In situ molecular composites of ladder polyphenylsilsesquioxane and polyisophthalamide and their electro-spinning fibers†
Zhongjie
Ren
a,
Ye
Tian
b,
Ping
Xie
b,
Shouke
Yan
*a and
Rongben
Zhang
*b
aState Key laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing, China. E-mail: skyan@mail.buct.edu.cn; Fax: + 861064455928; Tel: + 861064455928
bLaboratory of Polymer Science and Material, Beijing National Laboratory for Molecular Sciences (BNLMS), Institute of Chemistry, Chinese Academy of Sciences, Beijing, China. E-mail: zhangrb@iccas.ac.cn; Tel: + 861062565612
First published on 28th May 2010
Abstract
The molecular composites of ladder polyphenylsilsesquioxane (Ph-LPSQ) and alkoxy substituted polyisophthalamide (Cn-PA) are in situ prepared for the first time by supramolecular template directed synchronous two-step synthesis, mainly including (A) self-assembled ladder superstructure-based synchronous growth polymerization of the silylated diaminophenylene-bridged ladder polyphenylsiloxane (Ph-DLPS) and (B) isophthalylchloride (IPC)-induced synchronous cleavage of the silylated diaminophenylene-bridge and in situ dehydrochlorination condensation. Moreover, novel composite fibers with sub-μm level diameters are prepared by electro-spinning and their morphologies are investigated by SEM. In order to improve the spinning capability, alkoxy groups in different lengths are introduced into the benzene ring of IPC to produce alkoxy substituted polyisophthalamide (Cn-PA). The fiber-forming ability and the morphology of the resulting electro-spun fibers depend strongly on the solvents used, solution concentration and lengths of the alkoxy groups. Uniform continuous fibers of Cn-PA/Ph-LPSQ are spun from chloroform solution. The longer side chains can result in the formation of a fiber with a smaller diameter. The molecular composition mechanism of Ph-LPSQ and Cn-PA is, in particular, examined using FT-IR spectroscopy, indicating that the hydrogen bond between the amide groups of Cn-PA and Si–O–Si bonds of Ph-LPSQ plays a decisive role in their uniform compounding.
Introduction
Wholly aromatic polyamides (PA) are among the oldest members of the class of thermally stable polymers, and some of them have been commercialized as high temperature resistant fibers, high-strength and high-modulus fibers, and high-performance plastics.1 High molecular weight (Mw) ladder polyphenylsilsesquioxanes (Ph-LPSQs) have been especially emphasized due to their hybrid composite nature and excellent resistance to thermal and irradiation degradations caused by the stronger bond energy of Si–O linkages than that of the common C–C and C–O ones. 2,3The molecular composition of polyisophthalamide (PA) and ladder polyphenylsilsesquioxane (Ph-LPSQ) is currently attracting a great deal of interest due to the well-known advantages of combining the exceptional heat and flame resistance and high mechanical properties of wholly aromatic polyamides Cn-PA and superior heat-, irradiation- and oxidation-resistant stability of Ph-LPSQ. Thus, it can open a new way to composites of Cn-PA/Ph-LPSQ as promising structural or functional materials. However, it is impossible to realize the molecular composition merely by utilizing the traditional blending methods because the non-meltability of Ph-LPSQ and the limited miscibility of Ph-LPSQ and Cn-PA make both the melt and co-solvent blending methods infeasible.
The most usual method for the synthesis of polyaramide (PA) is the one starting from diamine-diacid chloride monomer pairs through either interfacial or low-temperature solution polycondensation.4 Recently, Lozano et al.5 prepared silylated diamines in situ by adding trimethylchlorosilane to the diamine solutions, and then adding diacid chloride to N-silylated diamines to give polyaramides. The synthesis of high Mw ladder Ph-LPSQ still has many straightforward difficulties, such as cyclization and gelation reactions resulting in formation of very complicated products with cyclic, ladder, cage and cross-linked structures.6 In 1960, Brown et al.7 first reported a high Mw ladder polyphenylsilsesquioxane (Ph-LPSQ). Unfortunately, its ladder structure was later refuted by Frye and Klosowski8 who indicated that the so-called Ph-LPSQ actually was “partially opened polycyclic cages”. As Bailey9 proposed, the most desirable type of reaction for preparing perfect ladder polymers is the one in which both sides of the ladder grow synchronously. To realize synchronous growth, we put forward a supramolecular template strategy named “supramolecular architecture-directed stepwise coupling and polymerization”, by which a series of ordered ladder polysilsesquioxanes were prepared.10,11
To prepare the target molecular composites of Cn-PA/Ph-LPSQ, a supramolecular template directed synchronous two-step synthesis was proposed as shown in Scheme 1. (A) Self-assembled ladder superstructure-based synchronous growth polymerization to diaminophenylene-bridged ladder polyphenylsiloxane (Ph-DLPS)12 and (B) isophthalylchloride (IPC)-induced synchronous cleavage of the silylated diaminophenylene-bridge and in situ dehydrochlorination condensation.11 The key point of the synthesis is step B. IPC can initially form a stable donor–acceptor complex with the silylated diamine-bridge of Ph-DLPS by hydrogen bond, benzene ring's π-stacking and so on. The complex would make the bridge synchronously cleave into two new ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–Cl groups, which can be converted to Si–O–Si ladder rung by controllable hydrolysis and in situ condensation. Meanwhile, the N-silylated phenylenediamine-bridge of Ph-DLPS can react with IPC to give aromatic polyamides.11 So the molecular composite of synchronously formed ladder polyphenylsilsesquioxane (Ph-LPSQ) and polyaramide (PA) was successfully prepared.
Si–Cl groups, which can be converted to Si–O–Si ladder rung by controllable hydrolysis and in situ condensation. Meanwhile, the N-silylated phenylenediamine-bridge of Ph-DLPS can react with IPC to give aromatic polyamides.11 So the molecular composite of synchronously formed ladder polyphenylsilsesquioxane (Ph-LPSQ) and polyaramide (PA) was successfully prepared.
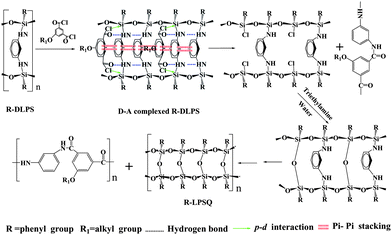 | ||
| Scheme 1 The synthetic route to Ph-LPSQ and Cn-PA. | ||
It is noteworthy that electrospinning technology could remove the continuous polymer nanofibers from polymer solutions or melts in high electric fields.13–15 Thus far, a wide variety of organic polymers nanofibers have been successfully prepared via this process.16,17 In particular, there has been an increased interest in the macroscopic alignment of electrospun fibers for their various applications, ranging from electrical and optical technologies to biological tissue engineering.18 Therefore, electrospinning technology can be a feasible solution that makes the molecular composites of Cn-PA/Ph-LPSQ to be promising functional materials.
In this study, the molecular composite fibers of Cn-PA/Ph-LPSQ were prepared by in situ synchronous synthesis firstly and then electro-spinning. In order to improve the spinning capability, alkoxy groups of different lengths were introduced into the benzene ring of isophthalylchloride (IPC) to produce alkoxy substituted Cn-PA. Moreover, the molecular composition mechanism of PA/Ph-LPSQ was examined using FT-IR spectroscopy.
Results and discussion
1. The preparation and characterization of Ph-LPSQ and Cn-PA
 | ||
| Fig. 1 (A) 29Si-NMR spectra of (a) Ph-DLPS (δ = −18.1); (b) Ph-LPSQ (δ = −21.3); (B). XRD spectra of (a) Ph-LPSQ; (b) Ph-DLPS and (C) MALDI-TOF MS of Ph-LPSQ. | ||
To characterize the Ph-LPSQ and Cn-PA, both of them were isolated from the composites respectively by the appropriate solvent extraction. In the 29Si-NMR spectra (Fig. 1A–b), the signal centered at δ = −21.5 ppm with Δ as small as ca. 1 ppm is assigned to the PhSiO3/2 of Ph-LPSQ unit, indicating that the polymer only consists of regular structure units without any noticeable branched moieties. The XRD profile of the Ph-LPSQ sample (Fig. 1B–a) exhibits two distinct peaks at 2θ = 7.2° and 19.8°, which stand for the ladder width w = 1.21 nm and the ladder thickness t = 0.45 nm, respectively. X-Ray photoelectron spectroscopy (XPS) analysis of Ph-LPSQ (ESI†) clearly demonstrates none of the N or Cl atoms derived from the Ph-DLPS remained, indicating the high purity of Ph-LPSQ. Moreover, it was well documented that matrix-assisted laser desorption ionization mass spectroscopy (MALDI-TOF MS) spectrum is much helpful in characterizing condensation polymers. MALDI-TOF MS spectrum of Ph-LPSQ (Fig. 1C) bears the characteristic shape of a condensation polymer. It is made up of clusters of isotopic peaks. The nominal separation between these alternate major clusters, 258 Daltons, is exactly equal to the Ph–Si (O)2/2–O–Si(O)2/2-Ph repeat unit, indicating that the synthesis proceeded as expected to give a double chain ladder structure without other side reactions. The displacement of the major clusters, i.e. the two indicated 258 Daltons in Fig. 1C, is attributed to the Me3SiO- and HOSiO-capped Ph-LPSQ respectively. As mentioned in the experimental section, the Ph-DLPS was capped by trimethylchlorosilane. Therefore, the Ph-LPSQ could be terminated either by Me3SiO– or HOSiO– groups, which made the molecular weight slightly different and caused the cluster displacement. All of the above-mentioned results demonstrate that real ladder Ph-LPSQ has been successfully synthesized. Concerning the molecular weight of the resultant polymer, generally, the gel permeation chromatograph (GPC) with polystyrene as standard is useful to determine the molecular weight of polymers. It is, however, only suitable for flexible single-chain polymers. For this reason, the molecular weight of the resultant Ph-LPSQ was determined by VPO (Mn = 3.87 × 104) in toluene solution as shown in Table 1. Nevertheless, GPC was used here to determine the polydispersity index (PDI) of the Ph-LPSQ, showing its low PDI.
Fig. 2a–d shows the MALDI-TOF MS spectra of Cn-PA. The overall spectra also bear the characteristic shape of a condensation polymer. We only get the upper limit molecular weight about 7000 Daltons because of ionization, detector effect and so on.22 However, we also could deduce the purity of polymer and the proceeding manner of polymer chain from the spectrum of the oligomer. The nominal separation between major clusters of every spectrum, 238, 339, 422, 506 Daltons, is equal to the repeat unit of C0-PA, C6-PA, C12-PA and C18-PA respectively, indicating that aromatic polyamide with and without alkoxyl substitutes are produced successfully. The molecular weights (MW) of all alkoxyl substituted aromatic polyamide were determined by GPC as shown in Table 1. They have high Mw with > 104 Daltons and the low DPI.
 | ||
| Fig. 2 MALDI-TOF MS of (a) C0-PA; (b) C6-PA; (c) C12-PA; (d) C18-PA. | ||
The glass transition temperature (Tg) of the composites Ph-LPSQ/Cn-PA was investigated by DSC. As shown Fig. 3A, all the composites of Ph-LPSQ/Cn-PA displayed only one Tg at high temperature, indicating good miscibility of Ph-LPSQ and Cn-PA. Moreover, Tg was gradually decreased with the increase of the substituted alkoxyl chain of aromatic polyamide because the alkoxyl chain could promote the mobility of PA chain segment.
 | ||
| Fig. 3 (A) DSC curves of (a) Ph-LQSQ/PA (231 °C); (b) Ph-LQSQ/C6-PA (201 °C); (c) Ph-LQSQ/C12-PA (190 °C); (d) Ph-LQSQ/C18-PA (174 °C) and (B) TGA curves of (a) C0-PA/Ph-LPSQ; (b) C6-PA/Ph-LPSQ; (c) C12-PA/Ph-LPSQ and (d) C18-PA/Ph-LPSQ. | ||
Thermogravimetric investigations on all the composites of Ph-LPSQ/C0-PA revealed its high thermal stability with a thermal decomposition temperature about 580 °C and the weight loss of 40 mass % of the initial amount at 800 °C. However there were two stages of weight loss for Ph-LPSQ/Cn-PA.(Fig. 3B) The first one corresponds to thermal decomposition processes of alkoxyl groups which stability decreased with the increase of the substituted alkoxyl group. Comparatively, the composites Ph-LPSQ/C6-PA with short alkoxy group possesses better thermo-stability. The second one is attributed to thermo-destruction of the backbone of Ph-LPSQ/PA, as shown by the weight loss of 52 mass % of the initial amount at 800 °C for Ph-LPSQ/C6-PA.
2. Electro-spinning of the composites Cn-PA/Ph-LPSQ
According to the literature,25–27 spinning solution parameters (solvent, solution concentration, solution viscosity, surface tension, molecular weight of the polymer, etc), and electro-spinning parameters (applied voltage, solution feeding rate, tip-to-collector distance, etc.) are the main factors that influence the electrospinning process. By changing the electrospinning conditions, either fiber or bead morphologies of the Ph-LPSQ/C12-PA composites has been observed.
The selection of solvents depending on boiling point, volatility and polarity, solubility parameter, dielectric constant, density, etc. is a key factor which may have a significant influence on the morphology and continuous fiber-forming ability. Chloroform, toluene, dimethylforamide (DMF) and mixed solvent of DMF and chloroform were tested in this study. Medium alkoxyl group substituted C12-PA and Ph-LPSQ were chosen to conduct electro-spinning. When toluene or DMF was used as solvent, we can only get some spheres with diameter ranging from 200 nm to 1 μm (Fig. 4A–B). Continuous fiber can be spun from the chloroform solution or mixed solvent of chloroform–DMF (8/1) in suitable concentration. But if the solution concentration is too low, it is impossible to obtain uniform continuous fiber and the beaded fiber structures were produced, (Fig. 4C–F) indicating that a high viscosity is required to obtain uniform fibers. Below the critical viscosity, application of voltage results in electrospraying or bead formation primarily due to a Rayleigh instability. This is consistent with the previous findings in the literature.28,29
 | ||
| Fig. 4 SEM images of Ph-LPSQ/C12-PA by electro-spinning in different solvent: (A) toluene 30%; (B) DMF 30%; (C) chloroform–DMF (8/1) 30%; (D) chloroform 18%; (E) chloroform 21%; (F) chloroform 23%. | ||
It is also found that the electrospinning of composite Ph-LPSQ/PA without substituted alkoxyl group can't be conducted to get the fiber even in any kinds of solvents used. On one hand, Shenoy et al.30 have reported that chain entanglement is one of many parameters that can significantly influence fiber-forming capacity during the electrospinning. Sufficient entangled network of polymer chains is required to obtain the continuous electron-spun fiber. The Mw of PA maybe is very low, so PA/Ph-LPSQ can't reach the critical chain entanglement; on the other hand, both PA and Ph-LPSQ are very rigid, resulting in a great difficulty in stretching them. So the alkoxyl groups should be introduced into the PA molecules, which increased the solubility and flexibility of PA and Ph-LPSQ. The electro-spun fibers of the various composites of Ph-LPSQ/C6-PA, Ph-LPSQ/C12-PA and Ph-LPSQ/C18-PA were collected under the optimal electrospinning condition. Fig. 5 shows a series of SEM images of the composite fibers in diameters of 400 nm–1μm electrospun from chloroform solution of Cn-PA/Ph-LPSQ. The appearance of the composites fibers is uniform and smooth. In addition, comparing the composites C18-PA/Ph-LPSQ with C6-PA/Ph-LPSQ and C12-PA/Ph-LPSQ, it is found that long side chains would result in the uniform fibers with the smaller diameters because the longer substituted alkoxyl chains of C18-PA make the composites more extendable in the electric field.31
 | ||
| Fig. 5 SEM images of Ph-LPSQ/Cn-PA in chloroform by electro-spinning: (A) Ph-LPSQ/C6-PA 24%; (B) Ph-LPSQ/C12-PA 23%; (C) Ph-LPSQ/C18-PA 21%. | ||
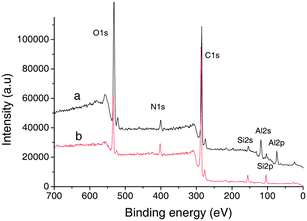 | ||
| Fig. 6 XPS spectra of (a) electrospinning fiber of C12-PA/Ph-LPSQ and (b) C12-PA/Ph-LPSQ. | ||
In addition, SEM images show (Fig. 7) that the crack section of Ph-LPSQ/C6-PA fiber quenched by liquid nitrogen displays the obvious fragile break because of its low flexibility (Fig. 7A). No phase separation can be recognized from the SEM image. Also the fibers of the composites C12-PA/Ph-LPSQ and C18-PA/Ph-LPSQ show the uniform section by ductile break without any phase separation (Fig. 7B–C). This case is attributed to increase of the flexibility of Ph-LPSQ/Cn-PA with the increasing lengths of substituted alkoxyl groups. According to the literature,32 the hydrogen bonding can exists between Si–O–Si bonds of polyhedral oligomeric silsesquioxane (POSS) and N–H bond of polypeptides. Therefore, there should be the intermolecular hydrogen bonding between Si–O–Si bonds of Ph-LPSQ and N–H bond of Cn-PA, which leads to the formation of the uniform fibers. To elucidate this inference, FT-IR analysis was conducted to determine the relationship between the hydrogen-bonded N–H stretching vibration peak of PA and the content of Ph-LPSQ. As shown in Fig. 8, the absorption peak assigned to the hydrogen-bonded N–H stretching vibration peak of PA shifts from 3269 cm−1 to 3280 cm−1 as the content of Ph-LPSQ increases from 0 (Fig. 8a) to 85% (Fig. 8g). This phenomenon can be attributed to the fact that the intermolecular hydrogen bond between carbonyl groups and amine groups of PA has been partially replaced by the hydrogen bond between the amine groups of PA and Si–O–Si bonds of Ph-LPSQ after adding Ph-LPSQ into PA. The hydrogen bond between Si–O–Si bonds and N–H bonds is weaker than that between C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds and N–H bonds of PA itself, which would result in the red shift of wave-number.33,34 This result indicated that the hydrogen bond formation between the Si–O–Si groups of Ph-LPSQ and the N–H groups of PA plays an important role for the molecular composites of Ph-LPSQ/Cn-PA.
O bonds and N–H bonds of PA itself, which would result in the red shift of wave-number.33,34 This result indicated that the hydrogen bond formation between the Si–O–Si groups of Ph-LPSQ and the N–H groups of PA plays an important role for the molecular composites of Ph-LPSQ/Cn-PA.
 | ||
| Fig. 7 SEM images of Ph-LPSQ/Cn-PA fibres: (A) Ph-LPSQ/C6-PA; (B) Ph-LPSQ/C12-PA; (C) Ph-LPSQ/C18-PA. | ||
 | ||
Fig. 8 IR spectra of the N–H stretching region: (a) PA; (b) Ph-LPSQ/PA (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :90 wt%); (c) Ph-LPSQ/PA (25:75 wt%); (d) Ph-LPSQ/PA (40:60 wt%); (e) Ph-LPSQ/PA (55:45 wt%); (f) Ph-LPSQ/PA (70:30 wt%); (g) Ph-LPSQ/PA (85:15 wt%); (h) Ph-LPSQ. :90 wt%); (c) Ph-LPSQ/PA (25:75 wt%); (d) Ph-LPSQ/PA (40:60 wt%); (e) Ph-LPSQ/PA (55:45 wt%); (f) Ph-LPSQ/PA (70:30 wt%); (g) Ph-LPSQ/PA (85:15 wt%); (h) Ph-LPSQ. | ||
Conclusions
The molecular composites of ladder polyphenylsilsesquioxane (Ph-LPSQ) and polyaramide (Cn-PA, n = 0, 6, 12, 18) is in situ prepared by the following synchronous synthesis: (A) self-assembled ladder superstructure-based synchronous growth polymerization to silylated diaminophenylene-bridged ladder polyphenylsiloxane (Ph-DLPS); and (B) isophthalylchloride (IPC)-induced synchronous cleavage of the silylated diaminophenylene-bridge and in situ dehydrochlorination condensation. In addition, the fibers of the composites Ph-LPSQ/Cn-PA are made by electro-spinning. The electro-spun fiber-forming ability and the morphologies of the resultant fibers are tuned upon by the solvents, solution concentration and lengths of side chains. Moreover, the FT-IR spectroscopy analysis indicates that hydrogen bond formed between amine groups of Cn-PA and Si–O–Si promotes the molecular composition of Ph-LPSQ and Cn-PA.Experimental
Materials
Toluene, 1,4-dioxane, hexane and tetrahydrofuran were distilled over sodium and benzophenone. Triethylamine was distilled over sodium before use. All other reagents were obtained from commercial sources and without further purification.Characterization
Fourier transform infrared (FT-IR) spectra were recorded using a NICOLET spectrophotometer in the range of 400–4000cm−1. 1H and 29Si nuclear magnetic resonance spectra were obtained with Bruker DX400 and Bruker DX300 spectrometer at 400 MHz and 59.6 MHz at room temperature respectively. For 29Si-NMR and 1H-NMR measurements tetramethylsilane was used as the reference. X-Ray diffraction (XRD) measurements were performed on a Rigaku D/max 2400 diffractometer with Cu-Kα radiation at a scanning rate 5°/min. Vapor pressure osmometry (VPO) analysis was measured in dry toluene at 40 °C on a Knauner VPO instrument at 20% relative humidity. Elemental analysis was conducted by Heraeus CHN-RAPID, DATEL System, Inc. (Germany). Matrix assisted laser desorption/ionization time-of-flight mass spectra (MALDI-TOF MS) were obtained with a Bruker Biflex III spectrometer using laser of 337 nm and sodium chloride and 4′-hydroxy-α-cyanocinnamic acid as ionizing and matrix reagents, respectively. X-ray photoelectron spectroscopy (XPS) data were obtained with an ESCALab220i-XL electron spectrometer from VG Scientific using 300 W Mg-Kα radiation. The base pressure was about 3 × 10−9 mbar. The binding energies were referenced to the C1s line at 284.8 eV from adventitious carbon. The fitting of the curves was made by Avantage 3.95. Thermogravimetric analysis (TGA) of TCLP was performed on Perkin-Elmer TGA7 thermal analyzer under a 50 mL min−1 nitrogen flow. The samples were heated from 25 to 800 °C at a rate of 10 °C min−1. The DSC investigation was carried out on a Mettler Toledo Star 822 differential scanning calorimeter with heating rate 10 °C min−1 in nitrogen atmosphere. Scanning electron microscopy (SEM) was conducted with a Hitachi S-4300 (Japan). Each sample was sputter-coated with the gold for analysis. Gel permeation chromatography (GPC) analysis was also performed by a set of a Hitachi/Merck L-7100 pump, a Waters 2414 refractive index detector and a Waters 486 ultraviolet detector, the combination of Hersteller MZ-Gel SDplus 5 μm, porosity 100 Å, 103 Å, 104 Å, and 106 Å. DMF with 1g L−1 LiBr was used as eluent at flow rate of 0.8mL min−1 at 35 °C. Polystyrene standards were used for the calibration. The electrospinning apparatus consisted of a metered flow pump, a high D.C. voltage supply and aluminium foils as targets for fiber collection. Electrospinning solutions were prepared by dissolving the required polymers on weight percentage basis in the solvent and stirring the solutions for 24 h to make a well mixed homogenous solution. The solution was then ejected through a syringe using a syringe flow pump at feed rate of 1 ml h−1 and applying a voltage of 15 kV and tip target distance of 20 cm.Synthesis
Acknowledgements
The financial support of NSFC [No.50833006, 50773088, 50973008] and Open Project of State Key Laboratory of Supramolecular Structure and Materials (SKLSSM 201016) are gratefully acknowledged.Notes and References
- P. E. Cassidy, Thermally Stable Polymers, Marcel Dekker, New York, USA 1980 Search PubMed.
- Q. Zhou, S. Yan, C. C. Han, P. Xie and R. Zhang, Adv. Mater., 2008, 20, 2970–2976 CrossRef CAS.
- C. Sanchez, H. Arribart and M. M. Guille, Nat. Mater., 2005, 4, 277–281 CrossRef CAS.
- P. W. Morgan, in Condensation Polymers by Interfacial and Sol & on Methods, Interscience, New York, USA 1965 Search PubMed.
- A. E. Lozano, J. de Abajo and J. G. de la Campa, Macromolecules, 1997, 30, 2507–2508 CrossRef CAS.
- Y. Abe and T. Gunji, Prog. Polym. Sci., 2004, 29, 149–182 CrossRef CAS.
- F. Brown Jr., L. H. Vogt, A. Katchman, J. W. Eustance, K. M. Kiser and K. W. Krantz, J. Am. Chem. Soc., 1960, 82, 6194–6195 CrossRef CAS.
- C. L. Frye and J. M. Klosowski, J. Am. Chem. Soc., 1971, 93, 4599–4601 CrossRef CAS.
- W. J. Bailey, in Concise Encyclopedia of Polymer Science & Engineering, ed. J. L. Kroschwitz, John Willey & Sons, Inc., New York, USA 1990, p. 516 Search PubMed.
- X. Zhang, P. Xie, Z. Shen, J. Jiang, C. Zhu, H. Li, T. Zhang, C. C. Han, L. Wan, S. Yan and R.B. Zhang, Angew. Chem., Int. Ed., 2006, 45, 3112–3116 CrossRef CAS.
- Z. Ren, X. Cao, P. Xie, R. Zhang, S. Yan and Y. Ma, Chem. Commun., 2009,(27), 4079–4081 RSC.
- T. Zhang, K. Deng, P. Zhang, P. Xie and R. Zhang, Chem.–Eur. J., 2006, 12, 3630–3635 CrossRef CAS.
- D. Li and Y. Xia, Adv. Mater., 2004, 16, 1151–1170 CrossRef CAS.
- Y. Dzenis, Science, 2004, 304, 1917–1919 CrossRef CAS.
- Y. Zhu, J. Zhang, Y. Zheng, Z. Huang, L. Feng and L. Jiang, Adv. Funct. Mater., 2006, 16, 568–574 CrossRef CAS.
- S. Kedem, J. Schmidt, Y. Paz and Y. Cohen, Langmuir, 2005, 21, 5600–5604 CrossRef CAS.
- S. Limmer, S. Seraji, Y. Wu, T. P. Chou, C. Nguyen and G. Z. Cao, Adv. Funct. Mater., 2002, 12, 59–64 CrossRef CAS.
- X. Lu, C. Wang and Y. Wei, Small, 2009, 5, 2349–2370 CrossRef CAS.
- Y. Oishi, M. Kakimoto and Y. Imai, Macromolecules, 1988, 21, 547–550 CrossRef CAS.
- Y. Oishi, M. Kakimoto and Y. Imai, J. Polym. Sci., Part A: Polym. Chem., 1991, 29, 1925–1932 CrossRef CAS.
- M. Brook, in Silicon in Organic, Organometallic, and Polymer Chemistry, John Wiley & Sons, Inc., New York, USA, 2000, p. 31 Search PubMed.
- C. McEwen, C. Jackson and B. Larsen, Int. J. Mass Spectrom. Ion Processes, 1997, 160, 387–394 CrossRef CAS.
- G. Taylor, Proc. R. Soc. London, Ser. A, 1964, 280, 383–397 CrossRef.
- Y. Shin, M. Hohman, M. Brenner and G. Rutledge, Appl. Phys. Lett., 2001, 78, 1149–1151 CrossRef CAS.
- S. Fridrikh, J. Yu, M. Brenner and G. Rutledge, Phys. Rev. Lett., 2003, 90, 144502–144504 CrossRef.
- N. Xuyen, E. Ra, H. Geng, K. Kim, K. An and Y. Lee, J. Phys. Chem. B, 2007, 111, 11350–11353 CrossRef CAS.
- S. Theron, E. Zussman and A. Yarin, Polymer, 2004, 45, 2017–2030 CrossRef CAS.
- J. Zheng, A. He, J. Li, J. Xu and C. Han, Polymer, 2006, 47, 7095–7102 CrossRef CAS.
- T. Uyar and F. Besenbacher, Polymer, 2008, 49, 5336–5343 CrossRef CAS.
- S. Shenoy, W. Bates, H. Frisch and G. Wnek, Polymer, 2005, 46, 3372–3384 CrossRef CAS.
- P. Gupta and G. Wilkes, Polymer, 2003, 44, 6353–6359 CrossRef CAS.
- S. Kuo, L. Fang, W. Huang, K. Jeong and F. Chang, Macromolecules, 2009, 42, 1619–1626 CrossRef CAS.
- M. Roberts and S. Jenekhe, Macromolecules, 1991, 24, 3142–3146 CrossRef CAS.
- K. Lee, K. Kim, A. Pesapane, H. Kim and J. Rabolt, Macromolecules, 2008, 41, 1494–1498 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR of Cn-IPC, Cn-PA and Ph-LPSQ; XPS of Ph-LPSQ and Ph-LPSQ; IR of Ph-LPSQ. See DOI: 10.1039/c0py00033g |
| This journal is © The Royal Society of Chemistry 2010 |