68% one or two AMS end-groups (Fig. S5†, peaks 3a, 3b and 7a). This results in a complex situation where a broad number of different hydroformylation products is formed, see Fig. 3B. Nevertheless, for the given reaction time of 24 h high conversion of the pS, p(S-co-AMS1) and p(S-co-AMS2) macromonomers is obtained with a high selectivity towards the aldehyde functionality.
 | ||
| Fig. 3 MALDI-ToF-MS spectrum for the p(S-co-AMS) functionalized macromonomer. Explanation of the spectrum: (6a) H–(STY)n–(AMS)1–CHO; (5a) H–(STY)n–CHO; (3a) H–(STY)n–CH2–C(C6H5)CH2; (6b) NC(CH3)2–(STY)n–(AMS)1–CHO; (5b) NC(CH3)2–(STY)n–CHO; (5c) H–(STY)n–H2; (5d) NC(CH3)2–(STY)n–H2; (3b) NC(CH3)2–(STY)n–CH2–C(C6H5)CH2 and (8a) H–(STY)n–(AMS)2–CHO. | ||
The selectivity towards the aldehyde functionality for the pS macromonomers (peaks 5a and 5b in Fig. 2D and 3B) under the given reaction conditions has proven to be lower than is the case for the p(S-co-AMS) macromonomers, where nearly complete selectivity towards the aldehyde functionality is observed. pS macromonomers carrying an AMS end-group can be readily obtained from CCTP and provide an efficient means of obtaining pS aldehyde functionalized polymers.
The reported hydroformylation of the pMMA, pS and p(S-co-AMS) macromonomers, using a [Rh(CO)2acac]/L1 catalyst, allows for the preparation of aldehyde functionalized polymers with nearly complete conversion, high selectivity and pre-determined structural positioning of the aldehyde functional group. Moreover, since CO2 is used as a solvent and CO and H2 as reactants, isolation of the polymer product is expected to be straightforward. As CO, H2, and CO2 are gases at room temperature and atmospheric pressure, only small amounts of these gases will stay dissolved in the hydroformylated product after depressurization. Although this has not been attempted yet, using CO2 to extract the catalyst after the hydroformylation could be another advantageous aspect of the method presented here. It is known that L1 has a significant solubility in scCO2.45,60 There are studies on the extraction of L1 from polymers using scCO2.61,62 The rhodium complex modified with L1 has proven to have a sufficient solubility in scCO2 to carry out very efficient hydroformylation catalysis.45 Therefore, in particular in the case of the higher molecular weight hydroformylation products it is to be expected that extraction with CO2 will allow for subsequent separation of L1 and rhodium from the hydroformylated polymer product. To enhance the extraction of rhodium with CO2 one could think of adding quantities of CO and H2 in order to convert all rhodium species into HRh(CO)4. Based on previous findings on hydroformylation catalysis in CO2 in our laboratory and by others, HRh(CO)4 should have a good solubility in CO2.45,47,63
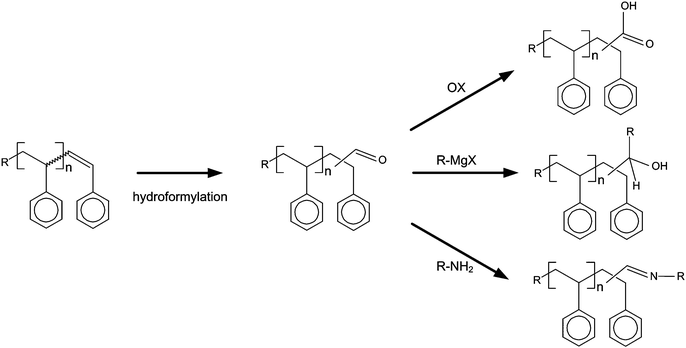
The prepared aldehyde functionalized polymers can be modified for a wide range of further applications. Although this is beyond the scope of the current manuscript, a selection of possible modifications is presented in Fig. 4. The preparation of surfactants is possible through oxidation of the aldehyde functionality towards a carboxylic acid. Block-copolymers and functionalized polymers64 can be obtained e.g. from a reaction of the aldehyde functionality with a primary amine or via a Grignard reaction.
Conclusions
The results of this work show that aldehyde end-functionalized polymers can readily be obtained from a combination of catalytic chain transfer polymerization (CCTP) and rhodium catalyzed hydroformylation. CCTP is a versatile technique for the preparation of a broad range of macromonomers, and offers the advantage of end-group control as the vinyl group will be present in the terminal unit of the polymer chain. Nearly complete conversion of the macromonomers was obtained with a high chemoselectivity for the aldehyde end-group functionality. Optimization of the presented synthesis route can further increase conversion and chemoselectivity. Moreover, since the hydroformylation is performed in CO2 using H2 and CO as reactants, subsequent purification of the hydroformylated polymer product by extraction with CO2 is expected to be straightforward. The combination of CCTP and rhodium catalyzed hydroformylation opens a synthetic route for well-defined aldehyde end-functionalized polymers that can be modified for a broad range of applications.References
- H. Gao and K. Matyjaszewski, Prog. Polym. Sci., 2009, 34, 317 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Acc. Chem. Res., 2008, 41, 1133 CrossRef CAS.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2008, 33, 93.
- D. Fournier, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1369 RSC.
- N. Hadjichristidis, H. Latrou, M. Pitsikalis and J. Mays, Prog. Polym. Sci., 2006, 31, 1068 CrossRef CAS.
- K. Matyjaszewski, Prog. Polym. Sci., 2005, 30, 858 CrossRef CAS.
- J. F. Lutz, H. G. Borner and K. Weichenhan, Macromol. Rapid Commun., 2005, 26, 514 CrossRef CAS.
- C. J. Hawker and K. L. Wooley, Science, 2005, 309, 1200 CrossRef CAS.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15 CrossRef CAS.
- A. B. Lowe, Polym. Chem., 2010, 1, 17 RSC.
- H. Willcock and R. K. O'Reilly, Polym. Chem., 2010, 1, 149 RSC.
- N. S. Enikolopyan, B. R. Smirnov, G. V. Ponomarev and I. M. Belgovskii, J. Polym. Sci., Part A: Gen. Pap., 1981, 19, 879 Search PubMed.
- J. P. A. Heuts, G. E. Roberts and J. D. Biasutti, Aust. J. Chem., 2002, 55, 381 CrossRef CAS.
- A. A. Gridnev and S. D. Ittel, Chem. Rev., 2001, 101, 3611 CrossRef CAS.
- L. V. Karmilova, G. V. Ponomarev, B. R. Smirnov and I. M. Belgovskii, Russ. Chem. Rev., 1984, 53, 223 Search PubMed.
- T. P. Davis, D. M. Haddleton and S. N. Richards, J. Macromol. Sci., Rev. Macromol. Chem., 1994, C34, 243 Search PubMed.
- T. P. Davis, D. Kukulj, D. M. Haddleton and D. R. Maloney, Trends Polym. Sci., 1995, 3, 365 CAS.
- A. A. Gridnev, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1753 CrossRef CAS.
- D. M. Haddleton, C. Topping, D. Kukulj and D. Irvine, Polymer, 1998, 39, 3119 CrossRef CAS.
- D. M. Haddleton, C. Topping, J. J. Hastings and K. G. Suddaby, Macromol. Chem. Phys., 1996, 197, 3027 CrossRef CAS.
- K. J. Abbey, D. L. Trumbo, G. M. Carlson, M. J. Masola and R. A. Zander, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 3417 CAS.
- P. Cacioli, D. G. Hawthorne, R. L. Laslett, E. Rizzardo and D. H. Solomon, J. Macromol. Sci., Part A: Pure Appl. Chem., 1986, 23, 839 Search PubMed.
- L. M. Muratore, T. P. Davis and K. Steinhoff, J. Mater. Chem., 1999, 9, 1687 RSC.
- L. M. Muratore and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 810 CrossRef CAS.
- D. Kukulj, J. P. A. Heuts and T. P. Davis, Macromolecules, 1998, 31, 6034 CrossRef CAS.
- S. C. J. Pierik, D. Masclee and A. M. van Herk, Macromol. Symp., 2001, 165, 19 CrossRef.
- J. P. A. Heuts, D. A. Morisson and T. P. Davis, Controlled/Living Radical Polymerization, in ACS Symposium Series, ed. K. Matyjaszewski, American Chemical Society, Washington, 2000, vol. 768, p. 313 Search PubMed.
- J. J. J. Chiefari, J. Krstina, C. L. Moad, G. Moad, A. Postma, E. Rizzardo and S. H. Thang, Macromolecules, 2005, 38, 9037 CrossRef CAS.
- T. P. Davis, M. D. Zammit, J. P. A. Heuts and K. Moody, Chem. Commun., 1998, 2383 RSC.
- A. A. Gridnev and S. D. Ittel (E. I. DuPont de Nemours and Co.), PCT Int. Pat. Appl., WO 0035960 A2 20000622, 1998; Chem. Abstr. 2000, 133, 59233 Search PubMed.
- D. A. Morrison, L. Eadie and T. P. Davis, Macromolecules, 2001, 34, 7967 CrossRef CAS.
- C. D. Frohning, C. W. Kohlpaintner and H.-W. Bohnen, in Applied Homogeneous Catalysis, ed. B. Cornils and W. A. Herrman, Wiley-VCH, Weinheim, 2nd edn, 2002, vol. 1, pp. 31–103 Search PubMed.
- T. W. G. Solomons, in Solomons Organic Chemistry, ed. T. W. G. Solomons, Wiley, New York, 6th edn, 1996 Search PubMed.
- B. Breit and W. Seiche, Synthesis, 2001, 1–36 CrossRef CAS.
- P. W. N. M. van Leeuwen, Homogeneous Catalysis: Understanding the Art, Kluwer Academic Publishers, Dordrecht, 2004, pp. 139–174 Search PubMed.
- D. J. Cole-Hamilton, Science, 2003, 299, 1702 CrossRef CAS.
- P. G. Jessop, J. Supercrit. Fluids, 2006, 38, 211–231 CrossRef CAS.
- S. Bektesevic, A. M. Kleman, A. E. Marteel-Parrish and M. A. Abraham, J. Supercrit. Fluids, 2006, 38, 232–241 CrossRef CAS.
- S. Kainz, D. Koch, W. Baumann and W. Leitner, Angew. Chem., Int. Ed. Engl., 1997, 36, 1628–1630 CrossRef CAS.
- P. G. Jessop and W. Leitner, in Chemical Synthesis Using Supercritical Fluids, ed. P. G. Jessop and W. Leitner, Wiley-VCH, Weinheim, 1999, pp. 1–36 Search PubMed.
- P. G. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1999, 99, 475 CrossRef CAS.
- A. Baiker, Chem. Rev., 1999, 99, 453 CrossRef CAS.
- E. J. Beckman, J. Supercrit. Fluids, 2004, 28, 121–191 CrossRef CAS.
- D. J. Heldebrant and P. G. Jessop, J. Am. Chem. Soc., 2003, 125, 5600 CrossRef CAS.
- D. Gourgouillon, H. Avelino, J. Fareleira and M. N. da Ponte, J. Supercrit. Fluids, 1998, 13, 177 CrossRef CAS.
- D. J. Forster, J. P. A. Heuts, F. P. Lucien and T. P. Davis, Macromolecules, 1999, 32, 5514 CrossRef CAS.
- G. Zwolak and L. P. Lucien, Macromolecules, 2008, 41, 5141 CrossRef CAS.
- G. Zwolak, N. S. Jayasinghe and F. P. Lucien, J. Supercrit. Fluids, 2006, 38, 420 CrossRef CAS.
- B. Bonavoglia, G. Storti and M. Morbidelli, Macromolecules, 2005, 38, 5593 CrossRef CAS.
- A. C. J. Koeken, N. E. Benes, L. J. P. van den Broeke and J. T. F. Keurentjes, Adv. Synth. Catal., 2009, 351, 1442 CrossRef CAS.
- A. Bakač, M. E. Brynildson and J. H. Espenson, Inorg. Chem., 1986, 25, 4108 CrossRef CAS.
- A. C. J. Koeken, S. J. M. de Bakker, H. M. Costerus, L. J. P. van den Broeke, B.-J. Deelman and J. T. F. Keurentjes, J. Supercrit. Fluids, 2008, 46, 47 CrossRef CAS.
- G. E. Roberts, C. Barner-Kowollik, T. P. Davis and J. P. A. Heuts, Macromolecules, 2003, 36, 1054 CrossRef CAS.
- J. P. A. Heuts, D. J. Forster, T. P. Davis, B. Yamada, H. Yamazoe and M. Azukizawa, Macromolecules, 1999, 32, 2511 CrossRef CAS.
- G. Montaudo, F. Samperi and M. S. Montaudo, Prog. Polym. Sci., 2006, 31, 277 CrossRef CAS.
- G. Montaudo, M. S. Montaudo and F. Samperi, in Mass Spectrometry of Polymers, ed. G. Montaudo and R. P. Lattimer, CRC Press, Boca Raton, 2002, ch. 2 and 10 Search PubMed.
- F. J. Cox, M. V. Johnston and A. Dasgupta, J. Am. Soc. Mass Spectrom., 2003, 14, 648 CrossRef CAS.
- C. Puglisi, F. Samperi, R. Alicata and G. Montaudo, Macromolecules, 2002, 35, 3000 CrossRef CAS.
- A. van Rooy, E. N. Orij, P. C. J. Kamer and P. W. N. M. van Leeuwen, Organometallics, 1995, 14, 34 CrossRef CAS.
- H. J. Vandenburg, A. A. Clifford, K. D. Bartle, J. Carroll, I. Newton, L. M. Garden, J. R. Dean and C. T. Costley, Analyst, 1997, 122, 101R–115R RSC.
- For example: N. J. Cotton, K. D. Bartle, A. A. Clifford and C. J. Dowle, J. Appl. Polym. Sci., 1993, 48, 1607 Search PubMed.
- D. Koch and W. Leitner, J. Am. Chem. Soc., 1998, 120, 13398 CrossRef CAS.
- T. Davis and C. Erkey, Ind. Eng. Chem. Res., 2000, 39, 3671 CrossRef CAS.
- R. C. Li, R. M. Broyer and H. D. Maynard, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5004 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: SEC chromatograms and MALDI-ToF-MS spectra of the prepared macromonomers. See DOI: 10.1039/copy00111b |
| ‡ Current address: Department of Chemical Engineering, Queen's University, Kingston, Ontario, Canada, K7L 1R3. |
| § Current address: Laboratory of Inorganic Chemistry and Catalysis, Utrecht University, Sorbonnelaan 16, 3584 CA, Utrecht, The Netherlands. |
| This journal is © The Royal Society of Chemistry 2010 |