Synthesis of nanogels/microgels by conventional and controlled radical crosslinking copolymerization
Nicolas
Sanson
a and
Jutta
Rieger
*b
aLaboratoire de Physico-chimie des Polymères et Milieux Dispersés, UMR 7615 UPMC-CNRS-ESPCI, Ecole Supérieure de Physique et de Chimie Industrielles ESPCI, 10 rue Vauquelin, 75231, Paris Cedex 05, France
bLaboratoire de Chimie des Polymères, UPMC Univ. Paris 6 and CNRS, UMR 7610, 4 place Jussieu, Tour 44-54, 75252, Paris Cedex 05, France. E-mail: jutta.rieger@upmc.fr; Tel: +33 1 44275137
First published on 26th April 2010
Abstract
This review compares conventional and controlled radical polymerization techniques and processes in preparing nano-/microgels. Special focus is made on the synthetic parameters that allow controlling their size, morphology, composition, and structural homogeneity.
 Nicolas Sanson | Nicolas Sanson completed his PhD in 2005 under the supervision of Corine Gérardin and François Fajula at the ENSC of Montpellier. He started a postdoctoral position with Prof. Eugenia Kumacheva at the University of Toronto, where he was introduced to the field of microgels. Subsequently, he worked as a postdoctoral fellow at the CRPP (Bordeaux) with Virginie Ponsinet. Since 2007, he is assistant professor at the University Pierre et Marie Curie (UPMC, Paris) and his research occurs at the ESPCI. His research focuses on assemblies of nanoparticles induced by stimuli-responsive polymers and on synthesis and characterization of microgels. |
 Jutta Rieger | Jutta Rieger is currently a CNRS researcher at the University Pierre et Marie Curie (UPMC, Paris). She completed her PhD at the University of Liège (Belgium) and CERMAV (France) under the supervision of Professors Robert Jérôme and Rachel Auzély-Velty in 2005. After briefly working in 2006 for Macopharma at ICMMO (France), she received a FNRS fellowship for a research project on stimuli-reponsive nanogels in collaboration with Dr Christine Jérôme. In 2007 she joined Professor Bernadette Charleux's group (UPMC) where her research focuses on the synthesis of functional self-assemblies by controlled radical polymerization in homogeneous or heterogeneous media. |
I Introduction
Polymers having a branched architecture can be classified as “star polymers”, “grafted polymers”, “dendrimers”, “branched polymers”, or “gels”, depending on their molar mass and size, functionality, and the number and relative arrangement of the branching points within the macromolecule. In contrast to “gels” i.e. macroscopic networks, their nanometric/micrometric counterparts, i.e. “nanogels” or “microgels”, may dissolve in solvents—just as linear polymers—however, preserving a nearly fixed conformation. They may swell and change their dimensions depending on the solvent and environmental conditions. Their structure is thus intermediate between branched and macroscopically crosslinked systems. “Microgels” are defined as gel particles of any shape with an equivalent diameter of approximately 0.1 to 100 µm, whereas the diameter of “nanogels” is approximately 1 to 100 nm both exhibiting network structures that swell in a suitable solvent.1 The polymers that they are composed of are synthetic or natural, and they are crosslinked either chemically or physically. Nano-/microgels are not new materials, but they have already been described in the mid-1930's2 (as a by-product), and again in the late 1940's.3 Since the nineties a great deal of work has been published, and the number of publications/year on this subject is steadily rising (Fig. 1).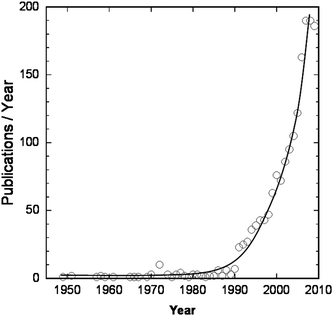 | ||
| Fig. 1 Evolution of the number of publications on nanogels/microgels since 1950. (Source: SciFinder Scholar, Keywords: “microgels” and “nanogels”. For 2009, the number of papers registered until October 2009 is plotted.) | ||
First syntheses were all performed in organic solvents at high dilution. Since then significant advances in the synthetic pathways (polymerization techniques and processes) have been made (cf. Section II), that allow not only tuning the chemical composition of those crosslinked polymer particles but also their size, morphology and functionality (cf. Section III). In the age of green chemistry today's syntheses tend to use environmental friendly processes, mainly aqueous heterogeneous polymerization processes.
Numerous groups have focused on the development of “smart” or “responsive” microgels that undergo structural or morphological changes such as volume transition in response to (environmental) stimuli such as pH, temperature, ionic strength, solvent, light, magnetic fields, enzymatic activities, or ligand binding. They pave their way towards new materials that not only have tunable dimensions, but also differ in the material's properties such as wettability, permeability, refractive index, flexibility, and viscoelasticity. Within the class of stimuli-responsive polymers, the most common responsive polymer is poly(N-isopropylacrylamide) (PNiPAm), which is a thermo-responsive polymer that undergoes a coil-to-globule transition in aqueous media at ∼32 °C, i.e. at its “lower critical solution temperature” (LCST). When those chains are crosslinked in a polymer network, the responsivity appears as a volume collapse arising from the expulsion of the solvent (water). This transition temperature is thus referred to as a “volume phase transition temperature” (VPTT), which is generally close to the LCST of the corresponding linear polymer. In the last decade, multiresponsive materials that are sensitive to multiple stimuli and show a more complex transition behavior have received increasing interest.4,5 Hydrophilic, hydrophobic, neutral or charged monomers can be introduced that allow the preparation of combined temperature–pH, amphoteric or double temperature-sensitive microgels.6–8 Furthermore, functional/reactive groups have been incorporated regioselectively in their core or in their shell and they are then available for the attachment (bioconjugation) of bioactive compounds, such as ligands or enzymes.9 Such functionalized nano-/microgels pave the way towards bioresponsive materials that enable—for instance—targeted and/or triggered drug delivery. In addition, certain functional groups may promote the complexation of metal ions/nanoparticles opening the door to a large spectrum of new sophisticated applications, e.g. the template-based fabrication of hybrid materials or separation and purification technologies.10,11
In addition to the intrinsic properties of the nano-/microgels given by the nature of the polymers/materials that they are composed of, their dimension and geometry are of crucial importance. Depending on the application, dimensions in the nanometre or in the micrometre range are targeted. Especially, optical applications based on the assembly of colloids in a lattice structure are very demanding in relation to size distribution (polydispersity). Microgels with polydispersity factors lower than 10%12,13 are generally required when photonic applications are targeted, such as their use as photonic crystals or sensors,14 or when they are used as model systems for fundamental studies in colloidal physics.6,15 In addition to the control over the size and size distribution of spherical gel particles, today's efforts tend to control the synthesis of nano-/microgels of more complex architectures, such as core–shell structures,16,17 hollow,18 or non-spherical, di- or trimeric structures.19–21 In Section III.1 and 2, the synthetic strategies to reach control over size/size distribution and morphology are discussed.
The applications of nanogels/microgels are manifold. For biomedical applications, hydrogel particles containing mostly water are especially interesting. Indeed, they may possess good biocompatibility, as the high water content results in low interfacial tension. However, supplementary studies on cytotoxicity and immunogenicity are still required. The potential application of nanogels/microgels as carrier systems relies primarily not only on their tunable size, their high loading capacity, thanks to their interior network structure, but also on their high stability (compared to micelles) and responsiveness to environmental factors (such as pH, ionic strength, light and temperature).22–24 Moreover, they possess large surface area allowing multivalent bioconjugation. Specific ligands have been introduced on the surface of nanogels allowing their accumulation in targeted tissues. The decoration of their surface by biocompatible polymers is also possible. It has for instance been demonstrated that poly(ethylene glycol) (PEG) coatings prolong considerably the circulation time of polymeric nanoparticles in the blood stream.25 Finally, the presence of such long stabilizing polymer chains at their surface may impart nano-/microgels with high stability and prevent them from aggregation.
Apart from the applications mentioned above other important fields are sensing,17 molecular imprinting,26 their application as emulsifiers or stabilizers in complex coatings, or their use as nanoreactors capable of modulating the catalytic activity of metal nanoparticles. The variety of applications makes clear the need for reliable and straightforward synthetic strategies towards the controlled synthesis of these complex materials. In contrast to existing reviews,7,24,27–32 this review is limited to syntheses pathways based on radical crosslinking copolymerization (RCC) of vinylic monomers with di- or multivinyl crosslinker (one-batch addition). In the following part, we will compare different polymerization processes and oppose synthetic strategies based on conventional radical polymerization (RP) to controlled radical polymerization (CRP) techniques. Depending on the targeted morphology, properties and application of the colloids, the limits and advantages/disadvantages of each technique are highlighted. Star polymers prepared in two steps by the arm-first or core first method,29i.e. polymer chains that are crosslinked at a central point and do not possess a gel-like core structure, and nanogels that are obtained in several steps through crosslinking of preassembled morphologies, are not discussed in this review. Consequently, core crosslinked (CCL) micelles33 and shell crosslinked (SCL) micelles34,35 where either the core or the shell of preformed amphiphilic block copolymer micelles is crosslinked through organic chemical reactions—such as amidification—are beyond the scope of this review.
II Synthesis of nanogels/microgels
II.1 Synthesis of nanogels/microgels by free radical crosslinking copolymerization (RCC)
Various synthetic strategies for the preparation of chemically crosslinked nanogels/microgels have been described. They include top-down nanotechnologies, such as template-assisted nanofabrication (imprint photolithographic techniques30,36), or bottom-up approaches such as micromolding37 and microfluidic methods.38,39 Synthetic pathways that require a minimum of specific equipment are crosslinking reactions carried out on preformed colloidal self-assemblies of synthetic or natural polymers, and finally “free radical crosslinking copolymerization” (RCC) of monovinylic monomers with di- or multifunctional comonomers (crosslinker).40 Typically, di(meth)acrylates, such as ethylene glycol di(meth)acrylate and 1,4-butanediol diacrylate, divinylbenzene, or degradable crosslinker (see p. 5 and 10) are used as crosslinking agents.29 This review is limited to the last approach, which may be considered as the simplest and most popular one. As colloidal networks, which are limited in size are targeted, the main challenge relies on identifying strategies that allow avoiding the formation of long-range networks. Several strategies have thus been developed in order to reach nanometric or micrometric gels instead of macroscopic networks, i.e. macroscopic gelation. They generally rely on the control of the distance between growing polymer chains.29 The first strategy is based on RCC performed in highly diluted solution. Decreasing the monomer concentration increases the distance between propagating chains, limits thus intermolecular crosslinking and increases the probability of intramolecular crosslinking (Fig. 2, schema for conventional RCC on the top); consequently macroscopic gelation can be prevented. Moreover, the use of low amounts of crosslinker or stopping the polymerization at low monomer conversion before the critical gel point also favors the preparation of soluble branched polymers instead of gels.29,41–43 Targeting the synthesis of colloidal gels, another efficient strategy is the application of heterogeneous polymerization processes, where the polymerization is performed in a confined nanometric/micrometric space. Here, the size of the gels will be limited by confining the crosslinking to intraparticle rather than interparticle crosslinking. Whereas the first approach, i.e. RCC in highly diluted homogeneous conditions, leads to soluble branched polymers, this second strategy may lead to more dense nano- or micrometric particles possessing an internal structure comparable to that of macroscopic networks.44 It includes (inverse) emulsion, (inverse) miniemulsion and (inverse) microemulsion polymerization processes, or precipitation and dispersion polymerization processes.30,45 The choice of the heterogeneous polymerization process clearly depends on the nature of the monomers. So, the synthesis of microgels composed of a high percentage of hydrophilic (ionic) monomers is generally performed (in aqueous droplets) in inverse (micro)emulsion in organic solvents (e.g. cyclohexane) or oils, whereas hydrophobic monomers can be polymerized in O/W emulsions in water. The process of polymerization directly impacts the (internal) structure and size of the gel particles and we consider important to clarify the main differences in the different processes: in the case of (mini)emulsion polymerizations, the monomer is not soluble but dispersed with the aid of a surfactant in the continuous phase—i.e. water in the case of O/W aqueous emulsions, or oil in the case of W/O emulsions (inverse emulsions). One main difference between miniemulsion and emulsion processes is the initial size of the dispersed phase. In the case of emulsion polymerizations, monomer droplets are formed by mechanical stirring and measure 1 to 20 µm. In contrast, for (O/W) miniemulsions an additive (hydrophobic) is generally added allowing stable droplets smaller than 500 nm to be formed by applying high shear stress, e.g. by ultrasonication or high-pressure homogenizer. Whereas (mini)emulsions are only kinetically stable, microemulsions are thermodynamically stable, thanks to large amounts of emulsifier, allowing the formation of initial monomer droplets of diameters below 20 nm. It is evident that the polymerization mechanism is essentially different depending on the localization of the radicals, monomers and crosslinker (which is determined by process). From a mechanistical point of view, micro/miniemulsions constitute the simplest system where the polymerization may proceed in “nanoreactors” in the dispersed phase. Here, each monomer-swollen micelle (droplet) is ideally converted into a polymer particle of similar size. On the contrary, emulsion polymerization is more complex: initiation takes place in the continuous phase, and particle nucleation and diffusion processes must be considered. Here, the size of the formed gel particles does not correspond to the initial size of the dispersed (monomer) phase at the beginning of the polymerization. In contrast to emulsion polymerizations, where the polymerization starts in heterogeneous conditions, dispersion or precipitation polymerizations are initially homogeneous, i.e. all compounds, the monomers, crosslinker, initiator and stabilizer, are initially soluble in the solvent. Upon polymerization, particle formation occurs as a result of polymer chains propagating to a critical chain length at which they are no longer soluble. The fundamental difference between precipitation and dispersion polymerization relies on the presence of a colloidal stabilizer in the case of dispersion polymerization yielding generally smaller particles.46,47 Actually, thermosensitive gel particles with narrow particle size distribution are generally synthesized by precipitation/dispersion48 polymerization processes performed at a temperature above the LCST, so that the forming gel particles/polymers undergo phase separation during synthesis. In this mechanism, water-soluble oligomers grow in the initially homogeneous medium until they reach a critical chain length. Beyond this length, the growing chains collapse to form precursor particles. The nuclei may then aggregate with other precursor particles or deposit onto existing colloidally stable preformed particles. Stability is typically achieved through the use of surfactants and/or by electrostatic stabilization originating from ionic groups of the initiator.28,31 For more specifications about polymerization mechanisms in dispersed media refer to recent reviews.46,49,50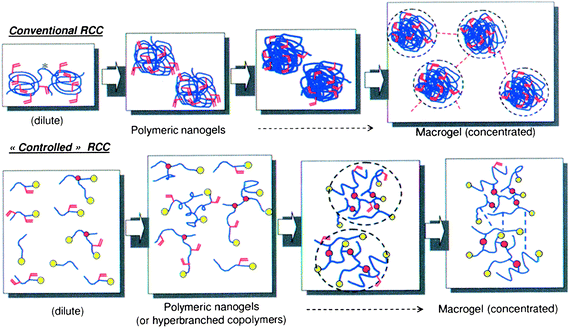 | ||
| Fig. 2 Schematic presentation of the crosslinking reactions in conventional and “controlled”/“living” radical polymerization systems. (Reprinted with permission from D. Taton, in Macromolecular Engineering: Precise synthesis, materials properties, applications, ed. K. Matyjaszewski, Y. Gnanou and L. Leibler, Wiley, Weinheim, 2007, vol. 2, ch. 8, pp. 1007–1056.) | ||
Finally, a smart strategy to reach nano-/microgels instead of macrogels (at quite high monomer conversion/concentration) relies on the limitation of the primary chain length by using high amounts of initiator or chain transfer agents.51 For the same purpose, controlled polymerizations, such as living ionic polymerizations52,53 or controlled/“living” radical polymerization (CRP)29 (cf. Section II.2), have been applied and their potential in the synthesis of well-defined/functional nanogels/microgels has been demonstrated.
II.2 Synthesis of nanogels/microgels using CRP techniques
The development of controlled/“living” radical polymerization (CRP) techniques since the mid-1990 can be considered as a breakthrough towards the easy synthesis of complex macromolecular structures with high degree of functionality and compositional variety. They can be performed under simple polymerization conditions and are tolerant to a wide variety of functional groups and solvents, for instance protic media such as water.54 The CRP techniques that have been implemented so far are mainly nitroxide-mediated polymerization (NMP),55 catalytic atom (group) transfer radical polymerization (ATRP),56–58 degenerative chain transfer polymerization represented by iodine-mediated polymerization (RITP),59,60 and reversible addition–fragmentation chain transfer (RAFT)61,62 polymerization/macromolecular design via the interchange of xanthates (MADIX).63 Each technique has its pros and cons, such as the synthetic ease of NMP—however, requiring high temperatures, the multitude of reaction conditions compatible with ATRP—but presenting purification issues because of the use of metal catalysts, and the versatility of RAFT (in terms of monomers and reaction conditions and compatibility to functional groups—necessitating no recourse to protecting chemistry)—yet leading frequently to colored or odorous polymers.62,64 The fundamental mechanism of CRP is the same as in conventional RP. It comprises four elementary reactions: initiation, propagation, transfer and termination. In contrast to conventional RP where the initiation process is slow and continuous, and propagation and termination reactions are fast leading to “dead” chains of broad molecular weight distributions with essentially no control over composition and macromolecular structure, CRP is essentially based on a fast initiation step (relative to propagation and termination) and a dynamic equilibrium between a low concentration of propagating radicals and a large amount of dormant reactivatable species. This results ideally in a nearly constant number of chains throughout the polymerization, which are initiated and grow at the same time with the same rate allowing control over molar mass distribution and architecture. It should be noted that—in contrast to NMP and ATRP—the RAFT mechanism is superimposed on a conventional free radical polymerization and the radicals are provided by a conventional initiator. Applying CRP, block copolymers can be easily prepared and the structure of the copolymers can be varied by simply changing the sequence of polymerization. Moreover, according to the used technique polymers with different reactive end groups can be obtained that are available for conjugation of functional molecules.Since the nineties, the beneficial effect of CRP has been demonstrated for the synthesis of branched polymers and especially macrogels prepared in homogeneous conditions (i.e. in the bulk or in solution).42,43,65–72 As schematized on the top of Fig. 2, in a conventional non-living RCC polymerization mechanism intramolecular crosslinking dominates at low conversions (at the left side) leading to the formation of dense/nodular “microgel domains” and a heterogeneous structure of the final macrogel (at the right side).29,65,73 In contrast, when CRP techniques are applied the kinetics is considerably slower than in conventional RP and dormant polymer chains have time to diffuse and relax before being reactivated to propagate. Consequently, crosslinking points are more homogeneously distributed within the networks—assuming equivalent reactivity of the monomer and crosslinker—as lately demonstrated by combined experimental and simulation data.74 It was demonstrated that for controlled radical crosslinking copolymerizations (cRCC) the number of primary (linear) chains is essentially determined by the concentration of control agent; their chain length and molar mass distribution depend directly on the monomer/control agent ratio.29,72,73,75–78 These experimental data were recently supported by Monte Carlo simulations based on a dynamic lattice liquid (DLL) model. They provide indeed a useful tool to understand the influence of various experimental parameters (such as dilution, monomer/control agent ratio and crosslinker concentration) on the onset of gelation.79
CRP techniques have thus successfully been applied to the controlled synthesis of nano-/microgels, i.e. networks which are limited in size and molar mass. Indeed, when nanogels were synthesized by ATRP or RAFT/MADIX in the presence of degradable crosslinker,29 such as disulfide-based di(meth)acrylate crosslinker,9,80–82 di(ethylene glycol) di(methacryloyloxy)ethyl ether,83 or N,N′-(1,2-dihydroxy ethylene) bisacrylamide84 degradation of the crosslinking points resulted in individual polymeric chains with quite narrow molar weight distribution and molar masses were close to those of the corresponding linear polymers prepared in equivalent conditions but in the absence of crosslinker.76,77,80–83 These experiments clearly demonstrate the effect of CRP on nano-/microgel formation. Applying CRP techniques, their synthesis can be carried out at much higher monomer and crosslinker concentration (or stopped at higher monomer conversion) as gelation is retarded.41–43,79 It is indeed possible to synthesize low molar mass branched polymers in solution at monomer concentrations as high as 20 wt% in the presence of up to 10 mol% crosslinker.76,85 Thanks to the living character of CRP, chain extensions can be performed after addition of a second monomer batch.75,85,91 Controlling the sequence of addition of (several) monomers or crosslinker, the structural composition and morphology of the nanogels/microgels can be controlled and a large variety of architectures becomes accessible (cf.Fig. 3 and Table 1). The use of functional initiators further allows the incorporation of functionalities in the core or at the surface of nano-/microgels, which are available for (bio)conjugation reactions (Fig. 3). Today the implementation of especially ATRP9,29,86,87 but also NMP68,88,89 and RAFT90–94/MADIX85 to the synthesis of nano-/microgels is well established (cf. Section III). It should also be noted that cRCC in heterogeneous (mostly aqueous) conditions45,46 has raised particularly interest for the reasons explained in Section II.1.
| Structural features | Conventional crosslinking radical polymerization (RCC) | Controlled radical crosslinking polymerization (cRCC) |
|---|---|---|
| Size/polydispersity | Parameters that influence size and size distribution are: (a) amount of crosslinker and initiator; (b) process of polymerization; (c) for heterogeneous aqueous polymerization processes: amount of surfactant/charged initiator, presence of a soluble comonomer, temperature, stirring speed. | Parameters that influence size and size distribution are: (a) crosslinker/CRP initiator (RAFT agent) and monomer/ CRP initiator (RAFT agent) ratios; (b) process of polymerization; (c) see (c) in RCC. |
| Highly monodisperse dispersions can be reached by heterogeneous polymerizations processes. | It is possible to reach well-defined nanogels in heterogeneous polymerization processes in the absence of surfactant using soluble or charged macroinitiators/macroRAFT agents that act as stabilizer and control agents. | |
| Polymerization in homogeneous conditions must be performed in high dilution (small amounts of monomer and crosslinker) or stopped at low conversion in order to avoid macrogelation. | High solids content (up to 20%) can be reached. | |
| Chain extension with a second monomer batch is possible. | ||
| Architectures | Importance of the process of polymerization. | Importance of the process of polymerization. |
| Core and shell crosslinked structures by multistage seeded polymerizations where the core and the shell are not covalently linked with each other and composed of a network structure. | Chain extension with a second monomer batch is possible ⇒ hairy shell or core might be composed of two different types of polymers. | |
| Multilayered microgels by multistage (precipitation) polymerizations. | Anisotropic gel particles are accessible by the formation of block copolymers. | |
| Hairy nano-/microgels where the hairy shell is formed by a macromolecular comonomer. | Hairy nano-/microgels where the core is covered by covalently linked polymer chains using macromolecular CRP initiators or macroRAFT agents. | |
| Hollow nano-/microgels by removal of a degradable core from core–shell nano-/microgels. | Hollow hairy nano-/microgels (nanocapsules) are accessible in one step in heterogeneous polymerization conditions. | |
| Hollow microspheres by inverse (mini)emulsion RCC. | ||
| Surface functionalization | Utilization of soluble functional comonomers in heterogeneous RCC. | Utilization of soluble functional (macro)initiators or (macro)RAFT agents in heterogeneous cRCC. |
| Functionalization of the core | Copolymerization with functional comonomers. | Copolymerization with functional comonomers (enhanced homogeneity). |
| ω-Chain end functionalities through the use of appropriate CRP initiators or RAFT agents. |
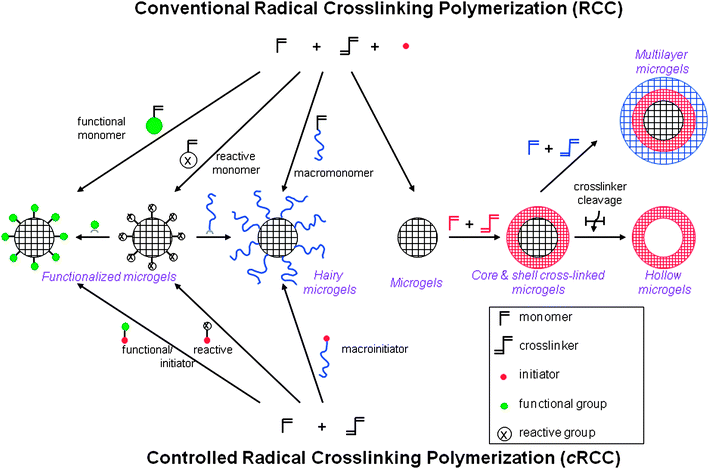 | ||
| Fig. 3 Synthetic approaches for the preparation of nano-/microgels of different morphologies and functionalities via conventional and controlled radical crosslinking polymerization. | ||
In the following part (Section III), we will discuss the synthetic parameters that allow one to tune the morphology, structure and size of nano-/microgels. Examples for both conventional radical crosslinking polymerization (RCC) and controlled radical polymerization (CRP) will be given and the different structures that are accessible by each technique are highlighted in Fig. 3 and in Table 1.
III Size, polydispersity, network homogeneity and architecture control
III.1 Synthesis parameters that influence size, size distribution and homogeneity of the network
Depending on the application, nano-/microgels of different dimensions and morphologies are targeted. As an example, for drug delivery application it has been demonstrated that the cellular uptake pathway of nanogels, their elimination by the mononuclear phagocyte (MPS) system and their accumulation in cancer cells (via the enhanced permeation and retention effect, EPR effect) are mainly governed by their morphology and size, which has generally to be less than 200 nm. Moreover, it should be noted that particle dimensions play an important role in the responsiveness since the rate of the volume phase transition is inversely proportional to the size of microgel.It is thus important to find synthetic approaches that allow tuning the size, architecture of the resulting particles while maintaining narrow particle size distribution (low polydispersity factors). The first and most important parameter that affects the dimensions of the resulting gel particles is the process of polymerization (heterogeneous/homogeneous) which is mainly determined by the solvent used. As described in Section II.1, network formation is fundamentally different in homogeneous conditions compared to heterogeneous ones and the resulting nano-/microgels differ in size, structure and properties. In solution, i.e. in homogeneous conditions, their molar mass essentially depends on the concentrations of the compounds: lower monomer concentrations yield gels of smaller dimensions (the formation of macroscopic gels can be avoided), as the probability of intramolecular crosslinking (loop formation) increases. At very low concentrations only “self-crosslinked molecules”, i.e. branched polymers are formed.44,95 The size of the macrogels can also be reduced by using decreased crosslinker concentration or enhanced initiator concentrations.75,76,96,97 As described before, chain transfer agents, e.g. thiols, and CRP control agents, i.e. NMP-/ATRP initiators or RAFT agents, have shown to allow the formation of low molar mass branched polymers instead of (macroscopic) gels by decreasing the molar mass of the primary chains.
Several authors studied the influence of the solvent in which RCC is performed.98–100 It was concluded that the dimensions of the resulting particles depend on the solubility of both the monomers and the polymers. Indeed, as the solvent properties change (temperature must also be considered), interfacial energies and the mechanism of the particle formation are modified. For instance, Kim et al.100 performed syntheses of PNiPAm nanogels either in water or in a water/THF mixture at 50 °C, i.e. above the LCST of PNiPAm in water. Gel particles obtained in the solvent mixture were bigger than those obtained in water (500 vs. 70 nm). The fundamental difference accounting for the variation in size must be found in the solubility of the growing polymer chains. With increased solubility, phase separation will be postponed and the critical length of the polymer chains at the phase separation becomes longer resulting in bigger colloidal gels. In a similar study, Stucky et al. performed surfactant-free RCC of methyl methacrylate (MMA) either in water (i.e. in emulsion conditions) or in 25 wt% acetone/water mixture. Here, bigger particles were obtained in water compared to the acetone/water mixture. This must be attributed to a different nucleation mechanism due to the difference in monomer solubility. In the solvent mixture, MMA is more soluble which might contribute to an enhanced number of nucleation seeds leading to smaller particles.99
In such heterogeneous polymerization conditions, particle stabilization must be considered. In fact, stability is given by repulsive electrostatic, electrosteric or steric interactions between particles generally provided by a surfactant. Under certain conditions, stable nano-/microgels can be prepared by surfactant-free emulsion or (aqueous) precipitation polymerization.32 Particles of low size distribution might be obtained, but their size is generally larger than in the presence of surfactant as an aggregation step generally occurs during particle growth. In surfactant-free aqueous heterogeneous polymerization, particles are stabilized by the charges provided by the initiator, such as ammonium persulfate (APS), potassium persulfate (KPS) or 2,2′-azobis(amidinopropane) dihydrochloride (V50). The particle size might slightly be diminished through the use of large amount of initiator. Typically, stable gel particles smaller than 300 nm could be achieved by aqueous precipitation polymerization of NiPAm in the presence of high amount of initiator,6 but their average size is generally larger compared to that of particles prepared in the presence of ionic surfactants.31 Apart from the initiator or the surfactant concentration, a third parameter may contribute to particle stabilization and affect the particle size. Actually, stabilizing (charged) comonomers, such as acrylic acid, may be employed in aqueous heterogeneous RCC.101 This approach does not only allow to tune the size of the particles (by changing the co-monomer/monomer ratio102) but also to impart additional functionalities or responsivity to the nanogels.
In addition, the application of CRP techniques to crosslinking polymerization in heterogeneous conditions opened up new surfactant-free synthesis pathways towards autostabilized hydrogel particles of very small dimensions. Indeed, the use of soluble or charged macromolecular CRP initiators (macroinitiators) or macromolecular RAFT agents (macroRAFT agents) allows small nanoparticles to be prepared in the absence of conventional surfactant.103,104 Such nanogels are (electro)sterically stabilized via a polymeric corona provided by the soluble macromolecular control agent (see Fig. 3). By changing the length and the nature (e.g. charged polymers vs. neutral polymer) of the stabilizing macromolecular control agent, nanogels of different dimensions could be obtained91 at rather high solids content.88,92 It should also be mentioned that in such heterogeneous conditions the stirring quality/speed, the application of microwave power99 and temperature are additional important parameters that impact the particle size and its distribution. It had been shown that a decrease of the temperature to 60 °C increased the particle size to 1.5 µm due to a prolonged nucleation stage.105 Further decrease of the temperature to 50 °C leads to less stable particles with an excessive amount of coagulum formed instead of monodisperse colloids. In order to achieve large >2 µm size PNiPAm microgels Lyon et al. performed the aqueous dispersion polymerization of NiPAm using a programmed temperature ramp from 45 to 65 °C (30 °C h−1) during the nucleation step. 2.5 to 5 µm large stable microgels could be afforded.106 It is proposed that initially, because of the slow decomposition rate of the thermal initiator, low nuclei concentrations are obtained and higher propagation rates (compared to initiation) favor particle growth in these particular conditions. The temperature ramp then compensates a decrease in propagation rate (because of monomer consumption) and maintains it approximately constant.
The particle size distribution (polydispersity) was shown to be intimately related to the process and kinetics of particle formation. It has been shown, that a short nucleation stage favors the formation of monodisperse samples,96,107 meaning that all particles are formed at low overall monomer conversion, and over most of the polymerization, particle growth dominates the kinetics instead of particle nucleation. Whereas nonliving dispersion polymerizations normally afford monodisperse particles, dispersion CRP—where the propagation kinetics is slowed down—often yields broad particle size distributions.46 Indeed, in a CRP system the nucleation stage is prolonged because high molar mass polymer is not formed instantaneously as it is in a nonliving system, resulting in broad particle size distributions. This issue has been overcome using a “two-stage”/seed strategy, whereby the control agent (RAFT agent) and the crosslinker were added after completion of the nucleation stage, resulting in monodisperse spherical micron-sized particles.108 It is in order to shorten the nucleation stage, that thermally initiated RCC of NiPAm in aqueous media is generally conducted at 70 °C, that is much higher than the LCST (∼32 °C). This relatively high temperature will not only induce rapid phase separation (as soon as the initiator adds some monomers), but also leads to an accelerated decomposition of the thermal initiator and thus induces a shorter nucleation period.
In addition to solvent and temperature effects, the influence of the crosslinker reactivity has also been studied, both experimentally99 and by Monte Carlo simulations.74 For instance, Stucky et al.99 studied the heterogeneous RCC of MMA in acetone/water mixture. It was found that the crosslinker of the lowest kp led to the biggest particles. They were indeed significantly bigger than reference particles prepared in the absence of crosslinker and the increase in dimension was attributed to the occurrence of interparticular crosslinking. In contrast, in the presence of N,N′-bismethylene acrylamide (MBA), i.e. the crosslinker of the highest reactivity, the particle size corresponded to that of the model particles meaning that interparticular crosslinking was prevented. Due to its high reactivity, MBA is indeed by far the most frequently employed crosslinker for the aqueous RCC of NiPAm, and leads to monodisperse particles possessing a gradient in crosslink density (from the core to the shell) because of difference in monomer reactivity.31 In order to foresee the structure of microgels, consumption of monomer and difunctional monomer can be calculated applying copolymerization kinetic models and the distribution of crosslinker and/or functional groups can be predicted.109,110 Actually, the homogeneity of crosslink distribution, i.e. the homogeneity of the polymer network, is an important issue of (hydro)gel particles that will influence their swelling properties and their mechanical properties. Generally, homogeneous networks are targeted where the crosslinking points are homogeneously distributed throughout the objects. As explained in part II.2, CRP methods may favor the formation of more homogeneous networks due to the mechanisms and kinetics that are inherent in them—assuming similar reactivity of the monomers and the crosslinker. To overcome reactivity issues, semi-continuous processes (semi-batch or starve-fed monomer feed strategies) can be applied where a crosslinker of enhanced reactivity is gradually added during polymerization.110,111
In emulsion or precipitation polymerization, it appears to be difficult to prepare microgels that have a completely uniform structure. In these heterogeneous conditions, not only differences in reactivity ratios between monomers and crosslinker but also differences in solubility (hydrophilicity/hydrophobicity) must be considered: for instance, when a hydrophilic monomer is copolymerized with a less hydrophilic (i.e. quite hydrophobic) crosslinker the formation of first crosslinks creates hydrophobic centers, and then partitioning of the crosslinking agent in these hydrophobic domains results in hydrogels with inhomogeneous distribution of crosslinks. Inverse miniemulsion polymerization might be a better method for achieving a uniform comonomer concentration as the polymerization mechanism does not involve diffusion between phases.32
III.2 Nano-/microgel architectures
As mentioned in Section II, conventional and controlled radical crosslinking copolymerization (RCC) in heterogeneous conditions can afford nano-/microgels with different morphologies. Indeed, architecture is a key parameter that has a strong influence on their physical properties and therefore great importance with regard to their applications. In general, the morphology of structured nano-/microgels is dictated by thermodynamic and kinetic considerations that afford multiple architectures depending on different parameters such as monomers and polymer solubility, reactivity of the vinyl and divinyl compounds, and reaction temperature.112 The aim of this part of the review is to give an overview on the different nano-/microgel morphologies obtained in the literature. Their synthesis by controlled or non-controlled radical crosslinking polymerization methods is described.III.2.1.1 Core and shell crosslinked nano-/microgels. Most of core–shell hydrogel particles studied in the literature are synthesized by two-step radical precipitation polymerization. In a typical synthesis of poly(N-isopropylacrylamide) PNiPAm core–shell microgels, the core is prepared first by (co)polymerization of NiPAm in the presence of a bi-functional comonomer as crosslinker (see Section II). In the second stage, a second monomer batch (the same or different from the core monomer) is added for the shell synthesis (seed and feed process). The polymerizations are carried out above the lower critical solution temperature (LCST) of PNiPAm, so that the collapsed core microgels serve as nuclei/seed for the shell synthesis. At this temperature the surface of the core microgel behaves as an “active surface” and growing oligomers constituting the shell adsorb on the surface of the preformed core particles (seeds). For such gel particles prepared by a two-step precipitation polymerization, there is no chemical linking between the core and the shell, only a low interpenetration of the chains at the interface exists. They can for instance be used for the synthesis of hollow microgels (cf. Section III.2.2.). Comonomers that are sensitive to other stimuli than temperature can also be selectively introduced either in the core or in the shell generating multiresponsive core–shell materials. Lyon's group was the first to report the synthesis of core and shell crosslinked microgels with a temperature-responsive core and a pH-responsive shell (and vice versa).5 For instance, they prepared NiPAm based core–shell microgels where either the core or the shell was constituted by a copolymer of NiPAm and acrylic acid (AA).16,114 The volume phase transition temperature (VPTT) of these multiresponsive core–shell microgels strongly depends on the crosslinker content in the shell, the thickness of the shell, the pH and the spatial location (core or shell) of the pH-sensitive comonomer (AA). When AA is exclusively incorporated in the core, in basic medium and at temperatures above the LCST of PNiPAm, the collapsed PNiPAm shell forms a dense barrier around the periphery of the core inducing a swelling restriction to the core.114 In contrast for the reverse system (AA exclusively present in the shell), they observed up to three volume phase temperature transitions as a function of the temperature due to the structural heterogeneities in the core–shell microgel.5 A similar study has been performed on pH- and temperature-sensitive poly(2-vinylpyridine) (P2VP)/PNiPAm microgels.115,116 For insulin delivery, NiPAm-based core–shell microgels with a shell constituted of a copolymer of NiPAm and a glucose-responsive comonomer have been prepared. They have proved to be promising carrier systems for glucose-trigged insulin release.117 Recently, double pH-responsive amphoteric core and shell crosslinked microgels, where the shell or the core is either composed of poly(2-diethylaminoethyl methacrylate) or poly(methacrylic acid), have also been reported.118 Since core microgels can be used as seeds for multiple step precipitation polymerization, Lyon et al. used core–shell microgel as seeds for the synthesis of triple core–shell–shell microgels (see Fig. 3). These hierarchic thermosensitive microgels were used as templates for gold nanoparticle synthesis in the middle compartment of the microgel and then assembled to form tunable colloidal crystals.119 This multistage polymerization offers great opportunities in the synthesis of stimuli-responsive multilayer materials for optic applications.
Richtering and others studied in detail the internal structure of double thermosensitive core–shell microgel (synthesized by a two-step precipitation polymerization in the presence of surfactants) where the core and the shell are composed of two different temperature-responsive polymers, namely N-isopropylacrylamide (NiPAm) and N-isopropylmethacrylamide (NiPMAm) possessing LCSTs of ∼32 °C and ∼45 °C120 respectively.121 Dynamic light scattering (DLS) and differential scanning calorimetry (DSC) revealed that they exhibited two distinct VPTT that corresponded to the LCST of the core and the shell polymer.122 Their internal structure, such as the volume ratios between the core and the shell (giving access to the thickness of the shell) and the crosslinker density of the shell, were extensively studied by dynamic light scattering (DLS) and small angle neutron scattering (SANS).123–125 For PNiPAm core/PNiPMAm shell microgels the swelling behaviour was mainly governed by the thickness and the crosslinker density of the shell and it was found that the core and the shell influence each other. A suitable universal analytical form factor based on the radial scattering length density distribution of core–shell microgel was introduced allowing the internal structure of microgel with a diffuse and/or sharp interface to be described. For the reverse system, PNiPMAm core/PNiPAm shell, at an intermediate temperature between the LCST of the two polymers, the size of the core–shell microgel is smaller than that of the core alone indicating that the PNiPAm shell in its collapsed state restricts the swelling of the core. The swelling kinetics of core–shell microgels which is an important parameter in the comprehension and development of responsive materials have also been studied by light scattering methods, 1H NMR, differential scanning calorimetry,126 fluorescence spectroscopy127 or laser-induced temperature transition measurements.128
To the best of our knowledge, no papers have been reported where controlled radical crosslinking polymerization was applied to the synthesis of core and shell crosslinked microgels where both the shell and the core possess a hydrogel-like structure.
III.2.1.2 Hairy nano-/microgels. The second type of core–shell nano-/microgels has been called “hairy nano-/microgels”, as a crosslinked core is surrounded by covalently linked stabilizing polymer chains (hairs). For their synthesis conventional (RCC) but also controlled radical crosslinking polymerization (cRCC) techniques have been used (see Fig. 3). For instance, PEGylated nano-/microgels (i.e. coated by poly(ethylene glycol) chains) were intensively studied by research groups interested in biomedical applications due to the dispersing, protecting and biocompatibility properties of poly(ethylene glycol) (PEG). For more specifications on PEGylated microgels see recent reviews and cited references.23,24,129
Commonly, hairy nano-/microgels are prepared by conventional radical crosslinking polymerization (RCC) in heterogeneous conditions in the presence of stabilizing macromonomers (macromonomer approach) (Fig. 3). The latter are necessarily soluble in the continuous phase as they will form a stabilizing “shell” around the crosslinked core. For instance, PNiPAm microgels coated by a PEG shell were prepared by aqueous radical precipitation polymerization in the presence of poly(ethylene glycol) methacrylate macromonomers.130,131 At temperatures above the LCST of PNiPAm, the PNiPAm core collapses but no particle aggregation occurs as the macromonomer remains soluble and contributes to the stabilization of the microgel. Other groups used the same PEG macromonomer for the synthesis of hairy PEGylated microgels based on different monomers such as 2-(diethylamino)ethyl methacrylate (DEA),132,133 2-(diisopropylamino)ethyl methacrylate (DPA)132 and vinylpyridine (VP).134–140 For the surfactant-free synthesis of microgels in dispersed media, the use of macromonomers as reactive polymeric stabilizer allows to reach higher solids content (up to 7% compared to ∼1 to 3% for syntheses performed in the absence of surfactant and macromonomers) and better colloidal stability. The combination of macromonomers and charged surfactants allows the synthesis of microgels at very solids content.132 In addition to neutral macromonomers, such as PEG, charged (polyelectrolyte) macromonomers have also been used.132,141 Poly(2-vinylpyridine) microgels stabilized by cationic poly[2-(dimethylamino)ethyl methacrylate] (PDMAEMA) chains were synthesized in this way. As expected, the pH was shown to be a critical parameter for the synthesis and for the physico-chemical properties of the microgels. Due to the difference of pKa between the polymers in the core and in the shell, P2VP/PDMAEMA microgels can be consequently considered as double pH-sensitive microgels.
The main disadvantage of the macromonomer approach using heterogeneous polymerization processes is the presence of residual macromonomer in the continuous phase. One way to tackle this problem is the use of azo-macroinitiators that decompose thermally, such as PEG–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–PEG. In addition, the application of controlled radical crosslinking copolymerization (cRCC) techniques such as ATRP,100 RAFT91,92 or NMP88,89 has proved to be a very straightforward approach to reach hairy nanogels at high solids content (Fig. 3). For instance, PEGylated thermosensitive nano-/microgels were directly synthesized by ATRP or RAFT using reversible PEG macroinitiators (e.g. poly(ethylene glycol bromoisobutyrate))100 or PEG macroRAFT agents.92
N–PEG. In addition, the application of controlled radical crosslinking copolymerization (cRCC) techniques such as ATRP,100 RAFT91,92 or NMP88,89 has proved to be a very straightforward approach to reach hairy nanogels at high solids content (Fig. 3). For instance, PEGylated thermosensitive nano-/microgels were directly synthesized by ATRP or RAFT using reversible PEG macroinitiators (e.g. poly(ethylene glycol bromoisobutyrate))100 or PEG macroRAFT agents.92
Charleux and co-workers proposed an elegant surfactant-free approach to reach very small thermosensitive poly(N,N′-diethylacrylamide) (PDEAm) nanogels that are coated by a pH-responsive polymer brush of poly(acrylic acid); in a one-pot synthesis DEAm was copolymerized with MBA in the presence of a poly(sodium acrylate) (PNaA) SG1-based macroalkoxyamine that plays the role of both the macroinitiator and stabilizer. When the crosslinker was introduced from the beginning (one batch conditions), macrogelation was observed with ≥3% of crosslinker. In contrast, when the crosslinker was not added at the beginning of the polymerization but only in a second stage, nanogels could be obtained at high solids content (up to 20%) and with high amounts of crosslinker (up to 9%). Using NMP but in homogeneous conditions in DMF, Hawker et al. reported the synthesis of PEGylated poly(N,N-dimethylacrylamide) (PDMA) star nanogels comporting reactive N-hydroxysuccinimide (NAS) groups that are available for bioconjugation. Here, the crosslinked PDMA core is coated by a “double shell” composed of a PEG-b-P(DMA-co-NAS) block copolymers.89 RAFT has also been used for the direct synthesis of hairy nano-/microgels. Hydrosoluble poly(N,N-dimethylacrylamide) (PDMAm)92 or poly(ethylene glycol)-b-poly(N,N-dimethylacrylamide) (PEG-b-PDMAm)92 macromolecular RAFT agents were used that made possible the preparation of stable sub-100 nm thermosensitive gel particles in a surfactant-free aqueous dispersion polymerization process. For both examples, a minimum chain length for the stabilizing (macroRAFT) agent was necessary in order to avoid the formation of aggregates or macrogelation at high solids content. Indeed for short macroRAFT agents, the forming nanogels were not sterically stabilized enough and aggregation occurred during polymerization yielding in interparticle crosslinking and the formation of heterogeneous dispersions.
Many groups in different application fields and especially in the biomedical prepare hairy nano-/microgels by conventional radical precipitation polymerization—via the macromonomer approach—because of the easiness of this technique. However, controlled radical crosslinking polymerization (cRCC) allows the synthesis of stable hairy gel nanoparticles at high solids content (up to 20%) without the necessity of using surfactants, as reversible macroinitiators/macroRAFT agents may play the double role of polymerization controlling agent and colloidal stabilizers. Indeed, such conditions open the way to large scale productions and thus industrial applications.
Similar to the synthesis of core and shell crosslinked microgels (illustrated in Fig. 3) they can be prepared by two-step precipitation polymerizations using degradable crosslinkers for the synthesis of the core. A common method consists in preparing first degradable cores (composed of PNiPAm) that are synthesized by RCC using for instance N,N′-(1,2-dihydroxy ethylene) bisacrylamide (DHEA) as degradable crosslinker. The next step consists in the polymerization of a crosslinked hydrogel-like shell around the collapsed core (i.e. at a temperature above the LCST of NiPAm) using non-degradable crosslinkers, e.g. MBA or DVB. Then, the (degradable) core–crosslinker is cleaved, e.g. by NaIO4 in the case of DHEA, yielding short polymer chains that can diffuse out of the remaining shell in its swollen state at T < LCST. Hollow PNiPAm microgels (microcapsules) are thus obtained. Apart from thermosensitive PNiPAm18 hollow microgels, double-sensitive glucose- and thermoresponsive poly(N-isopropylacrylamide-co-phenylboronic acid)146 poly(NiPAM-PBA) microcapsules have also been synthesized with the same templating process.
An original approach based on interfacial polymerization was developed by Deng and Sun for the synthesis of hollow PNiPAm microgels.147 They were synthesized in inverse emulsion in the presence of surfactants as shown schematically in Fig. 4. The polymerization was initiated at the oil/water interface by using a redox initiation system containing benzoyl peroxide in the oil phase and tetraethylenepentamine in the aqueous phase. As the polymerization is carried out above the LCST of PNiPAm, the forming polymer chains collapse and confine at the interface. Thermosensitive microgels of several microns (however, with a broad size distribution) were prepared and the hollow structure with a wall thickness of ca. 100 nm was confirmed by freeze-fracture microscopy (Fig. 4).
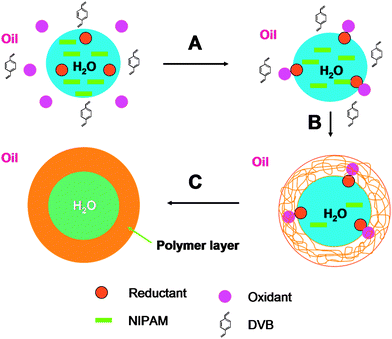 | ||
| Fig. 4 One step synthesis of poly(N-isopropylacrylamide) hollow microgels via interfacial polymerization with an inverse emulsion polymerization approach. (Reprinted with permission from ref. 147.) | ||
Finally, miniemulsion,148 microfluidic devices149 and membrane emulsification150 can also be used for the synthesis of monodisperse hollow microgels. In the two last approaches large microgels are obtained, where monomer and crosslinker are confined in the aqueous droplets while photoinitiator is located in the continuous oil phase. Then, polymerization initiated by UV-irradiation occurs at the oil/water interface. With microfluidic processes, the size of hollow microgels can be modulated from 50 to 80 µm depending on the surfactant content and the flow rates of the water and oil phases. Smaller capsules could be reached by combining miniemulsion polymerization processes with CRP. Thanks to the use of a PEO ATRP macroinitiator, Matyjaszewski et al. synthesized recently hairy nanocapsules made of crosslinked poly(butyl methacrylate) surrounded by a poly(ethylene glycol) corona.151 Similarly, Schork et al. used a PEO macroRAFT agent to reach thermosensitive PNIPAm nanocapsules in inverse miniemulsion, but in the absence of crosslinker.152
IV Conclusions and perspectives
Nano-/microgels have become a fascinating new class of polymeric materials that find application in a large variety of fields. They are easily obtained by radical crosslinking polymerization. In this review, special focus was made on the synthetic parameters that influence the structure of the resulting gel particles. Differences between conventional (uncontrolled) (RCC) and controlled radical crosslinking polymerization (cRCC) in synthesizing nano-/microgels were highlighted. It was shown that both polymerization techniques demonstrate advantages and drawbacks, and they do not necessarily allow the synthesis of nano-/microgels with the same structural features (Fig. 3 and Table 1). Depending on the targeted application and on the requested properties (size, polydispersity, architecture, functionality,…) RCC or cRCC is preferable. It was further stressed that the process of polymerization, i.e. polymerization in homogeneous conditions (in solution) or in heterogeneous conditions (such as (mini)emulsion or dispersion polymerization processes), is a key point in determining the size, polydispersity and morphology of the resulting nano-/microgels. Current developments tend to prepare crosslinked objects of more and more complex morphologies that are able to respond to various stimuli. The comprehension of the mechanisms that govern their formation is crucial and the development of reliable simulation models seems necessary in order to predict and develop new straightforward synthetic approaches that are feasible at large production scales in order to bring these fascinating products on the market.Acknowledgements
We thank Prof. B. Charleux for valuable discussion and proofreading.Notes and references
- A. D. McNaught and A. Wilkinson, IUPAC, Compendium of Chemical Terminology (The “Gold Book”), Blackwell Scientific Publications, Oxford, 2nd edn, 1997 Search PubMed.
- H. Staudinger and E. Husemann, Chem. Ber., 1935, 68, 1618–1634 CrossRef.
- W. O. Baker, Ind. Eng. Chem., 1949, 41, 511–520 CrossRef CAS.
- M. J. Snowden, B. Z. Chowdhry, B. Vincent and G. E. Morris, J. Chem. Soc., Faraday Trans., 1996, 92, 5013–5016 RSC.
- C. D. Jones and L. A. Lyon, Macromolecules, 2000, 33, 8301–8306 CrossRef CAS.
- B. R. Saunders and B. Vincent, Adv. Colloid Interface Sci., 1999, 80, 1–25 CrossRef CAS.
- M. Das, H. Zhang and E. Kumacheva, Annu. Rev. Mater. Res., 2006, 36, 117–142 CrossRef CAS.
- M. Das, N. Sanson and E. Kumacheva, Chem. Mater., 2008, 20, 7157–7163 CrossRef CAS.
- J. K. Oh, D. J. Siegwart, H.-i. Lee, G. Sherwood, L. Peteanu, J. O. Hollinger, K. Kataoka and K. Matyjaszewski, J. Am. Chem. Soc., 2007, 129, 5939–5345 CrossRef CAS.
- J. Zhang, S. Xu and E. Kumacheva, J. Am. Chem. Soc., 2004, 126, 7908–7914 CrossRef CAS.
- K. Min, W. Dayang, H. Bao, M. Gao, H. Möhwald and M. Jiang, Adv. Mater., 2005, 17, 267–270 CrossRef CAS.
- S. Auer and D. Frenkel, Nature, 2001, 413, 711–713 CrossRef CAS.
- A. S. J. Iyer and L. A. Lyon, Angew. Chem., Int. Ed., 2009, 48, 4562–4566 CrossRef CAS.
- K. Welser, M. D. Ayal, J. W. P. Aylott and W. C. Chan, Chem. Commun., 2009, 6601–6603 RSC.
- H. Senff and W. Richtering, J. Chem. Phys., 1999, 111, 1705–1711 CrossRef CAS.
- C. D. Jones and L. A. Lyon, Macromolecules, 2003, 36, 1988–1993 CrossRef CAS.
- J. Kim, N. Singh and L. A. Lyon, Angew. Chem., Int. Ed., 2006, 45, 1446–1449 CrossRef CAS.
- S. Nayak, D. Gan, M. J. Serpe and L. A. Lyon, Small, 2005, 1, 416–421 CrossRef CAS.
- T. He, D. J. Adams, M. F. Butler, C. T. Yeoh, A. I. Cooper and S. P. Rannard, Angew. Chem., Int. Ed., 2007, 46, 9243–9247 CrossRef CAS.
- T. He, D. J. Adams, M. F. Butler, A. I. Cooper and S. P. Rannard, J. Am. Chem. Soc., 2009, 131, 1495–1501 CrossRef CAS.
- J.-W. Kim, R. J. Larsen and D. A. Weitz, Adv. Mater., 2007, 19, 2005–2009 CrossRef CAS.
- J. K. Oh, D. I. Lee and J. M. Park, Prog. Polym. Sci., 2009, 34, 1261–1282 CrossRef CAS.
- K. Raemdonck, J. Demeester and S. De Smedt, Soft Matter, 2009, 5, 707–715 RSC.
- A. V. Kabanov and S. V. Vinbogradov, Angew. Chem., Int. Ed., 2009, 48, 5418–5429 CrossRef CAS.
- G. E. Francis, C. Delgado, D. Fisher, F. Malik and A. K. Agrawal, J. Drug Targeting, 1996, 3, 321–340 Search PubMed.
- G. Wulff, B.-O. Chong and U. Kolb, Angew. Chem., Int. Ed., 2006, 45, 2955–2958 CrossRef CAS.
- S. Nayak and L. A. Lyon, Angew. Chem., Int. Ed., 2005, 44, 7686–7708 CrossRef CAS.
- L. A. Lyon, Z. Meng, N. Singh, C. D. Sorrell and A. S. John, Chem. Soc. Rev., 2009, 38, 865–874 RSC.
- H. Gao and K. Matyjaszewski, Prog. Polym. Sci., 2009, 34, 317–350 CrossRef CAS.
- J. K. Oh, R. Drumright, D. J. Siegwart and K. Matyjaszewski, Prog. Polym. Sci., 2008, 33, 448–477 CrossRef CAS.
- R. Pelton, Adv. Colloid Interface Sci., 2000, 85, 1–33 CrossRef CAS.
- B. R. Saunders, N. Laajam, E. Daly, S. Teow, X. Hu and R. Stepto, Adv. Colloid Interface Sci., 2009, 147–148, 251–262 CrossRef CAS.
- A. Guo, G. J. Liu and J. Tao, Macromolecules, 1996, 29, 2487–2493 CrossRef CAS.
- K. B. Thurmond, T. Kowalewski and K. L. Wooley, J. Am. Chem. Soc., 1996, 118, 7239–7240 CrossRef.
- E. S. Read and S. P. Armes, Chem. Commun., 2007, 3021–3035 RSC.
- J. P. Rolland, B. W. Maynor, L. E. Euliss, A. E. Exner, G. M. Denison and J. M. DeSimone, J. Am. Chem. Soc., 2005, 127, 10096–10100 CrossRef CAS.
- J. Yeh, Y. B. Ling, J. M. Karp, J. Gantz, A. Chandawarkar, G. Eng, J. Blumling, R. Langer and A. Khademhosseini, Biomaterials, 2006, 27, 5391–5398 CrossRef CAS.
- S. Xu, Z. Nie, M. Seo, P. Lewis, E. Kumacheva, H. A. Stone, P. Garstecki, D. B. Weibel, I. Gitlin and G. M. Whitesides, Angew. Chem., Int. Ed., 2005, 44, 724–728 CrossRef CAS.
- R. K. Shah, J. W. Kim, J. J. Agresti, D. A. Weitz and L. Y. Chu, Soft Matter, 2008, 4, 2303–2309 RSC.
- W. Funke, O. Okay and B. Joos-muller, Adv. Polym. Sci., 1998, 136, 139–234.
- H. Gao, A. Miasnikova and K. Matyjaszewski, Macromolecules, 2008, 41, 7843–7849 CrossRef CAS.
- H. Gao, L. Wenwen and K. Matyjaszewski, Macromolecules, 2008, 41, 2335–3340 CrossRef CAS.
- I. Bannister, N. C. Billingham, S. P. Armes, S. P. Rannard and P. Findlay, Macromolecules, 2006, 39, 7483–7492 CrossRef CAS.
- M. Antonietti, Angew. Chem., Int. Ed. Engl., 1988, 27, 1743–1747 CrossRef.
- J. K. Oh, S. A. Bencherif and K. Matyjaszewski, Polymer, 2009, 50, 4407–4423 CrossRef CAS.
- P. B. Zetterlund, Y. Kagawa and M. Okubo, Chem. Rev., 2008, 108, 3747–3794 CrossRef CAS.
- G. Odian, Principes of Polymerization, Wiley, New Jersey, 4th edn, 2004 Search PubMed.
- Annotation of the authors: A controversial terminology is used for the polymerization of NiPAm in water above the LCST of PNiPAM. It is generally referred to as precipitation polymerization, even when the polymerization is conducted in the presence of a surfactant, i.e. according to polymerists' definition it should be called dispersion polymerization. For sake of clarity and in accordance with most of the authors we will here refer to the polymerization of NiPAm in water as precipitation polymerization.
- B. Charleux and F. Ganachaud, in Macromolecular Engineering: Precise Synthesis, Materials Properties, Applications, ed. K. Matyjaszewski, Y. Gnanou and L. Leibler, Wiley, Weinheim, 2007, vol. 1, ch. 14, pp. 605–642 Search PubMed.
- J. Qiu, B. Charleux and K. Matyjaszewski, Prog. Polym. Sci., 2001, 26, 2083–2134 CrossRef CAS.
- J. W. Stanbury, M. Trujillo-Lemon, X. Ding and S. M. Newman, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2006, 47, 825–826.
- O. Okay and W. Funke, Macromolecules, 1990, 23, 2623–2628 CrossRef CAS.
- L. Pille and D. H. Solomon, Macromol. Chem. Phys., 1994, 195, 2477–2489 CrossRef CAS.
- Handbook of Radical Polymerization, ed. K. Matyjaszewski and T. P. Davis, Wiley, Hoboken, 2002 Search PubMed.
- M. K. Georges, R. P. N. Veregin, P. M. Kazmaier and G. K. Hamer, Macromolecules, 1993, 26, 2987–2988 CrossRef CAS.
- J.-S. Wang and K. Matyjasewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963–5050 CrossRef CAS.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689–3745 CrossRef CAS.
- K. Matyjaszewski, S. Gaynor and J.-S. Wang, Macromolecules, 1995, 28, 2093–2095 CrossRef.
- P. Lacroix-Desmazes, R. Severac and B. Boutevin, Macromolecules, 2005, 38, 6299–6309 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- Handbook of RAFT Polymerization, ed. C. Barner-Kowollik, Wiley, Weinheim, 2008 Search PubMed.
- D. Charmot, P. Corpart, H. Adam, S. Z. Zard, T. Biadatti and G. Bouhadir, Macromol. Symp., 2000, 150, 23–32 CrossRef CAS.
- A. Favier and M.-T. Charreyre, Macromol. Rapid Commun., 2006, 27, 653–692 CrossRef CAS.
- N. Ide and T. Fukuda, Macromolecules, 1999, 32, 95–99 CrossRef CAS.
- N. Ide and T. Fukuda, Macromolecules, 1997, 30, 4268–4271 CrossRef CAS.
- S. Abrol, P. A. Kambouris, M. G. Looney and D. H. Solomon, Macromol. Rapid Commun., 1997, 18, 755–760 CrossRef CAS.
- S. Abrol, M. J. Caulfield, G. G. Qiao and D. H. Solomon, Polymer, 2001, 42, 5987–5991 CrossRef CAS.
- C. Jiang, Y. Shen, S. Zhu and D. Hunkeler, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3780–3788 CrossRef CAS.
- Q. Liu, Z. Ping, A. Qing, Y. Lan and M. Lu, Polymer, 2006, 47, 2330–2336 CrossRef CAS.
- H. Gao, K. Min and K. Matyjaszewski, Macromolecules, 2007, 40, 7763–7770 CrossRef CAS.
- A. R. Wang and S. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5710–5714 CrossRef CAS.
- K. Matyjaszewski and W. A. Braunecker, in Macromolecular Engineering: Precise Synthesis, Materials Properties, Applications, ed. K. Matyjaszewski, Y. Gnanou and L. Leibler, Wiley, Weinheim, 2007, vol. 1, ch. 5, pp. 161–215 Search PubMed.
- I. Bannister, N. C. Billingham and S. P. Armes, Soft Matter, 2009, 5, 3495–3504 RSC.
- B. Liu, A. Kazlauciunas, J. T. Guthrie and S. Perrier, Polymer, 2005, 46, 6293–6299 CrossRef CAS.
- J. Poly, D. J. Wilson, M. Destarac and D. Taton, Macromol. Rapid Commun., 2008, 29, 1965–1972 CrossRef CAS.
- J. Poly, D. Wilson, M. Destarac and D. Taton, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5313–5327 CrossRef CAS.
- B. Liu, A. Kazlauciunas, J. T. Guthrie and S. Perrier, Macromolecules, 2005, 38, 2131–2136 CrossRef.
- H. Gao, P. Polanowski and K. Matyjaszewski, Macromolecules, 2009, 42, 5925–5932 CrossRef CAS.
- J. Rosselgong, S. P. Armes, W. Barton and D. Price, Macromolecules, 2009, 42, 5919–5924 CrossRef CAS.
- J. K. Oh, C. Tang, H. Gao, N. V. Tsarevsky, D. J. Siegwart, G. Sherwood, L. Peteanu and K. Matyjaszewski, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2006, 47, 945–946 CAS.
- Y. Li and S. P. Armes, Macromolecules, 2005, 38, 8155–8162 CrossRef CAS.
- A. R. Wang and S. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5710–5714 CrossRef CAS.
- A. Cutivet, C. Schembri, J. Kovensky and K. Haupt, J. Am. Chem. Soc., 2009, 131, 14699–14702 CrossRef CAS.
- D. Taton, J. F. Baussard, L. Dupayage, J. Poly, Y. Gnanou, V. Ponsinet, M. Destarac, C. Mignaud and C. Pitois, Chem. Commun., 2006, 1953–1955 RSC.
- J. K. Oh, C. Tang, H. Gao, N. V. Tsarevsky and K. Matyjaszewski, J. Am. Chem. Soc., 2006, 128, 5578–5584 CrossRef CAS.
- J. K. Oh, D. J. Siegwart and K. Matyjaszewski, Biomacromolecules, 2007, 8, 3326–3331 CrossRef CAS.
- G. Delaittre, M. Save and B. Charleux, Macromol. Rapid Commun., 2007, 28, 1528–1533 CrossRef CAS.
- K.-I. Fukukawa, R. Rossin, A. Hagooly, E. D. Pressly, J. N. Hunt, B. W. Messmore, K. L. Wooley, M. J. Welch and C. J. Hawker, Biomacromolecules, 2008, 9, 1329–1339 CrossRef CAS.
- C.-D. Vo, J. Rosselgong and S. P. Armes, Macromolecules, 2007, 40, 7119–7125 CrossRef CAS.
- Z. An, Q. Shi, W. Tang, C.-K. Tsung, C. J. Hawker and G. D. Stucky, J. Am. Chem. Soc., 2007, 129, 14493–14499 CrossRef CAS.
- J. Rieger, C. Grazon, B. Charleux, D. Alaimo and C. Jérôme, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2373–2390 CrossRef CAS.
- Y. Chan, V. Bulmus, M. H. Zareie, F. L. Byrne, L. Barner and M. Kavallaris, J. Controlled Release, 2006, 115, 197–207 CrossRef CAS.
- G. Zheng and C. Pan, Macromolecules, 2006, 39, 95–102 CrossRef CAS.
- P. J. Flory, Principles of Polymer Chemistry, Cornell University Press, New York, 1953 Search PubMed.
- C. Pichot, A. Elaïssari, D. Duracher, F. Meunier and F. Sauzedde, Macromol. Symp., 2001, 175, 285–297 CrossRef CAS.
- T. Sato, N. Higashida, T. Hirano and M. Seno, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 1609–1617 CrossRef CAS.
- H. Kawaguchi, M. Kawahara, N. Yaguchi, F. Hoshino and Y. Ohtsuka, Polym. J. (Tokyo), 1988, 20, 903–909 Search PubMed.
- Z. An, W. Tang, C. J. Hawker and G. D. Stucky, J. Am. Chem. Soc., 2006, 128, 15054–15055 CrossRef CAS.
- K. H. Kim, J. Kim and W. H. Jo, Polymer, 2005, 46, 2836–2840 CrossRef CAS.
- C. D. Jones and L. A. Lyon, Macromolecules, 2000, 33, 8301–8306 CrossRef CAS.
- H. Kawaguchi, Y. Yamada, S. Kataoka, Y. Morita and Y. Ohtsuka, Polym. J. (Tokyo), 1991, 23, 955–962 Search PubMed.
- J. Rieger, G. Osterwinter, C. Bui, F. Stoffelbach and B. Charleux, Macromolecules, 2009, 42, 5518–5525 CrossRef CAS.
- C. Dire, S. Magnet, L. Couvreur and B. Charleux, Macromolecules, 2009, 42, 95–103 CrossRef CAS.
- Z. Meng, J. K. Cho, S. Debord, V. Breedveld and L. A. Lyon, J. Phys. Chem. B, 2007, 111, 6992–6997 CrossRef CAS.
- Z. Meng, M. H. Smith and L. A. Lyon, Colloid Polym. Sci., 2009, 287, 277–285 CrossRef CAS.
- X. Wu, R. H. Pelton, A. E. Hamielec, D. R. Woods and W. McPhee, Colloid Polym. Sci., 1994, 272, 467–477 CrossRef CAS.
- J. S. Song and M. A. Winnik, Macromolecules, 2006, 39, 8318–8325 CrossRef CAS.
- T. Hoare and D. McLean, J. Phys. Chem. B, 2006, 110, 20327–20336 CrossRef CAS.
- T. Hoare and R. Pelton, Curr. Opin. Colloid Interface Sci., 2008, 13, 413–428 CrossRef CAS.
- S. Meyer and W. Richtering, Macromolecules, 2005, 38, 1517–1519 CrossRef CAS.
- A. Musyanovych and K. Landfester, in Macromolecular Engineering: Precise Synthesis, Materials Properties, Applications, ed. K. Matyjaszewski, Y. Gnanou and L. Leibler, Wiley, Weinheim, 2007, vol. 2, ch. 13, pp. 1209–1247 Search PubMed.
- W. Xue, S. Champ and M. B. Huglin, Polymer, 2000, 41, 7575–7581 CrossRef CAS.
- C. D. Jones and L. A. Lyon, Langmuir, 2003, 19, 4544–4547 CrossRef CAS.
- X. Li, J. Zuo, Y. Guo and X. Yuan, Macromolecules, 2004, 37, 10042–10046 CrossRef CAS.
- M. Bradley and B. Vincent, Langmuir, 2008, 24, 2421–2425 CrossRef CAS.
- V. Lapeyre, C. Ancla, B. Catargi and V. Ravaine, J. Colloid Interface Sci., 2008, 327, 316–323 CrossRef CAS.
- K. E. Christodoulakis and M. Vamvakaki, Langmuir, 2010, 26, 639–647 CrossRef CAS.
- D. Suzuki, J. G. McGrath, H. Kawaguchi and L. A. Lyon, J. Phys. Chem. C, 2007, 111, 5667–5672 CrossRef CAS.
- D. Duracher, A. Elaissari and C. Pichot, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 1823–1837 CrossRef CAS.
- I. Berndt and W. Richtering, Macromolecules, 2003, 36, 8780–8785 CrossRef CAS.
- I. Berndt, C. Popescu, F.-J. Wortmann and W. Richtering, Angew. Chem., Int. Ed., 2006, 45, 1081–1085 CrossRef CAS.
- I. Berndt, J. S. Pedersen and W. Richtering, J. Am. Chem. Soc., 2005, 127, 9372–9373 CrossRef CAS.
- I. Berndt, J. S. Pedersen and W. Richtering, Angew. Chem., Int. Ed., 2006, 45, 1737–1741 CrossRef CAS.
- I. Berndt, J. S. Pedersen, P. Lindner and W. Richtering, Langmuir, 2006, 22, 459–468 CrossRef CAS.
- D. Gan and L. A. Lyon, J. Am. Chem. Soc., 2001, 123, 7511–7517 CrossRef CAS.
- D. Gan and L. A. Lyon, J. Am. Chem. Soc., 2001, 123, 8203–8209 CrossRef CAS.
- J. Wang, D. Gan, L. A. Lyon and M. A. El-Sayed, J. Am. Chem. Soc., 2001, 123, 11284–11289 CrossRef CAS.
- M. Oishi and Y. Nagasaki, React. Funct. Polym., 2007, 67, 1311–1329 CrossRef CAS.
- D. Gan and L. A. Lyon, Macromolecules, 2002, 35, 9634–9639 CrossRef CAS.
- N. Singh and L. A. Lyon, Colloid Polym. Sci., 2008, 286, 1061–1069 CrossRef CAS.
- J. I. Amalvy, E. J. Wanless, Y. Li, V. Michailidou, S. P. Armes and Y. Duccini, Langmuir, 2004, 20, 8992–8999 CrossRef CAS.
- D. Palioura, S. P. Armes, S. H. Anastasiadis and M. Vamvakaki, Langmuir, 2007, 23, 5761–5768 CrossRef CAS.
- D. Dupin, S. Fujii, S. P. Armes, P. Reeve and S. M. Baxter, Langmuir, 2006, 22, 3381–3387 CrossRef CAS.
- D. Dupin, J. Rosselgong, S. P. Armes and A. F. Routh, Langmuir, 2007, 23, 4035–4041 CrossRef CAS.
- P. A. Fitzgerald, D. Dupin, S. P. Armes and E. J. Wanless, Soft Matter, 2007, 3, 580–586 RSC.
- J. Yin, D. Dupin, J. Li, S. P. Armes and S. Liu, Langmuir, 2008, 24, 9334–9340 CrossRef CAS.
- D. Dupin, J. R. Howse, S. P. Armes and D. P. Randall, J. Mater. Chem., 2008, 18, 545–552 RSC.
- S. Fujii, D. Dupin, T. Araki, S. P. Armes and H. Ade, Langmuir, 2009, 25, 2588–2592 CrossRef CAS.
- W.-M. Wan and C.-Y. Pan, Macromolecules, 2007, 40, 8897–8905 CrossRef CAS.
- D. Dupin, S. P. Armes, C. Connan, P. Reeve and S. M. Baxter, Langmuir, 2007, 23, 6903–6910 CrossRef CAS.
- H. Tamber, P. Johansen, H. P. Merkle and B. Gander, Adv. Drug Delivery Rev., 2005, 57, 357–376 CrossRef CAS.
- G. Sukhorukov, A. Fery and H. Mohwald, Prog. Polym. Sci., 2005, 30, 885–897 CrossRef CAS.
- D. A. Hammer and D. E. Disher, Annu. Rev. Mater. Res., 2001, 31, 387–404 CrossRef CAS.
- L. X. Wen, L. A. Archer and Z. Yang, Adv. Mater., 2008, 20, 3987–4019 CrossRef CAS.
- Y. Zhang, Y. Guan and S. Zhou, Biomacromolecules, 2007, 8, 3842–3847 CrossRef CAS.
- Q. Sun and Y. Deng, J. Am. Chem. Soc., 2005, 127, 8274–8275 CrossRef CAS.
- K. Landfester, Angew. Chem., Int. Ed., 2009, 48, 4488–4507 CrossRef CAS.
- C.-H. Choi, J.-H. Jung, D.-W. Kim, Y.-M. Chung and C.-S. Lee, Lab Chip, 2008, 8, 1544–1551 RSC.
- C.-J. Cheng, L.-Y. Chu, P.-W. Ren, J. Zhang and L. Hu, J. Colloid Interface Sci., 2007, 313, 383–388 CrossRef CAS.
- W. Li, K. Matyjaszewski, K. Albrecht and M. Möller, Macromolecules, 2009, 42, 8228–8233 CrossRef CAS.
- F. Lu, Y. Luo, B. Li, Q. Zhao and F. J. Schork, Macromolecules, 2010, 43, 568–571 CrossRef CAS.
- D. Lee and D. A. Weitz, Small, 2009, 5, 1932–1935 CrossRef CAS.
- L.-Y. Chu, J.-W. Kim, R. K. Shah and D. A. Weitz, Adv. Funct. Mater., 2007, 17, 3499–3504 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |