NMR of Na+, glycine and HDO in isotropic and anisotropic carrageenan gels†
Christoph
Naumann
and
Philip William
Kuchel
*
School of Molecular Bioscience, University of Sydney, Australia, NSW 2006. E-mail: philip.kuchel@sydney.edu.au; Fax: +61 2 9351 4726; Tel: +61 2 9351 3709
First published on 1st June 2010
Abstract
The chemical-interaction environment of several gels made from carbohydrate polymers consisting of unmodified commercial-grade ι- and κ-carrageenan and varying in their NaCl and polysaccharide concentrations was studied by solution-state 1H, 2H, and 23Na NMR spectroscopy. All gels showed isotropic NMR spectra at low gel concentrations (1% ‘w/w’) inside 10 mm NMR tubes. Anisotropy was introduced by stretching the gels, and the degree of alignment depended on the extent of stretching as well as gel and NaCl concentration. For Na+ a strong binding component of the anisotropy in ι- and less in κ-carrageenan gels was found, in contrast to a partial binding of glycine, and a spatial and gel-concentration dependent anisotropic effect for deuterated water (HDO). This finding is explained by the possibility of electrostatic interaction between Na+ and ionic sulfate groups in the carrageenan polymer, HDO probably only interacts via hydrogen bonding, while glycine could interact by both means. The lifetime of bound Na+ is likely to be small but bigger than for hydrogen-bonded HDO; thus Na+ would be more exposed to the electric field gradient tensor (caused by stretching the gel) than HDO, resulting in larger anisotropic spectral splitting and explaining the strong NaCl-concentration dependence of 23Na+ NMR anisotropy, which was not evident with HDO in 2H NMR spectra.
1. Introduction
In living systems, second only to water and its constituent H+ and OH− ions, is the ubiquity of Na+ ions; but the interactions of this ion with polymeric systems in and around cells are poorly understood. However, this quadrupolar nucleus has important characteristics that manifest themselves in NMR spectra from anisotropic media such as some stretched gels.1 Likewise the quadrupolar deuterium nucleus in HDO, and the ubiquitous small amino acid glycine, yield NMR spectral features that report on their interactions with the polymeric matrix of stretched gels.1,2Carrageenan polysaccharides that in general strongly interact with Na+ (and other mono- and divalent cations) are widely distributed in marine plants and are of commercial value as gelling agents in processed food and confectionary. They are polymers that are based on alternating residues of 3-linked β-D-galactopyranose and 4-linked α-pyranose. The most common commercial types differ in the number of sulfate groups in the polymer chain (λ, three per unit, ι, two, and κ, one), and the presence (ι and κ) or the absence (λ) of an anhydride bridge between residues 3 and 6 in the 4-linked α-pyranose.3,4 The 3,6-anhydro functionality is thought to be essential for the formation of helices, and thus ι- and κ-carrageenan solutions become gels at low concentrations (less than 1% ‘w/w’), whereas λ-carrageenan solutions manifest, at most, soft-gel characteristics at much higher concentrations (∼10% ‘w/w’). ι-Carrageenan gels are generally softer than κ-carrageenan gels. The physical properties of ι- and κ-carrageenan gels strongly depend on Na+ and other mono- and divalent cations: the addition of Na+ increasingly leads to stronger and stiffer gels for ι- and κ-carrageenan. Many studies have focussed on these effects, and it is surmised that the physical differences arise from a change of individual disordered coils to ordered double helices, and the possible aggregation of these helices into a network to form gels (gelation on the superhelical level). Alternatively, coils may associate with each other to take part in more than one double helix, thus gelation occurs through helix formation (gelation on the helical level).5ι-Carrageenan gels are less sensitive to the exact kind of monovalent cation, whereas for κ-carrageenan gels Na+ is considered a weaker gelling agent, and the addition of K+, Rb+, and Cs+ ions results in stronger gels. It is also found that anions such as I− stabilize the double helix and reduce gelation.3–6 It is suggested that generally κ-carrageenan gels associate on the superhelical level, whereas ι-carrageenan and gelatin gels are likely to be formed by branching on the helical level through incomplete formation of double helices.5,7
Rigid and semi-rigid polymers may form ordered phases (liquid crystals) that show anisotropic behaviour detectable by spectroscopic means, such as solution-state NMR spectroscopy.8,9 Quadrupolar nuclei such as 23Na and 2H yield characteristic residual quadrupolar splittings. An alternative and often-competing state for many polymers is gelation, which is generally not associated with liquid crystalline behaviour and shows isotropic (Lorentzian) lines in the NMR spectrum for the aforementioned nuclei. Partly degraded (lower molecular weight) κ-carrageenan can form a liquid crystalline state, whereas identically prepared ι-carrageenan does not.9 The key to controlling the transition between ordered and gel states in κ-carrageenan is to increase or decrease charge repulsion between the double helices by a judicious choice of ion binding to the helices.9
A recent paper reported a special NMR-spectral effect for bound Na+ in 1% ‘w/w’ ι-carrageenan gels.10 The authors proposed a three-stage, two-state (bound/unbound) model for the gelation mechanism of ι-carrageenan in the presence of Na+. In stage 1 (0–0.06 mol L−1 added exogenous NaCl) when the ι-carrageenan chains are in a coil conformation there is no ‘bound’ Na+; the soft gel is isotropic as shown by single-quantum 23Na NMR spectra being Lorentzian without residual quadrupolar splittings. In stage 2 (0.06–0.17 mol L−1) the ι-carrageenan strands form a double-helix conformation and there is more ‘bound’ Na+, the gel becomes harder and increasingly anisotropic, and residual quadrupolar splittings are resolved. Finally, in stage 3 (0.17–0.58 mol L−1) all ‘binding sites’ are occupied, and additional Na+ ions do not interact with the ι-carrageenan helices; the gel does not change its rheological properties and becomes increasingly isotropic for Na+. Stage 2 corresponds to the Na+ concentration range where the transition from a soft to a hard gel is most pronounced. To account for the observed anisotropy, the authors hypothesized an Na+-dependent alignment of the ι-carrageenan chains in the NMR tube with respect to the B0 static magnetic field “like one observed in stretched gels of gelatin”.1,10,11
The former argument is very likely to be flawed, because in the present paper we show that although the observed 23Na+ anisotropy can be real, the reason for it is an unwanted flow-induced alignment of the viscous ι-carrageenan gels that arises during sample preparation. The relatively small alignment was noticeable because of the large 23Na+ anisotropy in stretched carrageenan gels, as is shown below (Results). Unstretched 1% ‘w/w’ ι-carrageenan gels that were transferred into a 10 mm NMR tube yielded a single Lorentzian peak in 23Na NMR spectra without exception, in the range of added NaCl concentration of 0–1.0 mol L−1. These and other carrageenan gels were, however, made anisotropic by stretching the gels in a manner similar to that done with gelatin.1,2,12 As with gelatin gels we were able to reversibly vary the degree of anisotropy by stretching the sample.13 However, in contrast to stretched gelatin gels, varying the concentration of the carbohydrate or salt (NaCl) resulted in different slopes when the NMR splitting of solute/ion versus the residual quadrupolar splitting of deuterated water (HDO) was plotted against each other. Na+ differs from glycine, alanine, and HDO in that there are recognized binding sites within the carrageenan polymers (the sulfate groups), thus it yields large anisotropies that may serve as a tool that sensitively reports on gel–polymer structure and organization. In this paper we discuss the large 23Na+, and smaller HDO (2H NMR), and glycine (1H NMR) anisotropies of the environments of reporter nuclei/solutes that are inferred from the NMR spectra, obtained from the same samples as a function of carrageenan concentration, and the concentration of added NaCl.
2. Experimental section
2H and 23Na NMR spectra were recorded at 61.422 and 105.842 MHz, respectively, on a Bruker DRX 400 Avance III spectrometer (Bruker, Karlsruhe, Germany) with an Oxford Instruments (Oxford, UK) 9.4 T, vertical, wide-bore magnet. 1H, 2H, and 23Na NMR spectra were recorded using a single pulse-acquire sequence (‘zero memory-and-go’, zg) with the sample thermostatted at 15 °C to be able to contrast spectra with those from comparable gelatin samples, with the exception of the variable temperature and T1 section. T1 measurements were carried out using standard inversion recovery experiments. Carrageenans were purchased from Aldrich: ι-carrageenan, C1138, batch number 067K1490 containing 4% K+, 4% Ca2+, 1% Na+ according to the certificate of analysis provided by the manufacturer, which closely correlated to our 23Na NMR estimate of endogenous Na+ concentration of 6.1 mM; κ-carrageenan, C1013, batch number 117K0050 containing 11% K+, 2% Ca2+, 0.5% Na+, and a moisture content of 10% according to the certificate of analysis (endogenous Na+ concentration 4.2 mM as determined by NMR); λ-carrageenan, 22049, batch number 0001408463, certificate of analysis not provided. 1H NMR spectra of the carrageenan solutions were recorded at 65 °C to measure the chemical shift of the anomeric protons.14 This ι-carrageenan sample did not contain 1H NMR signals ascribed to κ-carrageenan and vice versa. Carrageenan and gelatin gels were prepared as described previously.12,13 Carrageenan gels that were poured into 10 mm NMR tubes were subsequently reheated to 90 °C to eliminate flow-induced anisotropy. The gels once prepared were stored at 4 °C and only subsequently were heated for a short time (to melt the gel) if the samples needed to be transferred into NMR tubes, or silicone tubes for stretching. Our stretching set-up has been described previously.1,12 The minimum gel concentration used was 1% (weight of dry gel per weight of added solvent, ‘w/w’) for carrageenans, and 5% ‘w/w’ for gelatin. The maximum useable gel concentration depended on gel viscosity: ι-carrageenan up to 10% ‘w/w’, κ-carrageenan up to 8% ‘w/w’, whereas gelatin gels were not prepared close to their maximum concentration. All samples contained ∼1% ‘v/v’ added D2O. Samples of a gel concentration >1% ‘w/w’ also contained glycine between 0 and 0.5 mol L−1. When ι- and κ-carrageenan gels were prepared with several buffers containing various amounts of NaCl, after heating, the gels clearly differed in their mechanical properties. Less salt resulted in softer gels, conversely more salt gave more rigid gels. κ-Carrageenan gels were harder than ι-carrageenan gels, whereas λ-carrageenan gels could only be obtained at relatively high polysaccharide concentrations (>5% ‘w/w’) and were relatively soft. All samples discussed here were gels and did not flow when the sample tube was inverted, but the visual appearance of the gels ranged from almost liquid to almost rigid solid. Gelatin gels did not show any visible changes within the range of NaCl added for the carrageenan studies. The physical properties of ι- and κ-carrageenan gels have been described in many studies, so are not repeated here.6,103. Results and discussion
3.1. NMR of unstretched carrageenan samples
Eleven ι- and κ-carrageenan gel samples (1% ‘w/w’) that were prepared from solutions containing from 0.0 to 1.0 mol L−1 NaCl, and differing in their physical appearances from almost solution-like soft gel to hard gel, did not show any anisotropic features in the 23Na or 2H NMR spectra (a quadrupolar triplet in single-quantum 23Na NMR, or a quadrupolar doublet for HDO in 2H NMR) as long as the gels were transferred hot, and heated for a short time to 90 °C once they were in the 10 mm NMR tube (Fig. 1 for 1.0% ‘w/w’ ι-carrageenan gels). When the gels were transferred at lower temperatures (∼60 °C), without subsequent reheating, or into NMR tubes of a smaller diameter; or even more so into silicone rubber tubing; or when gels of a higher concentration were used (e.g., 3.0% ‘w/w’); the 23Na+ signal in single-quantum NMR spectra often showed evidence of sample anisotropy. This, usually unresolved, splitting was particularly evident with the more viscous gels that contained more NaCl (particularly >0.25 M NaCl) and/or higher concentrations (>2% ‘w/w’) of ι- and κ-carrageenan. | ||
| Fig. 1 Stacked plot of 23Na NMR (105.842 MHz) spectra of 1% ‘w/w’ ι-carrageenan gels with varying concentrations of added NaCl. The gels were transferred into 10 mm NMR tubes at 90 °C, and then heated to 90 °C for 10 min to reverse any flow-induced alignment. All spectra corresponded to ideal Lorentzian signals and were able to be closely superimposed when scaled to be of equal height (left inset). The right inset shows a stacked plot of 23Na NMR spectra of the sample prepared with 528 mmol L−1 added NaCl, transferred to silicone rubber tubing, and stretched at six different extensions, thus reversibly varying the anisotropy of the sample. Note the small flow-induced alignment at zero stretching. The maximum degree of HDO anisotropy was a residual quadrupolar splitting of less than 3 Hz. | ||
We could not reproduce an observation presented recently10 involving the addition of Na+ ions that putatively caused an alignment of the ι-carrageenan chains in the NMR tube, with respect to the B0 static magnetic field of the spectrometer, “like one observed in stretched gels of gelatin”.10 While the addition of Na+ reduces the electrostatic repulsion of the sulfate groups, which facilitates the binding of the ι-carrageenan helices, and consequently increases the rigidity of the gel, without stretching there was no anisotropy detectable by NMR (see Fig. 1). The 23Na NMR spectra for each sample were almost perfectly superimposed (see left inset in Fig. 1). The same was observed for κ- and λ-carrageenan gels.
We also measured the longitudinal relaxation times of 23Na+ in isotropic carrageenan samples of 1.0 and 3.0% ‘w/w’ (Table 1). The T1 values increased with increasing NaCl content for a given gel concentration and decreased when the gel concentration was tripled. Measuring the T1 values at a higher temperature (30 °C) led to an increase in the T1 values in a manner similar to the results obtained from free solution (Table 1). Contrary to what was claimed10 the sodium ions experienced increased mobility with higher Na+ content. The T1 data were fitted best by a double decaying-exponential function; this is probably due to competing processes influencing the T1 value. The higher viscosity associated with more Na+-containing carrageenan gels would decrease mobility of the Na+ ions and thus reduce the T1 values. But despite the apparent increased viscosity of the gels with increasing NaCl concentration the vast and growing majority of the Na+ ions were present in the bulk aqueous phase (thus the increasing T1 values) and in fast exchange with Na+ ions interacting with the carrageenan chains, as is shown in the anisotropic section.
| Added NaCl conc./mol L−1 | Solution | κ-Carrageenan | ι-Carrageenan | ||||
|---|---|---|---|---|---|---|---|
| Gel conc. (‘w/w’) | |||||||
| 15 °C | 30 °C | 1.0, 15 °C | 3.0, 15 °C | 1.0, 15 °C | 3.0, 15 °C | 3.0, 30 °C | |
| a Endogenous Na+ concentration was estimated by NMR to be 4.2 mmol L−1 for a 1.0% κ-carrageenan gel and 6.1 mmol L−1 for a 1.0% ι-carrageenan. | |||||||
| 0 | 30.4a | 25.0 | 29.2a | 24.1 | 35.8 | ||
| 0.010 | 47.7 | 65.5 | 31.2 | 30.6 | 24.7 | ||
| 0.025 | 31.6 | 32.3 | 25.8 | ||||
| 0.050 | 32.9 | 26.6 | 34.0 | 26.7 | |||
| 0.075 | 33.7 | 27.6 | 35.0 | 27.8 | |||
| 0.100 | 34.0 | 35.8 | 28.4 | ||||
| 0.150 | 34.9 | 28.7 | 37.1 | 30.2 | |||
| 0.250 | 36.1 | 38.7 | |||||
| 0.500 | 37.5 | 31.2 | 40.4 | 31.4 | 44.6 | ||
| 0.999 | 44.4 | 61.1 | 38.8 | 31.9 | 41.6 | ||
3.2. Anisotropic carrageenan samples
When the 1% ‘w/w’ ι- and κ-carrageenan gels, that showed isotropy with 10 mm NMR tubes, were transferred into 2 mm ID silicone rubber tubing for use in the stretching apparatus,12,13 the 23Na NMR spectrum at zero stretching already showed some degree of anisotropy (Fig. 1, right inset for a 1% ‘w/w’ ι-carrageenan gel, containing 528 mmol L−1 added NaCl). Subsequent stretching of the samples yielded a range of variously split 23Na spectra, without exception for each sample shown in Fig. 1. There was also a small 2H NMR anisotropy for HDO (from ∼1% ‘v/v’ added D2O) at the more pronounced stretching stages. 1% ‘w/w’ κ-carrageenan gels also showed anisotropic behaviour for 23Na+ when stretched, but less than for stretched ι-carrageenan gels. Because the maximum recorded HDO splitting was small (2.8 Hz) we used ι-carrageenan gels of higher concentration, typically between 2 and 5% ‘w/w’, to measure complete sets of HDO and 1H glycine methylene (3DHAHB) splitting in addition to 23Na+ splittings. For similar reasons, we generally used κ-carrageenan samples of 3% and 5% ‘w/w’ gel concentration. It was also noticeable that at identical extension factors, samples that contained the lowest NaCl concentration (the softest gels) had the largest 23Na NMR quadrupolar splitting. The anisotropic splittings increased almost linearly with extension factor12 (although not as uniformly as for stretched gelatin). The half-width of the satellites of the quadrupolar 23Na+ triplet increased linearly with 23Na+ and HDO quadrupolar splittings, whereas the half-width of the central 23Na+ signal only increased slightly (see Fig. S1, ESI†).Stretched λ-carrageenan gels showed a smaller degree of 23Na+ anisotropy at higher concentrations (10% ‘w/w’, Fig. 2).
 | ||
| Fig. 2 Stacked plot of 23Na NMR (105.842 MHz) spectra of a 10.0% ‘w/w’ λ-carrageenan (soft) gel prepared with 147 mmol L−1 added NaCl stretched to three different extensions. The maximum extent of HDO anisotropy was a residual quadrupolar splitting of 1 Hz. | ||
The observation of NMR-detected anisotropy in ι- and κ-carrageenan gels (unmodified, commercially available gel) can be explained by flow-induced alignment of the viscous (setting) gel: ι- and κ-carrageenan gels that contained small amounts of additional NaCl were more fluid than the harder gels produced by adding more NaCl. The latter, more viscous, gels were more susceptible to flow-induced alignment. After an apparent threshold NaCl concentration, when presumably the Na+ binding sites in the gels had largely been occupied, the residual quadrupolar 23Na+ splitting (being the weight-average of fast exchange between isotropic and anisotropic Na+ bound states) declined with more added NaCl. Hence the accidentally aligned gels obtained with higher salt concentrations displayed less anisotropy.
For this study we were not concerned with relatively small accidental alignments but sought to explore the dependence of anisotropy of 23Na+ and HDO on ι- and κ-carrageenan gels; while, the more fully characterized (to date) gelatin gels were used for comparison. The data recorded for stretched gelatin, and ι- and κ-carrageenan gels consisted of two parts: the first set was obtained by changing the gel concentration and using the same (∼150 mmol L−1) exogenous NaCl concentration; while the second entailed holding the gel concentration constant and varying the concentration of NaCl.
We used the stretching apparatus1,12 to extend gels in 4–7 steps to maximum extension (to an extension factor12 of generally less than 1 for the carrageenan gels, but more for gelatin gels), measuring 23Na, 2H, and 1H NMR spectra at each (anisotropic) stage. Each stage represented a separate extent of anisotropy, but a series of such stages yielded linear slopes when plotted against the extension factor, or one other of the measured NMR splittings. The slopes of such plots made easier the comparison of the effects of several conditions on the anisotropy of the gels.
To simplify the discussion of relative splittings on stretching a gel we define the particular slopes as the anisotropy ratio yAx with y being the ordinate and x the abscissa of the graph used to obtain the slope. The anisotropic ratio expresses the degree of anisotropy experienced by the monitoring molecules embedded in the gels (here Na+, HDO, and glycine) relative to each other and responding to each (gel) condition.
3.3. ι-Carrageenan gels
When the splittings in the NMR spectra, that showed evidence of anisotropy, (23Na+, HDO, 1H glycine) obtained for variably stretched ι-carrageenan gels (and κ-carrageenan and also gelatin) were plotted against the extension factor, the slope of the linear graphs depended on gel concentration. Generally the more concentrated the gel, the bigger was the anisotropic splitting. For e.g., doubling the gel concentration from 5% to 10% ‘w/w’ (while keeping the NaCl concentration constant) led to an increase in slope of 2.4 fold for HDO, 1.6 fold for 23Na+, and 1.8 fold for 1H glycine (Table 2).| Slopesa | 5% ‘w/w’ Gels | 10% ‘w/w’ Gels | ||||
|---|---|---|---|---|---|---|
| Gelatin | ι-Carrageenan | κ-Carrageenan | Gelatin | ι-Carrageenan | κ-Carrageenan | |
| a Relative slopes compared with the smallest value in each row. | ||||||
| HDO splitting vs. extension factor | 2 | 2 | 1 | 6 | 4 | — |
| 1H glycine splitting vs. extension factor | 1 | 5 | — | 3 | 9 | — |
| 23Na+ splitting vs. extension factor | 1 | 27 | 7 | 3 | 43 | — |
Stretched ι-carrageenan gels showed gel-concentration dependence when signals from one nucleus were plotted against another; e.g., the obtained slope of the graphs of 23Na+ residual quadrupolar spittings versus the corresponding HDO splittings (Na+AHDO), or alternatively versus the residual dipolar splitting of glycine (Na+AGly), were different when gel concentrations were varied.
Doubling the gel concentration (at constant NaCl concentration) decreased Na+AHDO by 34%; 8% for Na+AGly; and 28% for GlyAHDO. As can be seen in Fig. 3, for a range of ι-carrageenan gels prepared with the same NaCl medium, a quadrupolar HDO splitting of 15 Hz yielded different 23Na+ splittings for different gel concentrations. The smaller the gel concentration the larger was Na+AHDO. This does not mean that there was less quadrupolar splitting of 23Na+ at the higher gel concentration, in fact there was increased splitting as the plots of splitting versus extension factor showed.
 | ||
| Fig. 3 23Na+versus HDO quadrupolar splittings, recorded for variably stretched gels of different ι-carrageenan concentrations, prepared with the same buffer containing 147 mmol L−1 NaCl (including 99 mmol L−1 glycine). The slopes of the individual graphs correspond to Na+AHDO. Black squares, 1.0% ‘w/w’ ι-carrageenan concentration; red circles, 2.0%; green up-pointing triangles, 3.0%; blue down-pointing triangles, 5.4%; sky-blue diamonds, 7.6%; magenta triangles, 10.1%. | ||
However, it seemed that with increasing gel concentration the anisotropy detected by HDO increased significantly more than that of 23Na+ (see Table 2). The same general effect was evident with Na+AGly and GlyAHDO; viz., smaller slopes at higher concentrations. At higher gel concentrations the anisotropy detected by glycine increased relative to that of 23Na+ but decreased relative to HDO. The reduction of the isotropic space by an increase of ordered gel concentration enhanced the effect on HDO, most possibly because HDO has the highest mobility and least binding affinity for the matrix.
When gels of identical concentration but differing in the amount of added NaCl were variably stretched, and the obtained anisotropic NMR data plotted against the extension factor, it was noticed that the slopes of the graph of HDO versus the extension factor were mainly unchanged. The slopes for 23Na splitting decreased with increasing NaCl concentration, and also for 1H glycine, but less so than for 23Na+. Fig. 4 shows the outcome of 23Na+ and HDO quadrupolar splittings for gels of equal concentration (2% ‘w/w’) but differing NaCl concentration. Specifically, at a given HDO splitting, the corresponding 23Na+ splitting was largest for the samples with the least Na+.
 | ||
| Fig. 4 23Na+versus HDO quadrupolar splittings recorded for variably stretched ι-carrageenan (2.0% ‘w/w’) prepared with buffers containing different concentrations of NaCl (including 99 mmol L−1 glycine). The slopes of the individual graphs correspond to Na+AHDO. NaCl concentrations (mmol L−1): black squares, 0.0; red circles, 11.0; green up-pointing triangles, 20.0; blue down-pointing triangles, 50.0; sky-blue diamonds, 104; magenta triangles, 147; yellow grey-rimmed triangles, 248; dark-brown hexagons, 488. | ||
The anisotropic ratio Na+AHDO decreased with increasing NaCl concentration in the gel almost exponentially (Fig. 4, inset). This was also seen for the other anisotropic ratios, Na+AGly and GlyAHDO. The distribution (spread range) of slopes decreased with increasing gel concentration (Fig. 5). Again, concentration dependence was found, this time depending on the NaCl concentration. The addition of an increasing amount of NaCl to a fixed gel concentration changed not only the physical properties of the resulting gels (i.e., they became mechanically stronger) but also decreased the anisotropies for 23Na+ (relatively less is bound) and to a lesser extent for glycine, but hardly affected HDO anisotropies measured with a given extension factor.
 | ||
| Fig. 5 Slopes of 23Na+versus HDO quadrupolar splittings, Na+AHDO (a), and graph of slopes of glycine dipolar versus HDO quadrupolar splitting, GlyAHDO (b), for ι-carrageenan gels prepared with buffers containing different concentrations of NaCl versusι-carrageenan gel concentration ‘w/w’. NaCl concentration (mmol L−1): black squares, 0.0; red circles, 11.0; green up-pointing triangles, 20.0–22.0; blue down-facing triangles, 50.0–52.0; sky-blue diamonds, 104–110; magenta triangles, 144–147; yellow grey-rimmed triangles, 248–253; dark-brown hexagons, 483–488. | ||
At higher ι-carrageenan concentrations the spread of Na+AHDO values was smaller (Fig. 5a), and as discussed above their absolute values declined with higher gel concentration. In addition, Na+AHDO for the lower NaCl concentrations became more similar the higher the gel concentration was made.
The spread of the anisotropy ratio yAx depending on gel and NaCl concentration was also seen when the residual dipolar 1H glycine splitting versus HDO splitting was graphed (Fig. 5b). The inset in Fig. 5b shows GlyAHDO obtained from the splittings produced in 3% ‘w/w’ ι-carrageenan gels without added NaCl; there was little difference in anisotropy between samples that contained 99 mmol L−1 and 520 mmol L−1 added glycine. Also, there was little difference in slopes for 1H glycine versus HDO of ι-carrageenan samples that had not been subjected to heating to more than 42 °C, and identical samples that had been heated to 90 °C. However, only samples that had been heated to 90 °C, cooled to 15 °C and then stretched, showed the usual 23Na triplets. In the absence of heating to 90 °C, the 23Na+ satellites were broadened almost into the baseline. Heating to 90 °C converted a soft and almost liquid gel to a hard and firm one, but this physical transformation did not affect the anisotropic splittings for glycine and HDO. The 23Na+ and HDO splittings for stretched ι-carrageenan were not noticeably affected by glycine. In the absence of glycine similar slopes were obtained for the relative splitting expressed as Na+AHDOversus NaCl concentration (Fig. S2, ESI†).
The major factor determining HDO anisotropy in ι-carrageenan was the gel concentration. HDO anisotropy at similar extents of ordering was presumably increased by a reduction of the isotropic regions in the sample. In contrast, the effects of Na+ concentration on the 23Na NMR spectra indicated a high sensitivity of this ion to the physical changes occurring in the gel; and 23Na+ anisotropy was more affected by the NaCl concentration in the gel than by the concentration of the gel itself. This effect may have arisen because of existing binding sites for Na+ in ι-carrageenan helices, especially the sulfate groups; as a consequence, the reduction of the isotropic regions for Na+ in more highly concentrated gels was presumably less important.
The anisotropy data for glycine lay between the two extremes of 23Na+ and HDO. There was some sensitivity of the 1H-glycine 3DHAHB splitting to physical changes in the gel; a 2.5 fold reduction of the ratio of slope for 1H-glycine versus HDO splitting (GlyAHDO) from low to high NaCl concentration at constant gel concentration (2% ‘w/w’) was evident, but also to higher gel concentration (40% reduction of GlyAHDO from 1% to 10% gel concentration at 144–147 mmol L−1 NaCl, Fig. 5b).
3.4. κ-Carrageenan gels
Stretched κ-carrageenan gels showed much less gel-concentration dependence of splittings than similar ι-carrageenan samples. The anisotropy ratios Na+AHDO were very close for different κ-gel concentrations prepared with the same buffer (Fig. S3, ESI†). However, as Fig. 6a illustrates, for a 3% ‘w/w’ gel, the spread of Na+AHDO values for κ-carrageenan, having the same gel concentration but different Na+ concentrations, was clearly visible in a pattern like that of ι-carrageenan gels, but of reduced magnitude. This spread of slopes depending on NaCl concentration was also greatly reduced with 5% ‘w/w’ gel (data not shown). | ||
| Fig. 6 23Na+versus HDO quadrupolar splittings, Na+AHDO, for variably stretched κ-carrageenan (3.0% ‘w/w’) gel (a), and 30% ‘w/w’ gelatin (b), prepared with buffer containing different concentrations (mmol L−1) of NaCl (including 99 mmol L−1 glycine): black squares, 0.0; red circles, 11.0; green up-pointing triangles, 20.0; blue down-pointing triangles, 50.0; sky-blue diamonds, 104; magenta triangles, 147; yellow grey-rimmed triangles, 248; dark-brown hexagons, 488. | ||
Overall, the sensitivity of the anisotropic ratio Na+AHDO to NaCl concentration for κ-carrageenan gels was less pronounced (a factor of two between lowest and highest NaCl concentrations) than for ι-carrageenan (a factor of five), which points to a lesser degree of Na+ binding (only one sulfate group per unit cell in the κ-carrageenan polymer).
3.5. Gelatin gels
As for ι-carrageenan gels, plotting the anisotropy data (23Na+, HDO, and 1H glycine splittings) obtained from variably stretched gelatin gels against the extension factor, the slopes of the resulting linear plots depended on gel concentration. Doubling the gel concentration (while keeping the NaCl concentration constant) from 5% to 10% w/w led to a three-fold increase in slope for the splittings of HDO, 23Na+, and 1H glycine (Table 2). Although stretching a more concentrated gel sample yielded larger anisotropies at any given extension factor, almost the same splitting could be obtained by stretching a less concentrated sample more than a more concentrated one.12 In contrast to ι-carrageenan, but in a manner similar to κ-carrageenan, the anisotropic ratio (e.g., Na+AHDO) was changed little by gel concentration; stretched gelatin gels showed very linear behaviour for all three studied nuclei, and very little concentration dependence was evident as seen previously for DMSO and alanine (Fig. S4 in ESI†).1,2,12,13 Preparing gels of the same concentration with different NaCl concentrations did not change Na+AHDO very much either (Fig. 6b). The increases in anisotropy at higher gel concentration were most likely to have been caused by a reduction in isotropic regions. The amount of added NaCl did not greatly influence slopes of relative splittings versus extent of stretching of 30% ‘w/w’ gelatin gels, indicating that the gel structure was not changed by changes in gel and NaCl concentration. A higher gel concentration increased the measured anisotropy linearly. This was equally true for 23Na+, HDO and 1H-glycine (data not shown).3.6. Temperature dependence of the anisotropy for ι- and κ-carrageenan gels
1H, 2H and 23Na NMR spectra of ι- and κ-carrageenan containing 1H-glycine, HDO and 23Na+, were recorded at temperatures from 15 °C to 45 °C. At similar extension factors there was a reduced splitting due to anisotropy when the temperature was raised (probably implying a lessened anisotropic contribution from the exchange), but very similar anisotropic ratios Na+AHDO were found. Fig. 7 shows that the observed NaCl concentration dependence of Na+AHDO in ι-carrageenan samples was retained at least between 15 °C and 45 °C. Higher temperatures revealed smaller width-at-half-height values for the satellites of the 23Na+ quadrupolar triplet possibly indicating faster exchange between isotropic and anisotropic sodium states (while the quadrupolar triplet maintained integral ratios very close to the theoretical 3 : 4 : 3). | ||
| Fig. 7 23Na+versus HDO quadrupolar splittings recorded at different temperatures for variably stretched ι-carrageenan (2.0% ‘w/w’) gels prepared with buffers containing three different concentrations of NaCl. The slopes of the individual graphs correspond to Na+AHDO. Black squares, 15 °C; red circles, 25 °C; green up-pointing triangles, 35 °C; blue down-pointing triangles, 45 °C. | ||
κ-Carrageenan samples also showed reduced NMR splittings (hence inferred anisotropy) at higher temperatures, as well as unchanged Na+AHDO values (Fig. 8 for a typical sample and Fig. S5 in ESI†). The inset in Fig. 8 shows that when the sample was heated from 35 °C to 45 °C inside the NMR sample holder, the resulting 23Na NMR spectra showed a loss of anisotropy and a sharpening of the satellite peaks; and the reverse effect occurred on lowering the temperature from 45 °C to 15 °C. At all measured temperatures ι- and κ-carrageenan gels showed reversible increases in the widths of the satellite 23Na+ signals with increased stretching (and hence inferred anisotropy).
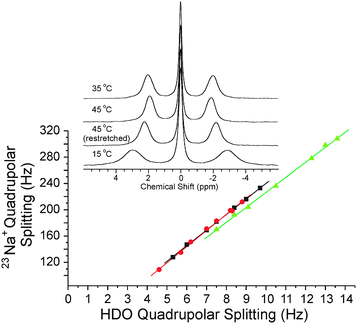 | ||
| Fig. 8 23Na+ quadrupolar splitting versus HDO quadrupolar splittings recorded at different temperatures for a variably stretched κ-carrageenan (3.0% ‘w/w’) gel prepared with 147 mmol L−1 NaCl (including 99 mmol L−11H-glycine). The slopes of the individual graphs corresponded to Na+AHDO. Black squares, 35 °C; red circles, 45 °C; green up-pointing triangles, 15 °C. The inset shows the 23Na NMR spectra obtained at the different temperatures using the same extension factor of 0.58. | ||
4. Conclusions
Carrageenan (ι and κ) gels (as well as gelatin) were shown to be isotropic by NMR, but these gels became anisotropic when stretched. The data obtained for equal gel concentration of the three different anisotropic polymers showed comparable effects for HDO in stretched gelatin, ι- and κ-carrageenan gels, but there was a large difference in relative anisotropies (e.g., Na+AHDO) between the carrageenan gels (ι > κ) and gelatin for 23Na+ (Table 1). It appeared that HDO anisotropy was dominated by fast exchange between isotropic and anisotropic regions. In contrast, the differences for the anisotropy of Na+ in ι-, and κ-carrageenan gels versus gelatin gels indicated a contributing electrostatic interaction between Na+ and, most likely, sulfate groups in the carrageenan gels.In summary, from our data we suggest a strong binding component in the anisotropy for Na+ in ι- and less in κ-carrageenan gels, a partial binding of glycine, and a mainly spatial anisotropic effect for HDO. The reason for the larger anisotropic effect for Na+ in stretched carrageenan gels probably lies with the electrostatic nature of Na+ binding to sulfate groups. HDO probably only interacts via hydrogen bonds. The lifetime of bound Na+ is probably small but bigger than for hydrogen-bonded HDO, thus Na+ would be relatively more exposed to the electric field gradient tensor (caused by stretching the gel) than HDO, resulting in larger anisotropic NMR-spectral splitting. The fact that Na+ is considered a weak gelling agent worked in our favor since a strong binder would likely exhibit broad signals. Currently we are exploring the effects that different ions have on the anisotropy of stretched carrageenan gels.
Our introduction of ‘anisotropy ratios’ as a means of expressing the order of stretched carrageenan gels will be expanded to include other anisotropic systems, with regard to understanding the factors that make a stretched polymeric gel anisotropic; and this, amongst other things, should illuminate understanding of the interaction of Na+, and metabolites with carbohydrate polymers, even in vivo.
Acknowledgements
Funding by an Australian Research Council Discovery Project grant to PWK (DP0877789) is gratefully acknowledged.Notes and references
- P. W. Kuchel, B. E. Chapman, N. Müller, W. A. Bubb, D. J. Philp and A. M. Torres, J. Magn. Reson., 2006, 180, 256–265 CrossRef CAS.
- G. Kummerlöwe, F. Halbach, B. Laufer and B. Luy, Open Spectros. J., 2008, 2, 29–33 Search PubMed.
- M. Rinaudo, Polym. Int., 2008, 57, 397–430 CrossRef CAS.
- V. L. Campo, D. F. Kawano, D. B. da Silva, Jr and I. Carvalho, Carbohydr. Polym., 2009, 77, 167–180 CrossRef CAS.
- C. Viebke, L. Piculell and S. Nilsson, Macromolecules, 1994, 27, 4160–4166 CrossRef CAS.
- For instance: L. Piculell, S. Nilsson and P. Strom, Carbohydr. Res., 1989, 188, 121–135 Search PubMed; J. Borgström, L. Piculell, C. Viebke and Y. Talmon, Int. J. Biol. Macromol., 1996, 18, 223–229 CrossRef CAS; A.-S. Michel, M. M. Mestdagh and M. A. V. Axelos, Int. J. Biol. Macromol., 1997, 21, 195–200 CrossRef CAS; Y. Yuguchi, H. Urakawa and K. Kajiwara, Food Hydrocolloids, 2003, 17, 481–485 CrossRef CAS.
- F. Van de Velde, H. S. Rollema, N. S. Grinberg, T. V. Burova, V. Y. Grinberg and R. H. Tromp, Biopolymers, 2002, 65, 299–312 CrossRef CAS.
- J. W. Emsley and J. C. Lindon, NMR Spectroscopy Using Liquid Crystal Solvents, Pergamon, Oxford, 1975 Search PubMed.
- J. Borgström, P.-O. Quist and L. Piculell, Macromolecules, 1996, 29, 5926–5933 CrossRef; J. Borgström, M. Egermayer, T. Sparrman, P.-O. Quist and L. Piculell, Langmuir, 1998, 14, 4935–4944 CrossRef.
- M. Gobet, M. Mouaddab, N. Cayot, J.-M. Bonny, E. Guichard, J.-L. Le Quéré, C. Moreau and L. Foucat, Magn. Reson. Chem., 2009, 47, 307–312 CrossRef CAS.
- K. Kobzar, H. Kessler and B. Luy, Angew. Chem., Int. Ed., 2005, 44, 3145–3147 CrossRef.
- C. Naumann and P. W. Kuchel, J. Phys. Chem. A, 2008, 112, 8659–8664 CrossRef CAS; C. Naumann, W. A. Bubb, B. E. Chapman and P. W. Kuchel, J. Am. Chem. Soc., 2007, 129, 5340–5341 CrossRef CAS.
- C. Naumann and P. W. Kuchel, Chem.–Eur. J., 2009, 15, 12189–12191 CrossRef CAS.
- F. van de Velde, L. Pereira and H. S. Rollema, Carbohydr. Res., 2004, 339, 2309–2313 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures referred to in the main text. See DOI: 10.1039/c0py00038h |
| This journal is © The Royal Society of Chemistry 2010 |