Self-healing and self-mendable polymers
Jay A.
Syrett
,
C. Remzi
Becer
and
David M.
Haddleton
*
Department of Chemistry, The University of Warwick, Coventry, CV4 7AL, UK. E-mail: d.m.haddleton@warwick.ac.uk; Tel: +44 (0)2476523256
First published on 13th May 2010
Abstract
Smart materials with the ability to repair themselves have been the focus of different fields of science and engineering. This mini-review provides an insight into the rapidly expanding area of research into smart materials with self-healing properties and also discusses both chemical (reversible and polymeric) and non-chemical (irreversible and microvascular) systems, with emphasis focused on the recent reports in the field.
 Jay A. Syrett | Jay A. Syrett completed his MChem studies at the University of Warwick in 2007. During this time he conducted his final year research project in the area of Single Electron Transfer Living Radical Polymerisation, under the supervision of Professor David M. Haddleton. Remaining within the Haddleton research group, Jay began his PhD studies in October 2007. He is sponsored by Lubrizol Ltd, and his research is focused on developing mendable polymers prepared by Living Polymerisation techniques. |
 C. Remzi Becer | C. Remzi Becer was born in 1980 in Izmir, Turkey. He received his BSc degree in 2003 at the Chemistry Department of the Istanbul Technical University (ITU). In 2005, he received his MSc degree in Polymer Science and Technology at the ITU. He completed his PhD study titled as “Controlling Polymer Architectures” in 2009 under the supervision of Ulrich S. Schubert at the Eindhoven University of Technology (The Netherlands) and the Friedrich-Schiller-University Jena (Germany). Since late 2009, he is a Marie Curie Research Fellow in the University of Warwick, UK. His research interests include controlled living polymerization techniques, click reactions and glycopolymers. |
 David M. Haddleton | David Haddleton has been at the University of Warwick since 1993 after moving from ICI/Zeneca. Currently he is Head of the Inorganic and Materials Section of the Department of Chemistry in the University of Warwick. His research group work in the general area of living and controlled radical polymerization. He is currently Editor in Chief of Polymer Chemistry. |
1 Introduction
One of the nature's most striking features is its ability to self-repair minor damage that is inflicted in one way or another, such as cuts or bruises which are often healed with full restoration. Engineering materials, including polymers, often have a limited lifespan due to unavoidable degradation and unexpected damage from constant stress and strain. In order to overcome these issues, research in the field of self-healing polymers, defined as a “material where damage automates a healing response,” is currently an active field of study.1 This research is driven by the possibility that future materials may not have to be replaced, which would result in cost and efficiency savings in many applications. Work in this area is diverse, ranging from automotive industry to airplane wings; computer circuit boards and lubricants which could all take advantage of this technology to improve performance.It is a challenge for polymer and materials chemists to develop smart materials which can undergo different responses upon an external stimulation. Smart materials have been prepared by combining state-of-the-art engineering techniques with efficient chemical reactions, often called click chemistry.2–4 Several concepts have been developed to provide the self-healing property to the material, such as microcapsule based systems, microvascular systems and nanoreservoirs, Fig. 1.39 Composite systems often require the contact of the crack to the nano/microdomains that contain a crosslinker, catalyst or monomer and initiates a polymerization that blocks or heals the crack.5 However, the irreversible systems can heal only once, which is a potential drawback. Therefore, there is an increasing interest on reversible systems which can break and heal repeatedly upon external stimuli.6
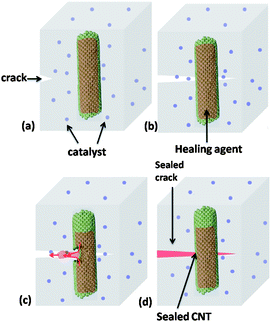 | ||
| Fig. 1 The self-healing concept based on carbon nanotube reservoirs. (a) Material is damaged at the site of impact. (b) Healing agent is released into the matrix. (c) An in situ polymerisation occurs where the crack was formed. (d) Fully restored crack, with full mechanical properties restored. Reproduced with permission from ref. 39. | ||
Reversible systems have been based on either covalent interactions, such as Diels–Alder (DA) and retro Diels–Alder (RDA) processes, or non-covalent interactions such as hydrogen bonding or π–π stacking. The concept of the rational design of stimuli responsive materials is based on the interaction of chemical functionalities with various forms of energy such as thermal, electrical, pressure, mechanical and electro-chemical radiation.7–10
In this current review, we focus on systems that should repair the damage at the microscopic level, as this is where damage often goes unnoticed and enables the material to restore its full mechanical properties, as opposed to macroscopic repair with adhesives. We highlight the selected examples of the irreversible systems in the first part, and the second part focuses on the reversible chemical reaction mechanisms.
2 Irreversible systems
It is appropriate to begin with irreversible systems, as these were the first developed systems for self-healing purposes. These require healing agents to repair damage in the material. Once healed, they cannot be reverted back to the appropriate monomers.2.1 Self-healing composites
Whilst work by White et al. was not the very first example of a self-healing polymer (SHP), it was ground breaking11 and stimulated further work in this area. White et al. focused on self-healing polymer composites, where a monomer healing agent is stored in microcapsules and dispersed through an epoxy matrix. Upon damage, the microcapsules are punctured, releasing dicyclopentadiene (DCP) monomer into the matrix. On contact with the first generation Grubbs catalyst ring opening metathesis polymerisation (ROMP) of the DCP heals the crack thus restoring the mechanical properties of the material, Fig. 2.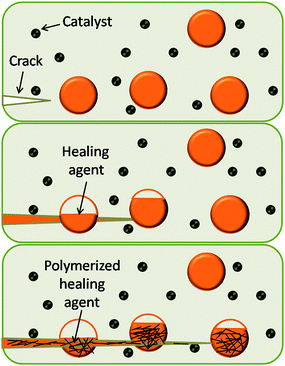 | ||
| Fig. 2 A crack forms in the matrix where the damage occurs (top), the crack ruptures the microcapsule, releasing the healing agent (middle), the healing agent contacts the catalyst, and the ROMP proceeds (bottom). Reproduced with permission from ref. 11. | ||
The matrix used to store the monomer microcapsules consisted of bisphenol based epoxide to diethylenetriamine (12 : 100) curing agent. The Grubbs catalyst used was a bis(tricyclohexylphosphine) benzylidine ruthenium(IV) dichloride, and this was embedded in wax microspheres that are solubilised by the DCP. The DCP was encapsulated in poly(urea-formaldehyde) microcapsules. A range of healing efficiencies is reported, peaking at 67% recovery of the virgin fracture.12 When the temperature of the system was increased to 80 °C the average healing efficiencies rose above 70% and peaking at 80%.12 Microencapsulation has been shown to exhibit better healing properties than silica microspheres and solid urea formaldehyde (UF) particles,13 and it has also been suggested that melamine–urea–formaldehyde (MUF) polymer shells14 show a much greater thermal stability, robustness and improved synthetic and handling properties over UF.13,15
Specific effects of three crystal morphologies of the Grubbs catalyst to tailor the dissolution kinetics16 and thermal properties of the self-healed polymer have been investigated.17 It was shown that the more rapidly dissolving polymorph gives increased healing efficiency.17 Moore et al. reported in 2007 that both the exo and endo isomers of DCP have relative advantages.18 The endo isomer is commercially available, has a long shelf life, low viscosity, and exhibits polymers with good mechanical properties. In contrast, the exo isomer polymerises 150 times faster and has superior low temperature properties.19 The exo isomer polymerises so fast that it gels blocking the release of further catalyst and a mixture of the two isomers was shown to be the best monomer composition, Fig. 3.18
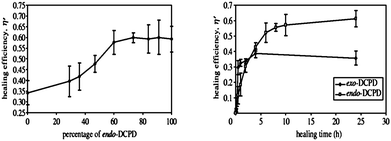 | ||
| Fig. 3 Whilst the exo isomer polymerises much faster, the healing efficiency was 50% than with pure endo isomer (left). A mix of the two isomers can improve healing efficiency (right). Reproduced with permission from ref. 18. | ||
Other work has shown that using a different monomer, a blend of DCP with 5-ethylidene-2-norbornene in a ratio of 1 : 3 can improve crack repair even further,20 with faster reaction times at a lower catalyst loading. However, this was limited to small cracks and showed an inability to repair multiple cracks.21 First generation Grubbs catalysts can be more efficient and catalyse ROMP faster than 2nd generation catalysts in this type of resins and curing agents.22 A computational model of White et al.'s original system has been produced,11 showing that a rest period during healing retards the crack propagation. White and co-workers continue to develop the methodology and have improved it further by adding “Shape Memory Alloy” (SMA) wires to the composite.23,24 The SMA wires are activated so that they close upon self-healing, thus making the crack smaller and greatly improving healing efficiency. The scope of the system was varied further using a tin catalysed polycondensation of phase separated droplets containing hydroxyl end functionalised polydimethylsiloxane (PDMS), Fig. 4.25 This type of self-healing polymer can withstand harsher conditions and the phase separated tin catalysed approach is far more air and water sensitive in comparison to the conventional microcapsule system. The tin catalyst can also withstand much higher temperatures of greater than 100 °C, but healing efficiencies are sacrificed. A similar reasoning was used by Yang and co-workers, who reported a catalyst-free self-healing system encapsulating isophorone diisocyanate monomer for use in coatings.26 The microcapsules showed good resistance against water, air and high temperature and these are now being used in new self-healing technologies. Further cost savings can be made by using a tungsten(VI) catalyst, for the ROMP of exo-DCPD.27 Whilst it is a more environmental friendly catalyst, healing efficiency in this initial study only reached up to 20%, and is so far limited to one isomer of DCPD.
 | ||
| Fig. 4 Schematic of the self-healing process: (a) self-healing composite consisting of microencapsulated catalyst (yellow) and phase-separated healing-agent droplets (white) dispersed in a matrix (green); (b) crack propagating into the matrix releasing catalyst and healing agent into the crack plane; (c) a crack healed by polymerized PDMS (crack width exaggerated). SEM images of (d) the fracture surface, showing an empty microcapsule and voids left by the phase-separated healing agent, and (e) a representative microcapsule showing its smooth, uniform surface. Reproduced with permission from ref. 25. | ||
2.2 Microvascular polymer composites
In a similar way to the aforementioned composite systems based on microspheres, fibre reinforced composites with intraply matrices are prone to cracks and damage, despite fibres imparting extra mechanical strength to the material. Cracks, especially in industries where these are used e.g. aerospace, are often difficult to detect and observe. Traditional cosmetic repair approaches are costly and difficult and thus these systems have several advantages over microcapsulated healing agents. For example, it is not possible to replenish new materials to the damaged site once the healing agent in the capsule has been used. However, microvascular systems have been designed to mimic the vascular system in the body. They do not provide a one time solution for the damaged materials, instead providing a continuous flow of healing agent to the site. This unique property makes them good candidates for future smart materials. | ||
| Fig. 5 Schematic diagram of smart repair concepts considered for polymer matrix composites. Reproduced with permission from ref. 34. | ||
The ideal scenario is a resin that can be detected by absorption or emission spectroscopy such as UV, allowing the sight of damage to be easily detected as well as the self-healing properties.28,29 The earliest publications by Pang and Bond showed that HGF could be produced that are UV active, with the fibres filled with 913 epoxy resin and a fluorescent dye. The self-healing mechanism was unclear; however, it was reliant upon the uncured resin combining with the hardener post-impact. Two other systems showed early signs of promise, exhibiting up to 97%28 and 93%29 restoration of original mechanical strength. The inclusion of a UV dye enabled the detection the location and the extent of the damage, Fig. 6.
 | ||
| Fig. 6 Optical micrograph of cross-section through impact damaged hybrid solid glass/hollow glass/epoxy laminate. Reproduced with permission from ref. 28. | ||
It was shown that adding these fibres to a composite can reduce the initial strength of the material, however, the increased damage tolerance and subsequent repair mechanisms compensate for this.32 The applications of these types of self-repair technologies are focused on transport,30 and work is now concentrating on making HGFs commercially viable by incorporation of resins with low viscosity, long shelf lives and no reliability on stoichometry.31
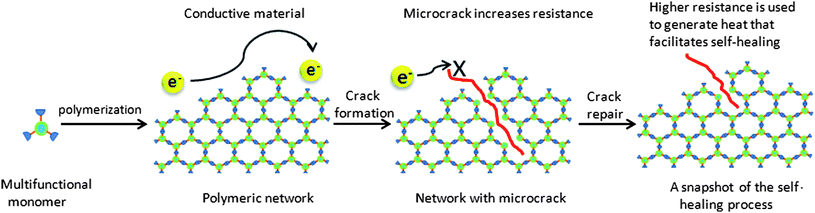 | ||
| Fig. 7 Concept of an electrically conductive, self-healing material. Electrical resistance increases upon formation of a microcrack, as the total number of electron percolation pathway decrease. As the microcrack is the source of the resistance increases, the generation of heat as localised at the fracture point. Reproduced with permission from ref. 35. | ||
A series of multitopis N-heterocyclic carbenes (NHCs) were synthesised as good electronic conductors. As a proof of principle, the polymerised NHCs were scored with a razor blade, and a noticeable smoothing was noticed after heating the polymer to 200 °C for 25 minutes. Other work simulated and quantified the extent of rehealing, and directed the heat source to the exact location where healing was required.36 However, limiting the study to NHCs and the need for solvent be present for the healing to occur has meant that no further publications on this topic have emerged. Indeed, resistance heating36 and the fact that the principle could be applied to carbon fibres have led to further research in the field.37,38 The polymers themselves were not electrically conductive, instead based on Diels–Alder type linkages that will be described in more detail in the next section.
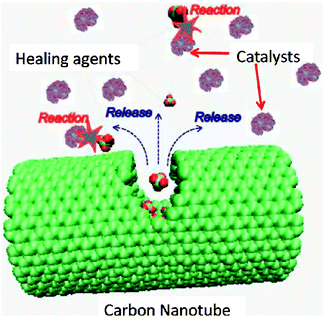 | ||
| Fig. 8 Molecular model of carbon nanotube and organic molecules for the self-healing system. Reproduced with permission from ref. 39. | ||
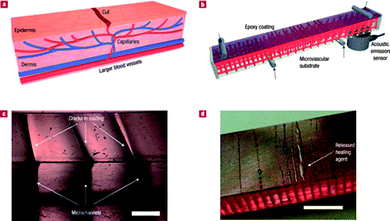 | ||
| Fig. 9 (a) Schematic diagram of a capillary network in the dermis layer of skin with a cut in the epidermis layer. (b) Schematic diagram of the self-healing structure composed of a microvascular substrate and a brittle epoxy coating containing embedded catalyst in a four-point bending configuration monitored with an acoustic-emission sensor. (c) High-magnification cross-sectional image of the coating showing that cracks, which initiate at the surface, propagate towards the microchannel openings at the interface (scale bar = 0.5 mm). (d) Optical image of self-healing structure after cracks are formed in the coating (with 2.5 wt% catalyst), revealing the presence of excess healing fluid on the coating surface (scale bar = 5 mm). Reproduced with permission from ref. 42. | ||
3 Reversible self-healing smart materials
Irreversible smart materials were the first type of self-healing polymers reported, however, their applications are limited, and involve relatively complex systems. Reversible systems are synthetically more applicable, and are able to undergo multiple repair cycles even upon damage at the same site. These do require an external stimulus, and as a consequence do not undergo repair autonomously. The reversible systems are reported in covalent and non-covalent sections.3.1 Thermo-responsive Diels–Alder based polymeric smart materials
Diels–Alder (DA) cycloadditions are a convenient route for the formation of carbon–carbon bonds via a facile reaction under undemanding conditions, a “click” reaction.43–45 Diels–Alder cycloadditions are thermoreversible reactions, which is ideal for the synthesis of mendable polymers, as it does not require delivery of an additional chemical, such as a catalyst. This feature has been exploited in the preparation of self-healing polymers carrying functionalities, either as the polymer chain-end (i.e. pendant ends) or in the monomeric repeating units. Furthermore, the reverse DA reaction does not involve free radicals, thus limiting side reactions that might prevent reformation.46 There are a number of examples where Diels–Alder (DA) moieties, a diene and dieneophile have been incorporated into polymers, perhaps the most common being the furan–maleimide based DA system.47 Other work in the 1990s concentrated on using bis-furans and bis-maleimides to prepare crosslinked thermally reversible polymeric networks, however, these systems had low degrees of polymerisation and poor reversible properties.48–53 The incorporation of furan–maleimide moieties into polymers by Stevens and Jenkins,54 and independently by Craven was presented prior to the technology being used for self-healing. Since this time there has been a series of publications using a similar concept,55–58 and have eventually developed into practical chemistry of mendable systems using DA technology.59,60 Despite the initial principle being published in 1979, it was not recognized until 1990 that Saegusa and co-workers developed a thermally “mendable” polymeric DA network.55Initial work56 was not targeted for the specific use of synthesising self-healing polymers, instead the formation of polyoxazoline hydrogels.56 These were prepared by formulating furan and maleimide pendant end groups on the polymeric chains and then an intermolecular DA reaction between the furan-modified poly(N-acetylethylenimine) (PAEI) and maleimide-modified PAEI proceeded,55Fig. 10.
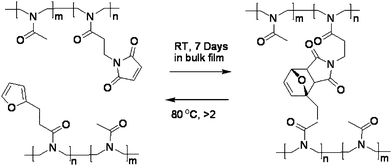
Stevens also varied the system by crosslinking polystyrene with maleimide based end groups with di-2-furfuryl adipate.61 This posed problems when attempting the reverse DA reaction at 150 °C as the furfuryl adduct is thermally unstable at this temperature.61 Gandini and co-workers discovered the same problem investigating the DA/RDA process using N,N′-methylenediphenylbismaleimide (MDPBM) with copolymers of methyl methacrylate and 2-furfuryl acrylate.62 Changing the diene to pendant 2-furfuryloxy moieties with a MDPBM cross-linking agent, and using 2-methylfuran as a trap to capture released MDPBM, enabled the reverse DA to proceed at 130 °C with no side reactions.58 Gandini and co-workers followed up these initial studies by making thermally reversible elastomers using maleimide pendant PDMS, crosslinked with a difuran derivative.
A DA crosslinked polymer synthesized for the specific use of self-healing was prepared by Wudl et al. in 2003, using furan- and maleimide-based monomers crosslinking along the polymer backbone.63–65 It was shown that fractured polymer materials, heated to 120 °C, exhibited 83% recovery of the polymers original strength. Crucially, this fracture/repair cycle could also be repeated, making this the first real DA polymeric system exhibiting thermally responsive self-healing behaviour. This and other work from various authors,66–70 for example self-healing dendrimers,69 polyamides,70 and film lithograpthy,66 have paved the way for a number of recent articles with specific applications. Zhang et al. utilized Diels–Alder technology in polyketones and bis-maleimides,59 the first example of Diels–Alder chemistry being used for a specific purpose: recycling thermoset polymers, Fig. 11. The systems employed exhibited 100% healing efficiencies with multiple cycles, Fig. 12.59 Similar work used DA chemistry to synthesise self-healing epoxy-amine thermosets, but whilst the cycles could be repeated on multiple occasions, unlike the earlier work, healing efficiencies only reached 21%.60
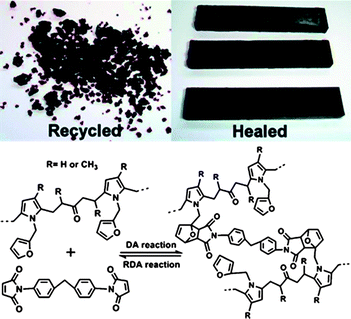 | ||
| Fig. 11 Schematic of the Diels–Alder (DA) and retro Diels–Alder (RDA) reactions between PK-furan and bis-maleimide. Reproduced with permission from ref. 59. | ||
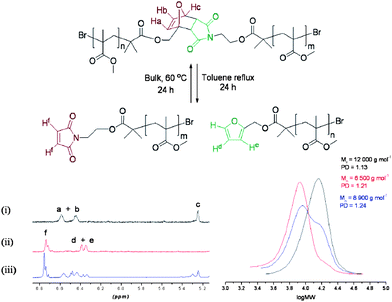 | ||
| Fig. 12 1H NMR of polymer (i) prior to heating, (ii) cleaved polymer following heating and (iii) reformed polymer. GPC data of (i) polymer (black), (ii) cleaved polymer (red) and (iii) the reformed polymer (blue). Reproduced with permission from ref. 78. | ||
Although the furan–maleimide DA reaction is popular other chemical reactions have also been exploited. Dicyclopentadiene (DCPD) based on DA polymeric networks have been under investigation for many decades. The advantage of this monomer is that it acts as both a diene and a dieneophile,6 and it also has varying properties depending on its stereochemistry, as previously mentioned. The sequence of events that has evolved since its first use as a monomer in a DA based network, by Stille and Plummer, has been relatively brief.71 Takeshita et al.72 incorporated DCPD monomers in a reversible crosslinked network whilst Kennedy has been the first to demonstrate DCPD in a thermally reversible crosslinked network. The retro Diels–Alder reaction on the CP terminated polymers occurred at 215 °C, and reformed its original shape after heating at 170 °C over three cycles.73,74 However, these reports were not specifically concerning SHPs and are thus outside the scope of this article with no further reports in the literature. Wudl and Bergman have reported DCPD polymers and their specific use of self-healing.6 The thermal stability of other DA linkers is much stronger than those previously mentioned, and as such the other DA based systems are rarely reported in the literature. Anthracene based crosslinked networks have been reported,75–77 but until 2009, none demonstrated self-healing capabilities.78
It is clear that DA chemistry is popular for self-healing systems and the number of reports in this area has grown very rapidly.79 To date the majority of reports have been focused on crosslinked networks formulating polymer gels and thermoset matrices. Recently, we have developed self-healing polymers that can be prepared by living radical polymerisation,78 so that the scope of industries and applications is broadened. It is shown in this report that it is possible to prepare SHPs that bring all the benefits of living polymerisation, monodisperse polymers with tailored architecture and molecular weight, Fig. 12.
In order to investigate the self-healing property, two polymerisation initiators and one crosslinker were prepared using DA chemistry. The linker shown exhibited excellent cleavage properties, with 50% reformation occurring upon the reheating cycle. However, the facile nature in which this linker cleaved can be problematic for practical applications. Changing the alcohol to 9-anthracenemethanol gave the Diels–Alder linkage higher thermal stability. The forward DA reaction is highly efficient when using 9-anthracenemethanol, and the small amount of cleavage occurring is reforming in the cooling cycle. To investigate other polymer architectures, the same principle was applied to arm first stars, however, this time it was the shear quantity of linkages that enabled the Diels–Alder bond to constantly cleave and reform throughout the heating process.
An alternative self-healing system based on living polymerisation techniques is microencapsulated glycidyl methacrylate (GMA) monomer healing agent that is contained within a poly(methyl methacrylate) (PMMA) matrix.80 The living nature of the PMMA means it can be reinitiated upon cracking of the matrix, releasing the GMA monomer forming a block copolymer which is covalently attached to the interface. In effect, the GMA acts as an initiator, thus no external catalyst is required. The polarity of the GMA is similar to that of PMMA, facilitating wetting on the surface of the matrix initiating the copolymerisation. Full healing of the test system was achieved after 21 hours at ambient temperature.80
3.2 Dynamic covalent transitions
Deng et al.81 reported a new type of self-healing “dynamic” covalent system, using polyacylhydrazones. These polymers, so-called “sol–gels,” are able to revert back their monomeric state, by changes in either temperature, pH, or solvent, Fig. 13. The authors carried out a condensation of acylhydrazines at either end of a poly(ethylene oxide) (PEO) (A2) with aldehyde groups in tris[(4-formylphenoxy)methyl]ethane (B3) to form a crosslinked network of acylhydrazone bonds. These undergo cleavage under mild acidic conditions and reformation upon increasing the pH. The dynamic character of these polymers was used to reshape the polymer gel back to its original state following degradation.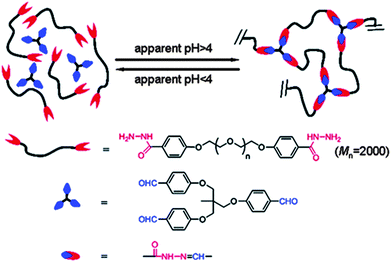 | ||
| Fig. 13 Construction of covalent cross-linked polymer gel based on reversible covalent acylhydrazone bond. Reproduced with permission from ref. 81. | ||
3.3 Hydrogen bonding
Covalent bonds utilize atoms as building blocks to form molecules and contain relatively high energy in comparison to non-covalent bonds, 35–135 kcal mol−1, 2–20 kcal mol−1, respectively. Therefore, a large amount of energy (heat or light) is required to break and reform covalent bonds, which is apparently a major drawback for other self-healing mechanisms, such as Diels–Alder. In contrast, non-covalent bonds, namely ionic, hydrophobic, metal coordination and hydrogen bonds, use molecules and ions to form assemblies with low kinetic stability.82 Therefore, low energy required reversible self-healing materials have been designed successfully in the last decade.The concepts of supramolecular polymerization and hydrogen bonding in between the macromolecules have been investigated by Meijer and Sijbesma et al.82–88,89 More than a decade ago, the formation of exceptionally stable DDAA dimers of a simple ureidopyrimidinone (Upy) derivative was reported. Ureidopyrimidinones may exist as dimers of 4[1H]-pyrimidinone and pyrimidinol tautomeric forms by a stable dimerization of the linear array of four hydrogen bonds. Dimerization via both DDAA (donor–donor–acceptor–acceptor) and DADA (donor–acceptor–donor–acceptor) array is possible, Fig. 14.90,91 These units can be incorporated into a polymer chain by various techniques, such as using a functional initiator or post-polymerization modifications.92,93 Hawker and Kramer et al. used the controlled incorporation of Upy units along the backbone to independently tune the macroscopic material properties such as stiffness and resistance to creep.
![Different tautomeric forms of 6[1H]-pyrimidinone monomer.](/image/article/2010/PY/c0py00104j/c0py00104j-f14.gif) | ||
| Fig. 14 Different tautomeric forms of 6[1H]-pyrimidinone monomer. | ||
A further approach has been demonstrated by Leibler et al. using the hydrogen bonding to construct self-healing elastomers without applying heat or catalyst.94–96 They designed a system which crosslinks via hydrogen bonds, Fig. 15. When the polymer is broken or cut, it can be simply repaired by bringing together the fractured ends for as little as few minutes to self-heal autonomously at ambient temperature.
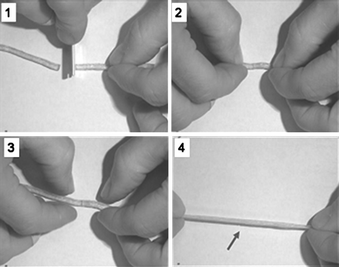 | ||
| Fig. 15 Self-healing polymers by gently pressing them towards each other for few minutes at room temperature. Reproduced with permission from ref. 93. | ||
This process is reproducible and the healed materials showed reasonably conserved mechanical properties after several repetitions. However, it is crucial to bring the ends of the material together as quick as possible to obtain sufficient self-healing as the hydrogen bonding units may react with the closest ones in their section. It is also noteworthy that this system is based on dimers and trimers of fatty acids, from bio-renewable resources, and their mixture prevents crystallization and warrant the rubbery property at high temperatures, Fig. 16.
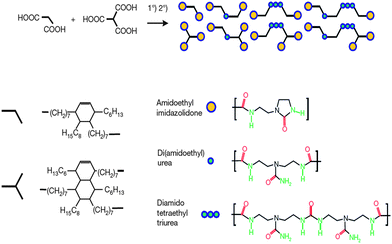 | ||
| Fig. 16 Schematic representation and molecular structures of materials used in Leibner's self-healing rubber. Reproduced with permission from ref. 93. | ||
3.4 Self-healing ionomers
Ionomers are thermoplastic ionic polymers, consisting of hydrocarbon chains bearing carboxylic acid groups that are either partially or completely neutralized with metal or quaternary, ammonium ions.97 It is usual for up to 20 mol% of ionic species to be incorporated into the structure of the organic polymer.1 Ionomers are advantageous since they can be synthesized using commercially available starting materials.1,6 These polymers are unique and their aggregates that have profound mechanical and physical properties, and are especially useful in ballistics1 and other commercial applications.98,99 These include sports equipment, coatings, ballistics and packaging.6,100–102 However, as with many such reversible interactions, there have been little publications specifically related to self-healing polymers.1,103The mechanism of healing is different from composite based systems, as there is no specific healing agent, the ionomer's morphology accommodates its self-healing capability, activated by high impact. It was reasonable to assume that the healing process occurred via thermal reversibility of the ionic interaction, and original hypothesis suggested this was the case.6,104 However, similar results and impact fractures were seen with Nurcel®,99 a product that is made via hydrogen bonding. The elastic nature of these polymers provides its self-healing process, Fig. 17.
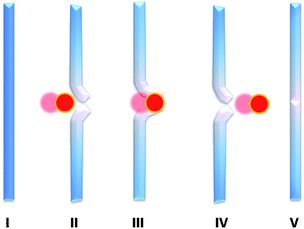 | ||
| Fig. 17 Friction causes melt. Elastically rebounds. Polymer still molten, able to reform at the damage site. Reproduced with permission from ref. 1. | ||
3.5 π–π Stacking
A further non-covalent interaction, π–π stacking, has been used in the synthesis of self-healing smart materials.105 A supramolecular polymer system of pyrenyl terminated polyamides (Mn = 6 kDa, Mw = 10 kDa) are intercalated into the chain folds of a polyimides (Mn = 16 kDa, Mw = 27 kDa). 1H NMR and viscometric studies used to show intercalation, against a variety of control experiments involving the single components. Films formation is a direct result of this interaction, with a measurable tensile strength. The viscosity melt fell 5 orders of magnitude after heating the film between 35 and 60, showing that the interaction, as expected, was thermoreversible much the same as hydrogen bonded and Diels–Alder systems. The cut film was reformed by pressing the pieces together and briefly heating to 80 °C; this exhibited full recovery of modulus strength so fast that the recovery mechanism could not be measured. Heating at 50 °C was required for the kinetics of the recovery to be monitored.105 The cycle was repeated on the same film 3 times, a key feature for industrially practical self-healing polymers, Fig. 18.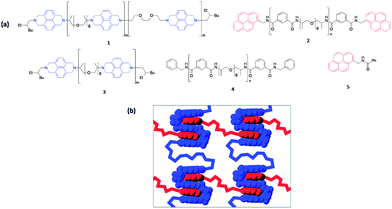 | ||
| Fig. 18 (a) Components 1 and 2 afford a π–π-stacked supramolecular polymer, while 3 and 4 are control materials lacking either, in 3, the chain-folding triethylenedioxy-diimide motif or, in 4, the terminal pyrenyl residues. (b) Schematic showing the potential for non-covalent cross-linking of polyimide chains by multiple intercalation of π-electron-rich pyrenyl end-groups (red) into designed polyimide chain-folds (blue). Reproduced with permission from ref. 105. | ||
4 Conclusions
The progress made on the fabrication of smart materials since the turn of the millennium has been remarkable. The original self-healing principle has expanded across the whole scientific world, from chemistry to engineering. The original systems developed in 2001 have been expanded upon and improved constantly. Similar types of composite based networks were reported, such as hollow glass fibres, and the field has gone from proof of principle to real life practical applications in the space of a decade.Chen and Wudl placed one of the most crucial milestones that really set the chemical and polymer based self-healing systems alight, as this showed chemists any reversible interaction could possibly be used to synthesise mendable polymers. Whilst Diels–Alder chemistry has been the most common, many other reversible networks have been developed, like hydrogen bonding and ionomers; commercial products have now been developed. We foresee that these types of networks will continue to be improved in the next decade.
However, there are a number of potential candidates for future work—in theory any reversible linker. The key factor for the successful self-healing polymers is to have a system that can reverse in nature, i.e. thermal and light.
The disulfide bond is a common and reversible interaction, with simple redox chemistry.106–108 The reversibility of the disulfide bond is evident in DNA replication. Matyjaszewski and Tsarevsky108,109 and Armes et al.110,111 have been developing disulfide containing polymers for work on biodegradable polymers. The first time they were used for crosslinked polymers was in 1993107 and since then Matyjaszewski108,109 has published papers showing the reversible nature of the disulfide bond in polymer networks. In reality, any system requires both reducing and oxidising agents to be present, seriously affecting the marketability of a disulfide based self-healing system. Nitroxide mediated polymerisation112 could be favoured, the formation of the stable free radicals is a thermal rather than chemically induced process.113,114 The radicals formed upon decomposition are stable and are readily reformed. Photocleavable polymers are also desirable, as the natural source of light could serve as the healing agent. Such photocleavable linkers have already been incorporated into polymers and shown their reversibility115,116 but not been used for specific use of SHP.
Acknowledgements
JAS is grateful to EPSRC and Lubrizol Ltd for their financial support. CRB is funded by European Union Marie Curie Fellowship scheme (proposal number 235999).Notes and references
- S. v. d. Zwaag, Self Healing Materials, Springer, Dordrecht, 2007 Search PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900–4908 CrossRef CAS.
- R. K. Iha, K. L. Wooley, A. M. Nyström, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS.
- D. Y. Wu, S. Meure and D. Solomon, Prog. Polym. Sci., 2008, 33, 479–522 CrossRef CAS.
- S. D. Bergman and F. Wudl, J. Mater. Chem., 2008, 18, 41–62 RSC.
- T. P. Russell, Science, 2002, 297, 964–967 CrossRef CAS.
- A. B. Descalzo, R. Martinez-Manez, R. Sancenon, K. Hoffmann and K. Rurack, Angew. Chem., Int. Ed., 2006, 45, 5924–5948 CrossRef CAS.
- C. R. Becer, S. Hahn, M. W. M. Fijten, H. M. L. Thijs, R. Hoogenboom and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7138–7147 CrossRef CAS.
- D. S. Bag and K. U. B. Rao, J. Polym. Mater., 2006, 23, 225–248 Search PubMed.
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. R. Sriram, E. N. Brown and S. Viswanathan, Nature, 2001, 409, 794–797 CrossRef CAS.
- M. R. Kessler, N. R. Sottos and S. R. White, Composites, Part A, 2003, 34, 743–753 CrossRef.
- E. N. Brown, S. R. White and N. R. Sottos, J. Mater. Sci., 2004, 39, 1703–1710 CrossRef CAS.
- X. Liu, X. Sheng, J. K. Lee and M. R. Kessler, Macromol. Mater. Eng., 2009, 294, 389–395 CrossRef CAS.
- R. G. Wang, H. Y. Li, H. L. Hu, X. D. He and W. B. Liu, J. Appl. Polym. Sci., 2009, 113, 1501–1506 CrossRef CAS.
- M. R. Kessler and S. R. White, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2373–2383 CrossRef CAS.
- A. S. Jones, J. D. Rule, J. S. Moore, S. R. White and N. R. Sottos, Chem. Mater., 2006, 18, 1312–1317 CrossRef CAS.
- T. C. Mauldin, J. D. Rule, N. R. Sottos, S. R. White and J. S. Moore, J. R. Soc. Interface, 2007, 4, 389–393 CrossRef CAS.
- J. D. Rule and J. S. Moore, Macromolecules, 2002, 35, 7878–7882 CrossRef CAS.
- X. Liu, J. K. Lee, S. H. Yoon and M. R. Kessler, J. Appl. Polym. Sci., 2006, 101, 1266–1272 CrossRef CAS.
- S. Maiti, C. Shankar, P. H. Geubelle and J. Kieffer, J. Eng. Mater. Technol., 2006, 128, 595–602 CrossRef CAS.
- X. Liu, X. Sheng, J. K. Lee, M. R. Kessler and J. S. Kim, Compos. Sci. Technol., 2009, 69, 2102–2107 CrossRef CAS.
- E. L. Kirkby, V. J. Michaud, J. A. E. Månson, N. R. Sottos and S. R. White, Polymer, 2009, 50, 5533–5538 CrossRef CAS.
- W. Xu and G. Li, Int. J. Solids Struct., 2010, 47, 1306–1316 CrossRef.
- S. H. Cho, H. M. Andersson, S. R. White, N. R. Sottos and P. V. Braun, Adv. Mater., 2006, 18, 997–1000 CrossRef CAS.
- J. Yang, M. W. Keller, J. S. Moore, S. R. White and N. R. Sottos, Macromolecules, 2008, 41, 9650–9655 CrossRef CAS.
- J. M. Kamphaus, J. D. Rule, J. S. Moore, N. R. Sottos and S. R. White, J. R. Soc. Interface, 2008, 5, 95–103 CrossRef CAS.
- J. W. C. Pang and I. P. Bond, Composites, Part A, 2005, 36, 183–188.
- J. W. C. Pang and I. P. Bond, Compos. Sci. Technol., 2005, 65, 1791–1799 CrossRef CAS.
- G. Williams, R. Trask and I. Bond, Composites, Part A, 2007, 38, 1525–1532 CrossRef.
- G. J. Williams, I. P. Bond and R. S. Trask, Composites, Part A, 2009, 40, 1399–1406 CrossRef.
- R. S. Trask, G. J. Williams and I. P. Bond, J. R. Soc. Interface, 2007, 4, 363–371 CrossRef CAS.
- M. J. Hucker, I. P. Bond, S. Haq, S. Bleay and A. Foreman, J. Mater. Sci., 2002, 37, 309–315 CrossRef CAS.
- S. M. Bleay, C. B. Loader, V. J. Hawyes, L. Humberstone and P. T. Curtis, Composites, Part A, 2001, 32, 1767–1776 CrossRef.
- K. A. Williams, A. J. Boydston and C. W. Bielawski, J. R. Soc. Interface, 2007, 4, 359–362 CrossRef CAS.
- N. Kwok and H. T. Hahn, J. Compos. Mater., 2007, 41, 1635–1654 CrossRef CAS.
- J. S. Park, H. S. Kim and H. Thomas Hahn, Compos. Sci. Technol., 2009, 69, 1082–1087 CrossRef CAS.
- J. S. Park, K. Takahashi, Z. H. Guo, Y. Wang, E. Bolanos, C. Hamann-Schaffner, E. Murphy, F. Wudl and H. T. Hahn, J. Compos. Mater., 2008, 42, 2869–2881 CrossRef CAS.
- G. Lanzara, Y. Yoon, H. Liu, S. Peng and W. I. Lee, Nanotechnology, 2009, 20, 335704 CrossRef CAS.
- K. S. Toohey, C. J. Hansen, J. A. Lewis, S. R. White and N. R. Sottos, Adv. Funct. Mater., 2009, 19, 1399–1405 CrossRef CAS.
- C. J. Hansen, W. Wu, K. S. Toohey, N. R. Sottos, S. R. White and J. A. Lewis, Adv. Mater., 2009, 21, 4143–4147 CrossRef CAS.
- K. S. Toohey, N. R. Sottos, J. A. Lewis, J. S. Moore and S. R. White, Nat. Mater., 2007, 6, 581–585 CrossRef CAS.
- C. K. Hartmuth, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- L. Nurmi, J. Lindqvist, R. Randev, J. Syrett and D. M. Haddleton, Chem. Commun., 2009, 2727–2729 RSC.
- V. Ladmiral, T. M. Legge, Y. Zhao and S. b. Perrier, Macromolecules, 2008, 41, 6728–6732 CrossRef CAS.
- F. R. Kersey, D. M. Loveless and S. L. Craig, J. R. Soc. Interface, 2007, 4, 373–380 CrossRef CAS.
- A. Gandini, Polímeros, 2005, 15, 95–101 CAS.
- G. C. Tesoro and V. R. Sastri, Ind. Eng. Chem. Prod. Res. Dev., 1986, 25, 444–448 CrossRef CAS.
- C. D. Diakoumakos and J. A. Mikroyannidis, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 2559–2567 CrossRef CAS.
- X. He, V. R. Sastri and G. C. Tesoro, Makromol. Chem. Rapid Commun., 1988, 9, 191–194 CrossRef CAS.
- C. D. Diakoumakos and J. A. Mikroyannidis, Eur. Polym. J., 1994, 30, 465–472 CrossRef CAS.
- N. Kuramoto, K. Hayashi and K. Nagai, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 2501–2504 CrossRef CAS.
- C. Gousse and A. Gandini, Polym. Int., 1999, 48, 723–731 CrossRef CAS.
- M. P. Stevens and A. D. Jenkins, J. Polym. Sci., Part A: Polym. Chem., 1979, 17, 3675–3685 Search PubMed.
- Y. Chujo, K. Sada and T. Saegusa, Macromolecules, 1990, 23, 2636–2641 CrossRef CAS.
- Y. Imai, H. Itoh, K. Naka and Y. Chujo, Macromolecules, 2000, 33, 4343–4346 CrossRef CAS.
- R. Gheneim, C. Perez-Berumen and A. Gandini, Macromolecules, 2002, 35, 7246–7253 CrossRef CAS.
- C. Gousse, A. Gandini and P. Hodge, Macromolecules, 1998, 31, 314–321 CrossRef CAS.
- Y. Zhang, A. A. Broekhuis and F. Picchioni, Macromolecules, 2009, 42, 1906–1912 CrossRef CAS.
- A. M. Peterson, R. E. Jensen and G. R. Palmese, ACS Appl. Mater. Interfaces, 2009, 1, 992–995 Search PubMed.
- S. E. Canary and M. P. Stevens, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 1755–1760 CrossRef CAS.
- H. Laita, S. Boufi and A. Gandini, Eur. Polym. J., 1997, 33, 1203–1211 CrossRef.
- X. Chen, F. Wudl, A. K. Mal, H. Shen and S. R. Nutt, Macromolecules, 2003, 36, 1802–1807 CrossRef CAS.
- C. Xiangxu, M. A. Dato, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698 CrossRef CAS.
-
F. Wudl and X. Chen, Thermally re-mendable crosslinked polymers, US Patent, 6
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 933361, 2005.
933361, 2005. - B. Gotsmann, U. Duerig, J. Frommer and C. J. Hawker, Adv. Funct. Mater., 2006, 16, 1499–1505 CrossRef CAS.
- M. Watanabe and N. Yoshie, Polymer, 2006, 47, 4946–4952 CrossRef CAS.
- Y. L. Liu and C. Y. Hsieh, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 905–913 CrossRef CAS.
- M. L. Szalai, D. V. McGrath, D. R. Wheeler, T. Zifer and J. R. McElhanon, Macromolecules, 2007, 40, 818–823 CrossRef CAS.
- Y. L. Liu and Y. W. Chen, Macromol. Chem. Phys., 2007, 208, 224–232 CrossRef CAS.
- J. K. Stille and L. Plummer, J. Org. Chem., 1961, 26, 4026–4029 CrossRef CAS.
-
Y. Takeshita, M. Uoi, Y. Hirai and M. Uchiyama, Reversibly crosslinked polymers, US Patent, 3890253, 1975.
- P. K. Joseph and F. C. Kenneth, J. Polym. Sci., Part A: Polym. Chem., 1979, 17, 2039–2054 Search PubMed.
- P. K. Joseph and F. C. Kenneth, J. Polym. Sci., Part A: Polym. Chem., 1979, 17, 2055–2070 Search PubMed.
- P. S. Malcolm, J. Polym. Sci., Part C: Polym. Lett., 1984, 22, 467–471 Search PubMed.
- G. Mircea and C. Georgiana, Polym. Int., 2001, 50, 1375–1378 CrossRef CAS.
- J. R. Jones, C. L. Liotta, D. M. Collard and D. A. Schiraldi, Macromolecules, 1999, 32, 5786–5792 CrossRef CAS.
- J. A. Syrett, G. Mantovani, W. R. S. Barton, D. Price and D. M. Haddleton, Polym. Chem., 2010, 1, 102–106 RSC.
- T. Paulöhrl, A. J. Inglis and C. Barner-Kowollik, Adv. Mater., 2010 DOI:10.1002/adma.201000361.
- H. P. Wang, Y. C. Yuan, M. Z. Rong and M. Q. Zhang, Macromolecules, 2010, 43, 595–598 CrossRef CAS.
- G. Deng, C. Tang, F. Li, H. Jiang and Y. Chen, Macromolecules, 2010, 43, 1191–1194 CrossRef CAS.
- L. J. Prins, D. N. Reinhoudt and P. Timmerman, Angew. Chem., Int. Ed., 2001, 40, 2382–2426 CrossRef CAS.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS.
- B. J. B. Folmer, R. P. Sijbesma, R. M. Versteegen, J. A. J. van der Rijt and E. W. Meijer, Adv. Mater., 2000, 12, 874–878 CrossRef CAS.
- K. E. Feldman, M. J. Kade, T. F. A. de Greef, E. W. Meijer, E. J. Kramer and C. J. Hawker, Macromolecules, 2008, 41, 4694–4700 CrossRef CAS.
- T. F. A. Greef and E. W. Meijer, Nature, 2008, 453, 171–173 CrossRef CAS.
- J. L. Wietor and R. P. Sijbesma, Angew. Chem., Int. Ed., 2008, 47, 8161–8163 CrossRef.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS.
- T. Park and S. C. Zimmerman, J. Am. Chem. Soc., 2006, 128, 11582–11590 CrossRef CAS.
- F. H. Beijer, H. Kooijman, A. L. Spek, R. P. Sijbesma and E. W. Meijer, Angew. Chem., Int. Ed., 1998, 37, 75–78 CrossRef CAS.
- F. H. Beijer, R. P. Sijbesma, H. Kooijman, A. L. Spek and E. W. Meijer, J. Am. Chem. Soc., 1998, 120, 6761–6769 CrossRef CAS.
- K. E. Feldman, M. J. Kade, E. W. Meijer, C. J. Hawker and E. J. Kramer, Macromolecules, 2009, 42, 9072–9081 CrossRef CAS.
- U. Mansfeld, M. D. Hager, R. Hoogenboom, C. Ott, A. Winter and U. S. Schubert, Chem. Commun., 2009, 3386–3388 RSC.
- P. Cordier, F. Tournilhac, C. Soulie-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS.
- D. Montarnal, P. Cordier, C. Soulie-Ziakovic, F. Tournilhac and L. Leibler, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7925–7936 CrossRef CAS.
- D. Montarnal, F. Tournilhac, M. Hidalgo, J. L. Couturier and L. Leibler, J. Am. Chem. Soc., 2009, 131, 7966–7967 CrossRef CAS.
- L. Holliday Ionic Polymers, John Wiley & Sons, New York, 1975 Search PubMed.
- http://www2.dupont.com/Surlyn/en_US .
- http://www2.%20dupont.com/Nucrel/en_US .
- H. J. Prosser and A. D. Wilson, Developments in Ionic Polymers, Applied Science Publishers, New York, 1983 Search PubMed.
- J. S. Kim and A. Eisenberg, Introduction to Ionomers, John Wiley & Sons, New York, 1998 Search PubMed.
- K. A. Mauritz, M. R. Tant, and G. L. Wilkes, Ionmers: Synthesis, Structure, Properties and Applications, Chapman and Hall, New York, 1997 Search PubMed.
- R. J. Varley and S. van der Zwaag, Acta Mater., 2008, 56, 5737–5750 CrossRef CAS.
- J. A. Hinkley and A. Huber, NASA Tech. Man., 2005, 213532 Search PubMed.
- S. Burattini, H. M. Colquhoun, J. D. Fox, D. Friedmann, B. W. Greenland, P. J. F. Harris, W. Hayes, M. E. Mackay and S. J. Rowan, Chem. Commun., 2009, 6717–6719 RSC.
- G. Wulff and I. Schulze, Angew. Chem., 1978, 90, 568–570 CrossRef CAS.
- Y. Chujo, K. Sada, A. Naka, R. Nomura and T. Saegusa, Macromolecules, 1993, 26, 883–887 CrossRef CAS.
- N. V. Tsarevsky and K. Matyjaszewski, Macromolecules, 2002, 35, 9009–9014 CrossRef CAS.
- N. V. Tsarevsky and K. Matyjaszewski, Macromolecules, 2005, 38, 3087–3092 CrossRef CAS.
- Y. Li and S. P. Armes, Macromolecules, 2005, 38, 8155–8162 CrossRef CAS.
- L. Wang, C. Li, A. J. Ryan and S. P. Armes, Adv. Mater., 2006, 18, 1566–1570 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS.
- Y. Higaki, H. Otsuka and A. Takahara, Macromolecules, 2004, 37, 1696–1701 CrossRef CAS.
- Y. Higaki, H. Otsuka and A. Takahara, Macromolecules, 2006, 39, 2121–2125 CrossRef CAS.
- Y. Chujo, K. Sada and T. Saegusa, Macromolecules, 1990, 23, 2693–2697 CrossRef CAS.
- Y. Chujo, K. Sada, R. Nomura, A. Naka and T. Saegusa, Macromolecules, 1993, 26, 5611–5614 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |