Novel polymer coupling chemistry based upon latent cysteine-like residues and thiazolidine chemistry
Joseph S.
Carlson
a,
Megan R.
Hill
a,
Taiga
Young
b and
Philip J.
Costanzo
*a
aCalifornia Polytechnic State University, Department of Chemistry and Biochemistry, 1 Grand Ave, San Luis Obispo, CA 93407-0402, USA. E-mail: pcostanz@calpoly.edu; Fax: + 1 805-756-5500; Tel: +1 805-756-2692
bCalifornia Polytechnic State University, Department of Materials Engineering, 1 Grand Ave, San Luis Obispo, CA 93407-0402, USA
First published on 24th August 2010
Abstract
Chain end functional polymers were prepared via reversible addition–fragmentation transfer (RAFT) polymerization techniques that were further chain extended with acrylonitrile. Under reducing conditions, latent cysteine-like residues were exposed at the chain ends. A variety of reduction conditions were explored and base polymers were then tethered together via thiazolidine chemistry.
The development of simple and efficient crosslinking techniques is sought after for a variety of applications including bioconjugation, material synthesis and design, and surface functionalization. The use of “Click” reactions is becoming increasingly popular because it offers excellent coupling efficiency and is applicable to many synthetic situations.1–6 One of the most popular methods is the copper catalyzed, 1,3-dipolar cycloaddition between an azide and an alkyne. A significant limitation of this methodology is that a metal catalyst, typically a CuI catalyst, is required. This catalyst can be difficult to remove which is detrimental to biological applications because the metal catalyst is not biocompatible.7 The Bertozzi group has developed specialty alkynes that undergo the “Click” reaction without the presence of a metal catalyst; however, these compounds are not commercially available and require multiple steps.8,9
Thiol–ene chemistry, which is a reaction between an alkene and a thiol, has become increasingly popular.10,11 Here, the thiol is added across the alkene via a radical mechanism that is initiated by either light or heat. Thiol–ene chemistry has been extensively utilized to prepare materials in the fabrication of microfluidic devices.12–15 Similar to “Click” chemistry, there are a few synthetic hurdles encountered when incorporating these functional groups into systems. This method can occur in the presence of oxygen and the absence of solvent. No metal catalyst is required, but an external stimulus such as light in the presence a photoinitiator or heat is necessary to generate a radical species.
The Grinstaff group developed a methodology focused upon the thiazolidine linkage, which occurs in the presence of a cysteine residue and an aldehyde (Fig. 1).16,17 Such a reaction occurs without a catalyst, is biocompatible, and proceeds without an external stimulus. The Grinstaff group was able to fabricate materials for replacement of sutures in cataract surgery. The significant limitation of the current methodology is the synthetic schemes utilized, which require the extensive use of peptide chemistry and protecting groups. This approach limits the incorporation of this technique to a smaller window of scientists who possess extensive synthetic capability. As such, the power and attractiveness of the thiazolidine mechanism are limited. The critical aspect of the reaction is the presence of a 1,2-amino-mercapto functionality. The amino functionality undergoes imine formation with an aldehyde and the mercapto group is in the correct proximity to cyclize. Therefore, any 1,2-amino-mercapto functionality will form a thiazolidine linkage in the presence of an aldehyde. This concept has been utilized in combinatorial chemistry, which demonstrates the wide versatility of the reaction.18
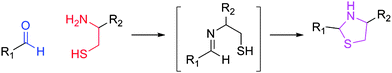 | ||
| Fig. 1 Formation of thiazolidine linkage between an aldehyde and a 1,2-amino-mercapto functionality. | ||
This work details the preparation of base polymers via reversible addition–fragmentation transfer (RAFT) polymerization techniques that were further chain extended with acrylonitrile. Under reducing conditions, latent cysteine-like residues were exposed and in the presence of isophthalaldehyde, polymeric dimers were prepared.
Scheme 1 illustrates the synthetic pathway for the preparation of latent cysteine-like chain end functionality. Compound 1 was synthesized and utilized as a RAFT initiator to prepare polystyrene base polymers. Next, a small block of acrylonitrile (∼2 to 5 repeat units) was chain extended. It was important to regulate the acrylonitrile block to only a few repeat units. Larger acrylonitrile blocks proved problematic for two reasons. First, isolation of the reduced material became extremely difficult due to the fabrication of an amphiphilic material, i.e. polystyrene-b-polyallylamine. Second, excess primary amine caused difficulty in completion of thiazolidine coupling reaction. Table 1 lists chain extension conditions attempted. The ability to control the length of the acrylonitrile block was achieved by employing high [AN], high [AIBN] and short reaction times, entry 8. Incorporation of acrylonitrile was observed by a clear shift in molecular weight as well as infrared (IR) analysis. The incorporation of such small blocks on all polymer chains is possible because the reactivity ratios of styrene and acrylonitrile are 0.29 and 0.020, respectively.19 This ensures that cross-propagation from a styrene unit to an acrylonitrile unit will be efficient and occur faster than acrylonitrile homopolymerization.
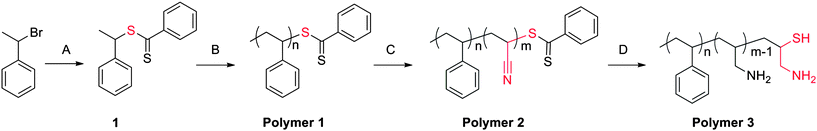 | ||
| Scheme 1 Synthetic route for preparation of cysteine-like chain-end functional polymers. Conditions: (A) CS2, phenyl magnesium chloride, THF, −78 °C to 80 °C, 20 h; (B) St, 110 °C, 24 h; (C) AN, AIBN, 60 °C, 15 min; (D) LAH. | ||
| Sample | M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :I:AIBN :I:AIBN |
Time/min | AN blocka | Coupling |
|---|---|---|---|---|
| a Determined by GPC analysis. | ||||
| 1 | 255:1:1.9 |
270 | Gel | Y |
| 2 | 255:1:1.9 |
15 | ∼26 units | Y |
| 3 | 210:1:1.7 |
15 | ∼9.4 units | N |
| 4 | 218:1:1.75 |
15 | ∼5.4 units | Y |
| 5 | 218:1:1.8 |
19 | ∼19.6 units | Y |
| 6 | 213:1:1.75 |
16 | ∼9.4 units | Y |
| 7 | 204:1:1.66 |
15 | ∼7.5 units | N |
| 8 | 175:1:1.4 |
15 | ∼2.6 units | N |
| 9 | 175:1:1.4 |
18 | ∼5 units | Y |
Next, several different reduction parameters were explored to expose the cysteine-like chain end functionality.
Preliminary efforts employed lithium aluminium hydride (LAH) to execute a reduction of the dithioester and the nitrile functionality simultaneously. Initial IR analysis of the reaction mixture showed a loss of the dithioester signal at 1728 cm−1 and a loss of the nitrile signal at 2210 cm−1. Fig. 2 displays gel permeation chromatography (GPC) analysis of the base polymers at different stages of the reaction. As previously described, Polymer 1 was prepared with pre-determined molecular weights and low polydispersity (PDI). Chain extension with acrylonitrile to prepare Polymer 2 proceeded with a clean shift in molecular weight without the presence of tailing or coupling.
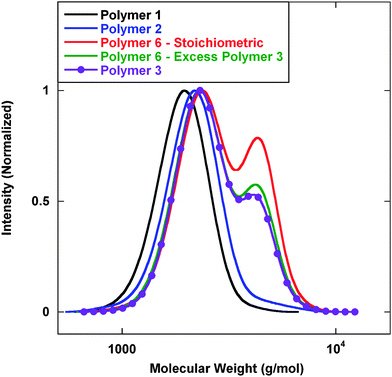 | ||
| Fig. 2 GPC analysis of polymers prepared viaScheme 1. Mn (g mol−1) (PDI): Polymer 1—1800 (1.09); Polymer 2—2000 (1.09); Polymer 6–stiochiometric—2700 (1.18); Polymer 6–excess Polymer 3—2500 (1.17); Polymer 3—2500 (1.16). | ||
It was then planned to add isophthalaldehyde to serve as a coupling reagent to prepare polymeric dimers; however, the preparation of polymeric dimers was met with limited success. GPC analysis of these reactions displayed increases in molecular weight indicating the formation of dimers. Unfortunately, there was significant residual starting material remaining regardless of the stoichiometric conditions utilized. It was initially assumed that LAH would reduce the dithioester first followed by reduction of the nitrile. Furthermore, disulfide formation would be inhibited under LAH reducing conditions, but analysis of Polymer 3 showed that a significant amount of polymer coupling had already occurred indicating the formation of disulfide bonds. It was also noted that there was a significant amount of unreactive polymer chains. This indicated that other side reactions were also occurring that were terminating polymer chains. Several potential side reactions could inhibit thiazolidine coupling chemistry. In particular, it is possible to cleave dithioesters with primary amines via aminolysis, and if the nitrile was reduced before the dithioester such a side reaction could occur between the newly formed allylamine repeat units and the RAFT chain end. Also any benzyl mercaptan that was generated from the cleaved chain end could form a disulfide bond and render a chain end inactive.
Several different reduction conditions were explored to eliminate this problem. First, it was proposed to selectively reduce the dithioester followed by reduction of the nitrile. Such a methodology would eliminate the possibility of the newly formed primary amine reducing the RAFT chain end. Second, disulfide formation is reversible, and it was proposed that the addition of β-mercaptoethanol or dithiothreitol (DTT) would cleave any formed disulfide bridges and inhibits the formation of additional disulfide bonds.
To selectively reduce the dithioester before the nitrile, NaBH4 was added followed by addition of LAH. This proved to be selective in targeting the dithioester before the nitrile as demonstrated by IR analysis and a change of sample color from pink to white; however, the addition of either β-mercaptoethanol or DTT after or during the reaction did not eliminate the formation of disulfide bonds.
To eliminate these issues, a new reduction scheme was developed (Scheme 2). Here, the dithioester was selectively reduced and subsequently protected by the formation of a methyl disulfide.20 IR analysis displayed a loss of the dithioester frequency while the nitrile signal was maintained. Furthermore, the sample displayed a distinct color change from pink to white also indicative of the removal of the dithioester. GPC analysis of this system demonstrated the inhibition of disulfide coupling, Fig. 3.
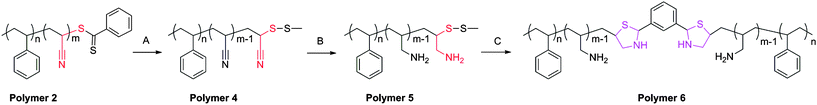 | ||
| Scheme 2 Modified synthetic route for preparation of cysteine-like chain-end functional polymers. Conditions: (A) propylamine, MTS, RT, 3 h; (B) LAH; (C) DTT, isophthalaldehyde. | ||
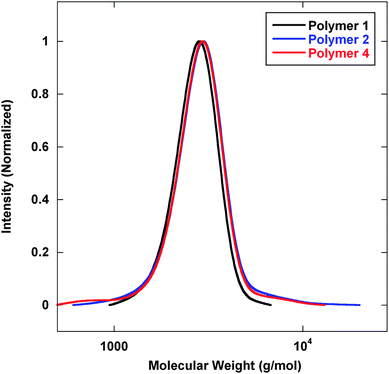 | ||
| Fig. 3 GPC analysis of polymers prepared viaScheme 2. Mn (g mol−1) (PDI): Polymer 1—2600 (1.07); Polymer 2—2700 (1.09); Polymer 4—2700 (1.09). | ||
Next, LAH was added to reduce the nitrile and IR analysis confirmed functional group transformation. To initiate polymer coupling, DTT was added to cleave the disulfide and expose the 1,2-amino-mercapto functional group followed by addition of isophthalaldehyde. Isophthalaldehyde was added under non-stiochiometric and stiochiometric conditions to demonstrate that coupling only occurs via the thiazolidine linkage and that each chain end was available for thiazolidine chemistry. Fig. 4 displays GPC analysis of the coupling experiment. In the presence of excess Polymer 5, it would be feasible for the mercapto functional groups to form disulfide linkages; however, the addition of DTT inhibited disulfide formation. Under stiochiometric conditions, the formation of polymeric dimers was essentially complete, as only very little residual starting material was observed by GPC. Furthermore, all observed coupling was completed via thiazolidine chemistry due to the presence of DTT.
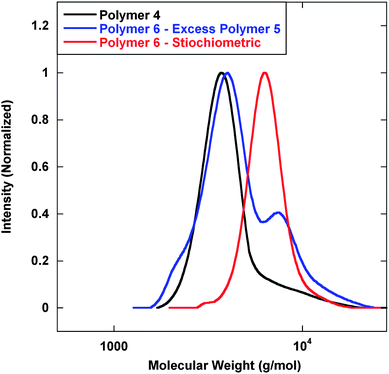 | ||
| Fig. 4 GPC analysis of polymeric dimers via thiazolidine coupling. Mn (g mol−1) (PDI): Polymer 4—3600 (1.09); Polymer 6–excess Polymer 5—4200 (1.14); Polymer 6–stiochiometric—6600 (1.06). | ||
In summary, base polymers were prepared and chain extended with acrylonitrile using RAFT polymerization techniques. Under reducing conditions, latent cysteine-like residues were exposed and in the presence of isophthalaldehyde efficient polymer coupling was observed.
Experimental
Methods and materials
All materials were purchased from commercially available sources. Styrene and acrylonitrile were dried over CaH2 and distilled to remove inhibitor. 1H NMR spectra were recorded on a 300 MHz varian instrument in CDCl3. Chemical shifts, δ (ppm), were referenced to the residual solvent signal. IR samples were prepared upon a ZnSe plate and analysis was conducted on an IR Galaxy Series FTIR 3000. GPC analysis was conducted in THF at 25 °C with flow rate of 1.00 mL min−1. Three Polymer Standards Service columns (100 Å, 1000 Å and linear) were connected in series to a Thermo Separation Products P-100 isocratic pump, autosampler, column oven, and Knauer refractive index detector. Samples with amino and mercapto functional groups were treated with an excess amount of acetic anhydride to cap active functional groups. Samples were calibrated against linear polystyrene.Synthesis of ethyl S-thiobenzoyl-2-thiobenzene (1)
A stir bar and 1-bromoethylbenzene (2.21 mL, 16.21 mmol) were loaded into a 50 mL Schlenk flask which was vacuum/backfilled three times with N2. In a 50 mL round bottom flask, a stir bar was loaded and then vacuum/backfilled with N2 (3×). Phenyl magnesium chloride (8.51 mL, 17.02 mmol) and THF (8 mL) were then added. The 50 mL round bottom flask was cooled to −78 °C, and while stirring CS2 (1.08 mL, 17.83 mmol) was added. The reaction stirred for 10 min at −78 °C and then for 45 min at room temperature. The contents of the round bottom flask were then transferred to the 50 mL Schlenk flask via cannula, and the reaction stirred for 20 h at 80 °C. The reaction mixture was partitioned after the addition of H2O (75 mL). The aqueous layer was then extracted with EtOAc (2 × 50 mL), and the organic layers were combined and dried over anhydrous MgSO4. Solvent was removed via rotary evaporation to yield 1 (3.25 g, 77%) as a red liquid. 1H NMR: δ (ppm) 7.94 (2H, d), 7.55–7.27 (10H, m, overlap of aromatic rings), 5.28 (1H, q), 1.79 (3H, d).Typical RAFT synthesis of polystyrene (Polymer 1)
Compound 1 (1.45 g, 5.61 mmol), styrene (32.1 mL, 281 mmol) and a stir bar were added to a 200 mL round bottom flask. The reaction mixture was purged with N2 for 30 min, and then heated at 110 °C for 24 h. The reaction was cooled to RT, and the polymer was isolated by precipitation into CH3OH to isolate Polymer 1 (16.5 g, 57%) as a pink powder. GPC: Mn—3300 g mol−1 (PDI—1.06).Typical RAFT synthesis of polystyrene-b-polyacrylonitrile (Polymer 2)
Polymer 1 (6.65 g, 2.02 mmol), azobisisobutyronitrile (AIBN) (463 mg, 2.82 mmol), acrylonitrile (22.5 mL, 343 mmol), and a stir bar were added to a 200 mL round bottom flask. The reaction mixture was purged with N2 for 30 min, and then heated at 60 °C for 15 min. The reaction was cooled to RT, and the polymer was isolated by precipitation into CH3OH to isolate Polymer 2 (5.81 g, 87%) as a pink powder. GPC: Mn—3800 g mol−1 (PDI—1.13). IR—2240 cm−1 (CN stretch).Typical preparation of polystyrene-b-polyacrylonitrile-S-SCH3 (Polymer 4)
Polymer 2 (5.06 g, 1.33 mmol), THF (15 mL), and a stir bar were added to a 50 mL round bottom flask. Next, methylmethanethiosulfonate (MTS) (2.51 mL, 26.6 mmol) and propylamine (1.10 mL, 13.3 mmol) were added to the reaction mixture and stirred at RT for 60 min. The polymer was isolated by precipitation into CH3OH to isolate Polymer 4 (4.82 g, 95%) as a white powder. GPC: Mn—3600 g mol−1 (PDI—1.10). IR—2240 cm−1 (CN stretch).Typical preparation of polystyrene-b-polyallylamine-S-SCH3 (Polymer 5)
Polymer 4 (1.05 g, 0.029 mmol), THF (10 mL), and a stir bar were added to a 50 mL round bottom flask, which was cooled to 0 °C. Next, lithium aluminium hydride (LAH) (110 mg, 2.9 mmol) was slowly added and the reaction was stirred at RT for 24 h. Excess LAH was quenched with ice. The polymer was isolated by precipitation into ice/CH3OH (50:50) to isolate Polymer 5 (857 mg, 82%) as a white powder. GPC: Mn—3500 g mol−1 (PDI—1.07). IR—no signal observed at 2240 cm−1.
Typical preparation of polymeric dimers (Polymer 6)
Polymer 5 (0.79 g, 0.22 mmol) and a stir bar were loaded into a 50 mL round bottom flask which was vacuum/backfilled three times with N2. Next, a purged solution of DTT in THF/MeOH (5:1) (18 mL, 0.1 M) was added and the reaction mixture was allowed to stir at RT for 24 h. Next isophthalaldehyde (14.7 mg, 0.11 mmol) was added and the reaction mixture was allowed to stir at RT for 24 h. The polymer was isolated by precipitation into ice/CH3OH (50:50) to isolate Polymer 6 (690 mg, 86%) as a white powder. GPC: Mn—6200 g mol−1 (PDI—1.06).
Acknowledgements
This research was supported by an award from the Research Corporation for Science Advancement. Additional funding and support provided by California Polytechnic State University via start-up funds.Notes and references
- B. Sumerlin and A. P. Vogt, Macromolecules, 2010, 43, 1–13 CrossRef CAS.
- W. H. Binder and C. Kluger, Curr. Org. Chem., 2006, 10, 1719–1815.
- C. J. Hawker, V. V. Fokin, M. G. Finn and K. B. Sharpless, Aust. J. Chem., 2007, 60, 381–383 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2004, 40, 2004–2021.
- C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- J. A. Prescher and C. R. Bertozzi, Nat. Chem. Biol., 2005, 1, 13–21 CrossRef CAS.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS.
- J. A. Codelli, J. M. Baskin, N. J. Agard and C. R. Bertozzi, J. Am. Chem. Soc., 2008, 130, 11486–11493 CrossRef CAS.
- K. L. Killops, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 5062–5064 CrossRef CAS.
- L. M. Campos, K. L. Killops, R. Saka, J. M. J. Paulusse, D. Damiron, E. Drockenmuller, B. W. Messmore and C. J. Hawker, Macromolecules, 2008, 41, 7063–7070 CrossRef CAS.
- T. Z. Cygan, J. T. Cabral, K. L. Beers and E. J. Amis, Langmuir, 2005, 21, 3629–3634 CrossRef CAS.
- T. Wu, Y. Mei, J. T. Cabral, C. Xu and K. L. Beers, J. Am. Chem. Soc., 2004, 126, 9880–9881 CrossRef CAS.
- A. E. Rydholm, C. N. Bowman and K. S. Anseth, Biomaterials, 2005, 26, 4495–4506 CrossRef CAS.
- E. C. Hagber, M. Malkoch, Y. Ling, C. J. Hawker and K. R. Carter, Nano Lett., 2007, 7, 233–237 CrossRef CAS.
- M. Wathier, C. S. Johnson, T. Kim and M. W. Grinstaff, Bioconjugate Chem., 2006, 17, 873–876 CrossRef CAS.
- M. Wathier, P. J. Jung, M. A. Carnahan, T. Kim and M. W. Grinstaff, J. Am. Chem. Soc., 2004, 126, 12744–12745 CrossRef CAS.
- C. Saiz, P. Wipf, E. Manta and G. Mahler, Org. Lett., 2009, 11, 3170–3173 CrossRef CAS.
- G. Odian, Principles of Polymerization, John Wiley and Sons, Hoboken, NJ, 2004 Search PubMed.
- P. J. Roth, D. Kessler, R. Zentel and P. Theato, Macromolecules, 2008, 41, 8316–8319 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |