Self-assembly and thermodynamic synthesis of rotaxane dendrimers and related structures
Ken Cham-Fai
Leung
* and
Kwun-Ngai
Lau
Center of Novel Functional Molecules, Institute of Molecular Functional Materials, Department of Chemistry, The Chinese University of Hong Kong, Shatin, NT, Hong Kong SAR, P. R. China. E-mail: cfleung@cuhk.edu.hk; Fax: (+852) 2603-5057; Tel: (+852) 2609-6342
First published on 24th May 2010
Abstract
The marriage between dendrimers and rotaxanes has been a topic of study. The use of noncovalent interactions to join dendritic fragments together to form rotaxane dendrimers could be more effective than traditional dendrimer synthesis using covalent bond formation reactions. Underpinning the fundamental study of rotaxane dendrimers would shed light on understanding and tuning the properties of mechanical bonds in a sterically hindered, dendritic environment. In this review article, we describe the classification and self-assembly of rotaxane dendrimers as well as the latest developments in thermodynamic synthesis of rotaxane dendrimers.
 Ken Cham-Fai Leung | Ken Cham-Fai Leung received his Bachelor and Doctorate degrees in chemistry from the Chinese University of Hong Kong in 1999 and 2003, respectively. Subsequently, he worked as a postdoctoral fellow for three years in the California NanoSystems Institute and the Department of Chemistry and Biochemistry, The University of California Los Angeles, USA. In September 2006, he joined the Center of Novel Functional Molecules at the Department of Chemistry, The Chinese University of Hong Kong as a research assistant professor. His current research is focusing on the synthesis and properties of polymeric nanomaterials for biological and drug delivery applications. |
1. Introduction
Dendrons and dendrimers are hyperbranched macromolecules with well-defined three-dimensional architectures.1 A dendron is a highly branched sector with surface groups at the periphery, branching units and a focal point. On the other hand, a dendrimer is another type of dendritic molecule with two or more dendrons attaching to a multivalent core molecule. The synthesis of this kind of macromolecule, with nanoscale size ranges, is commonly based on a bottom-up approach. That is, they can be synthesized using various molecular bits and pieces followed by linking and coupling with appropriate building blocks to afford desired dendritic assemblies. Dendrons/dendrimers are prepared mainly by either divergent2 or convergent3 synthetic approaches. In theory, dendrimers/dendrons are monodispersed rather than possessing molecular weight distributions as in the case of traditional polymers. Generally, the structures of dendrimers/dendrons change from the extended form to globular conformations with increasing generations due to steric crowding.4 Their properties also vary with functionalities attached at surface groups, interiors or cores/focal points, branching multiplicities (AB2versus AB3),1d polarities of dendrons/dendrimers themselves,5etc. Secondary dendritic structures can be furnished by subsequent dendrimer organizations and assemblies.6 These characteristics make it feasible to apply dendrons/dendrimers in nanomedicines,7 catalysis,8 light-harvesting,9 and as nanocapsules for drug or gene deliveries.10Rotaxanes (from the Latin rota and axis, meaning “wheel” and “axle”, respectively) contain a linear dumbbell-shaped component—bearing bulky end-groups or “stoppers”—around which one or more macrocycles are trapped.11 A pseudorotaxane is a rotaxane without its stoppers. One or more macrocycles on the pseudorotaxane are temporally encircled around an unstoppered thread through noncovalent interactions from which they are readily susceptible to dissociation. A common retro-synthetic disconnection shared by generic [2]rotaxanes invokes a [2]pseudorotaxane precursor, in which a linear molecule is threaded through a macrocyclic one. For [n]rotaxanes, n represents the number of separate components in the mechanically interlocked entities e.g., a [2]rotaxane consists of a dumbbell component and a macrocycle component. Post-assembly modification of the threaded [2]pseudorotaxane can proceed by end-capping of the linear component with sufficiently large groups (stopping), which results in the formation of a [2]rotaxane. This approach is termed as “threading-followed-by-stopping”. A complementary “clipping” strategy, in which an acyclic precursor is cyclized around a dumbbell-shaped template, has also been commonly used for rotaxane synthesis. An alternative approach to prepare rotaxanes, termed “slippage”, proceeds by heating the preformed macrocycle and dumbbell together in order to slip the macrocycle past the bulky stoppers to afford the thermodynamically favoured rotaxane. Recently, new approaches termed “threading-followed-by-shrinking”11b and “threading-followed-by-swelling”11c have been coined. The first approach involves threading a dumbbell-shaped unit through a relatively large macrocycle and then shrinking the free space within the macrocycle through coordination of other moieties (e.g., metal ions and their complexes).11b The latter approach involves threading a half-dumbbell unit through a macrocycle and then enlarging the smaller terminal group into an effective stopper.11c
The marriage between dendrimers and rotaxanes has been a topic of study.12 Intrinsic properties of dendrimer can be rapidly and reversibly altered through pseudorotaxane formation on dendrimer's periphery by direct self-assembly. The use of noncovalent interactions to join dendritic fragments together to form rotaxane dendrimers could be more effective than traditional dendrimer synthesis using covalent bond formation reactions. Moreover, underpinning the fundamental study of rotaxane dendrimers would shed light on understanding and tuning the properties of mechanical bond in sterically hindered, dendritic environment. In this review article, we will describe (1) the classification of rotaxane dendrimers, (2) selected recent examples of rotaxane dendrimers, (3) self-assembly and thermodynamic synthesis of rotaxane dendrimers, and (4) potential applications and perspectives.
2. Pseudorotaxane and rotaxane dendrimers
Classification
Dendrimers can be synthesized in a combination of various core, branching units and/or surface groups. In the same manner, rotaxane and rotaxane-like structures are utilized to build up dendrimers. In the past decade, pseudorotaxane dendrimers were synthesized and reported with many novel structures and properties. An excellent review about rotaxane dendrimers by Lee and Kim was published in 2003.12 In this review, leading and up-to-date examples will be included.As defined by Lee and Kim,12 dendrimers with a rotaxane core are regarded as type I rotaxane dendrimers. According to the positions of those dendron units, type I rotaxane dendrimers can be categorized into I-A, I-B, and I-C in which dendron units are attached to rod, ring, and both ring and rod components, respectively (Fig. 1). Type II dendrimers are pseudorotaxane dendrimers, from which pseudorotaxane features are incorporated at the dendrimer's periphery. Type II dendrimers are divided into two types, namely Type II-A and Type II-B, depending on whether the surface groups of the dendrimers are rod components or ring components, respectively (Fig. 2). Dendritic polyrotaxanes are defined as Type III rotaxane dendrimers in which rotaxane building units grow like a dendrimer. Type III-A dendrimers refer to those with ring components located on the branches, while Type III-B counterparts are those dendrimers with rotaxane units at the branching points (Fig. 3). Based on such definition, therefore, Type I and III dendrimers are either pseudorotaxane dendrimers or rotaxane dendrimers; while Type II dendrimers are pseudorotaxane dendrimers.
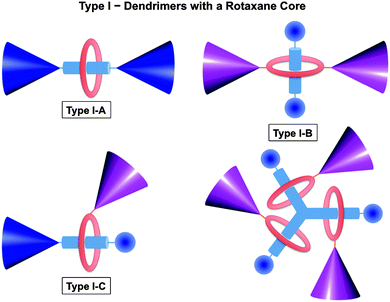 | ||
| Fig. 1 Classification of rotaxane dendrimers: Type I rotaxane dendrimers incorporate rotaxane-like features at the core. Type I rotaxane dendrimers have been sub-categorized into three different types: Type I-A: rotaxane dendrimers with dendron units attached to the rod component; Type I–B: rotaxane dendrimers with dendron units attached to the ring component; and Type I–C: rotaxane dendrimers with dendron units attached to both the ring and rod components. | ||
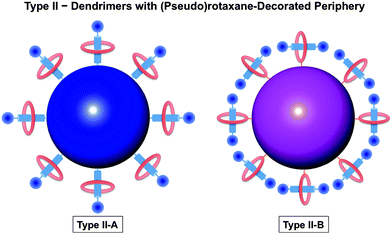 | ||
| Fig. 2 Classification of rotaxane dendrimers: Type II rotaxane dendrimers incorporate rotaxane-like features at the termini. Type II rotaxane dendrimers have been sub-categorized into two different types: Type II-A: (pseudo)rotaxane-terminated dendrimers with covalently-attached rod components at the periphery; and Type II–B: (pseudo)rotaxane-terminated dendrimers with covalently-attached ring components at the periphery. | ||
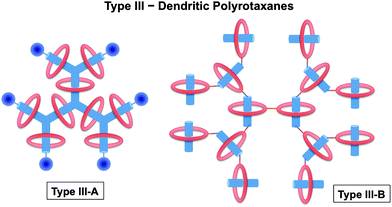 | ||
| Fig. 3 Classification of rotaxane dendrimers: Type III rotaxane dendrimers incorporate rotaxane-like features at the branches. Type III rotaxane dendrimers have been sub-categorized into two different types: Type III-A: dendritic polyrotaxanes incorporating ring components on the branches; and Type III–B: dendritic polyrotaxanes incorporating ring components at the branching points. | ||
In comparison, pseudorotaxane dendrimers and rotaxane dendrimers possess different characteristics in terms of their synthetic efficiency, ability for product isolation, stability, and properties. Pseudorotaxane dendrimers, which are formed by self-assembly of their separate components together, offer high efficiency, high specificity, and rapid property tunability. Multiple molecular recognition motifs which operate in a self-sorting manner,13 can be employed in one dendritic system simultaneously. However, the stability of self-assembled pseudorotaxane dendrimers depends on the binding affinities (equilibrium constants) of the recognition motifs, wherein they are subjected to disassembly at high temperature, low concentration and/or competitive binding guests. Furthermore, isolation and purification of pseudorotaxane dendrimers may be a difficult task. This situation can be partially solved by using high fidelity recognition motifs14 with an exact ratio of components in the self-assembling mixture. In contrast to pseudorotaxane dendrimers, rotaxane dendrimers are relatively stable in solution independent of concentration; and are able to be separated by chromatographic methods. One drawback is that the synthesis of rotaxane dendrimers may be time-consuming and give low yields since it involves a self-assembly process between separate components followed by covalent bond forming reaction, e.g., macrocyclization, stoppering.
Cucurbituril-based dendrimers
Cucurbituril (CB) refers to a series of macrocyclic compounds that bear a hydrophobic cavity with two identical carbonyl-fringed portals.15 Besides binding hydrophobic guests inside their cavities through hydrophobic effect in water, in addition, carbonyl groups at both ends can bind cationic species via hydrogen bonding and charge–dipole interactions. Numerous examples were reported for the use of CB in pseudorotaxane synthesis.16 Pseudorotaxane dendrimers with CB units have also been reported in the past decade.17 Recently, Wang and Kaifer have reported18 the synthesis of two different dendrons which bear (Fig. 4) either a para-dialkoxybenzene (1) or a 4,4′-bipyridinium (2) as the focal point unit. Three-component self-assembly between cucurbit[8]uril (CB[8]) host and the two different dendrons afforded a dumbbell-shaped [3]pseudorotaxane dendrimer CB[8]⊃1·2via charge-transfer interactions in water (∼6 mM) at room temperature. This type of [3]pseudorotaxane dendrimer which contains a rotaxane moiety as the core with dendritic rod structure, is defined as type I-A pseudorotaxane dendrimer. The CB[8] host possesses a hydrophobic internal cavity, which promotes the formation of the charge-transfer complex originated from electron-deficient bipyridinium ions (viologen) and electron-rich para-dialkoxybenzene moiety. 1H NMR spectroscopy revealed that the [3]pseudorotaxane dendrimers CB[8]⊃1·2 could be formed only in the presence of CB[8] host. Interestingly, the two different dendrons would not form a stable charge-transfer complex in the absence of CB[8]. This observation may be due to the massive steric hindrance and back-folding of dendrons, leading to a decreased stability of the dendritic charge-transfer complex. Clearly, CB[8] performs two roles. First, it acts as a promoter for the formation of CB[8]⊃1·2 complex and second, a protector for the resulting pseudorotaxane charge-transfer complex upon experiencing dendritic steric crowding. Furthermore, the [3]pseudorotaxane dendrimers CB[8]⊃1·2 exhibited electrochemical switching and redox control properties. Later, Kim and coworkers have extended this idea into the synthesis of new CB[8]-based, [3]pseudorotaxane dendrimers that possess structural modifications on the dendron and the charge-transfer molecular pair.19![[3]Pseudorotaxane dendrimer CB[8]⊃1·2 formation with CB[8] macrocycle ring, dendron with dialkoxybenzene (electron-rich π-donor) 1, and dendron with 4,4′-bipyridinium (electron-deficient π-acceptor) 2.](/image/article/2010/PY/b9py00380k/b9py00380k-f4.gif) | ||
| Fig. 4 [3]Pseudorotaxane dendrimer CB[8]⊃1·2 formation with CB[8] macrocycle ring, dendron with dialkoxybenzene (electron-rich π-donor) 1, and dendron with 4,4′-bipyridinium (electron-deficient π-acceptor) 2. | ||
Li and coworkers reported20 the synthesis of a series of polyamidoamine (PAMAM) dendrimers 3 from generation zero [G0] to generation three [G3] that bear ammonium naphthyl moieties at the periphery. These dendrimers were employed (Fig. 5) to form type II-A pseudorotaxane dendrimers 3⊂(CB[7])n through supramolecular assemblies between the peripheral naphthalene threads and CB[7] macrocycles in water. The interplay between the ammonium naphthyl and CB[7] relies on the hydrophobic effect offered by the naphthyl group and CB[7] cavity, as well as the [N+–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C] hydrogen bonds and ion-dipole interaction. The 1
C] hydrogen bonds and ion-dipole interaction. The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding of the molecular recognition pair—ammonium naphthyl⊂CB[7]—leads to a relatively high affinity constant (Ka) of 5 × 106 M−1. Furthermore, 1H NMR revealed that all the surface naphthyl groups of PAMAM dendrimers were encapsulated by one equivalent of CB[7] per naphthalene group, owing to the complete chemical shifts of the naphthyl proton signals and CB[7] proton signals when compared to their separated components. Furthermore, the partial and full dissociation of CB[7] macrocycles from the naphthyl dendrimers were feasible by a competitive binding mechanism using 1-aminoadamantane, based on the fact that 1-aminoadamantane binds strongly with CB[7], forming a 1:1 inclusion complex with Ka value of 4 × 1012 M−1.
:1 binding of the molecular recognition pair—ammonium naphthyl⊂CB[7]—leads to a relatively high affinity constant (Ka) of 5 × 106 M−1. Furthermore, 1H NMR revealed that all the surface naphthyl groups of PAMAM dendrimers were encapsulated by one equivalent of CB[7] per naphthalene group, owing to the complete chemical shifts of the naphthyl proton signals and CB[7] proton signals when compared to their separated components. Furthermore, the partial and full dissociation of CB[7] macrocycles from the naphthyl dendrimers were feasible by a competitive binding mechanism using 1-aminoadamantane, based on the fact that 1-aminoadamantane binds strongly with CB[7], forming a 1:1 inclusion complex with Ka value of 4 × 1012 M−1.
![Pseudorotaxane dendrimers based on naphthalene peripherally functionalized PAMAM dendrimers and CB[7].](/image/article/2010/PY/b9py00380k/b9py00380k-f5.gif) | ||
| Fig. 5 Pseudorotaxane dendrimers based on naphthalene peripherally functionalized PAMAM dendrimers and CB[7]. | ||
Kim and coworkers21 have recently reported (Fig. 6) an efficient way to obtain [10]pseudorotaxane (second generation, [G2]) dendrimers with 13 molecular components. The [10]pseudorotaxane dendrimer requires the formation of two different pseudorotaxanes with CB[6] and CB[8] macrocycles. To begin with, tetraammonium compound 4 was self-assembled with two CB[6] rings, leading to the formation of a stable [3]pseudorotaxane 4⊂(CB[6])2 through the complexation between CB[6] and butyl diammonium unit. On the other hand, a trifurcated hexapyridinium compound 5 was act as a core. It was self-assembled with three [3]pseudorotaxanes 4⊂(CB[6])2 in the presence of larger CB[8] rings to form the [10]pseudorotaxane superstructure 6 wherein charge-transfer complexes between 2,6-dialkoxynaphthalene and bipyridinium moieties were formed with the encapsulation of a CB[8] ring.22 Consequently, the first of its kind, a type III-A [10]pseudorotaxane dendrimer with pseudorotaxane units at every branch was furnished in high yield with the molecular weight confirmed by mass spectrometry.
![A [10]Pseudorotaxane dendrimer with CB-based pseudorotaxane units at every branch.](/image/article/2010/PY/b9py00380k/b9py00380k-f6.gif) | ||
| Fig. 6 A [10]Pseudorotaxane dendrimer with CB-based pseudorotaxane units at every branch. | ||
Cyclodextrin-based dendrimers
Cyclodextrin (CD) possesses a hydrophobic cavity and a hydrophilic exterior, such arrangement allows it to host hydrophobic guest under aqueous medium. A number of rotaxane, and polyrotaxane systems have been made with CDs.23 Therefore, CD-based rotaxane and polyrotaxane systems that are supported by dendrimers have also been reported.Reinhoudt and coworkers24 described the synthesis of biferrocene-functionalized poly(propylene imine) (PPI) dendrimers 7. Ferrocenes frequently form stable inclusion complexes (pseudorotaxanes) with a wide variety of macrocyclic ring compounds, such as CD23 and CB.16 Complexation of biferrocene-functionalized PPI dendrimers with β-CDs can lead to the formation of a series of Type II-A pseudorotaxane dendrimers (Fig. 7a). The higher association between β-CD and biferrocene as compared to that of ferrocene results in a complete coverage of β-CDs on each biferrocene of the PPI dendrimer 7⊂(β-CD)n (n = 4, 6, 8). On the other hand, the feasibility of biferrocene-functionalized PPI dendrimers 7 adsorbed onto a CD-modified, flat gold surface has also been demonstrated (Fig. 7b). A self-assembled monolayer (SAM) of heptathioether-functionalized β-CD was formed on a flat gold surface. Subsequently, this SAM of CD was used as a platform for self-assembling biferrocene-functionalized PPI dendrimers 7 through multivalent host–guest interactions between the biferrocene end groups of the dendrimer and the immobilized β-CD molecules. Oxidation of the iron centers in these complexes results in the dissociation of the host–guest complexes and desorption of the dendrimers from the host Au-CD surface. Furthermore, the use of these dendrimers as supramolecular reversible glues has been demonstrated in later literatures with high tunability in layer-by-layer SAM formations.25
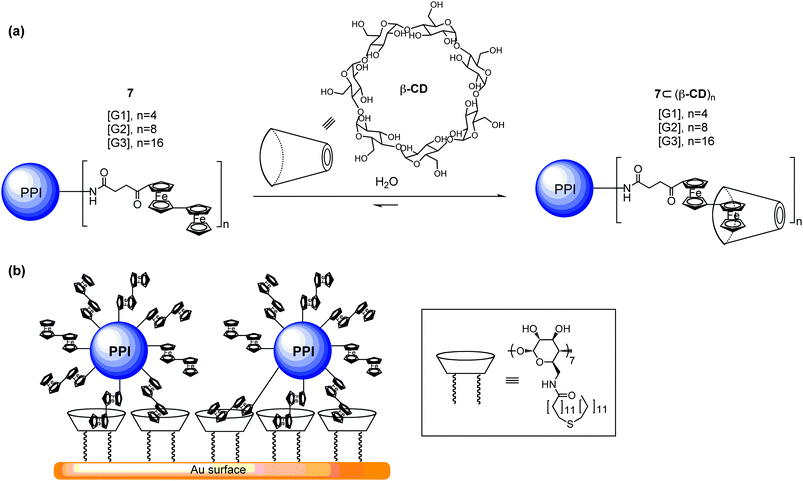 | ||
| Fig. 7 (a) Self-assembly of biferrocene-functionalized PPI dendrimers 7 with β-CDs. (b) Self-assembled monolayer formation between a CD-modified gold surface and the biferrocene-functionalized PPI dendrimer 7. | ||
Huskens and coworkers reported the synthesis of adamantane-functionalized [G5] PPI dendrimers 8 which were further used for the pseudorotaxane formation with β-CDs (Fig. 8) at the dendrimer's periphery.26 Adamantane-functionalized [G5] PPI dendrimers 8 have also been self-assembled onto gold and silicon dioxide nanoparticles with CD-derived SAMs. By using similar self-assembly protocols, layer-by-layer SAM formation on both flat surfaces and spherical particles have been demonstrated.26,27
![Adamantane-functionalized [G5] PPI dendrimers terminated with β-CD.](/image/article/2010/PY/b9py00380k/b9py00380k-f8.gif) | ||
| Fig. 8 Adamantane-functionalized [G5] PPI dendrimers terminated with β-CD. | ||
Kim et al. reported the synthesis of a series of amphiphilic amide dendron-pyrene conjugates 9, which self-organized into vesicles in water (Fig. 9).28 By taking advantage of the higher binding affinity between γ-CD (or β-CD) and pyrene, addition of extra CDs to the vesicle system rendered the transformation of vesicles into nanotubular structures with CD⊃pyrene pseudorotaxanes at the surface of the nanotubes. This outcome could be reversed with the addition of a poly(propylene glycol) (PPG) polymer to extract the CDs from the nanotubes, forming back the vesicles with the addition of a new polypseudorotaxanes CDn⊃PPG.
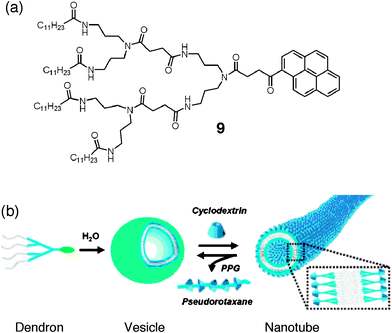 | ||
| Fig. 9 (a) Amphiphilic amide dendron-pyrene conjugates 9. (b) The amphiphilic dendron 9 (left) can be self-assembled into vesicular structures (middle) in water, while a nanotube (right) can be formed in the presence of γ-CD (or β-CD). The formation process can be forced to reverse by the addition of a PPG polymer whereas new CD-based polypseudorotaxane forms. Picture taken from Ref. 28. | ||
Crown ether- and cyclophane-based dendrimers
When multiple wedge-shaped dendritic fragments are attached to a multivalent polymeric core, dendronized polymers can be formed.29 Dendronized polymers may exhibit relatively rigid, cylindrical structures. Recently, dendronized polypseudorotaxanes (10)n⊂11 (Type I-A pseudorotaxane dendrimer) have been produced by supramolecular self-assembly between dendritic ammonium molecules 10 and multivalent dibenzo[24]crown-8-containing polymers 11 (Fig. 10).30a The multiple self-assembly of dendrons onto polymer relies upon the strong binding affinity between the dialkylammonium ion center and the [24]crown-8 recognition motif, resulting in formation of multiple hydrogen bonds, electrostatic interaction and additional π–π stacking. The formation of the dendronized polypseudorotaxanes is acid–base switchable to either rod-like dendronized polymer or flexible polymer state, leading to a reversible control over the conformation of polymer backbone. Recently, dendronized polymers with crown ether branching units have been synthesized30b with ion-induced stretching properties.![Formation of supramolecular dendronized polymers (10)n⊂11 through dendritic ammonium compound 10 and dibenzo[24]crown-8-containing polymer 11.](/image/article/2010/PY/b9py00380k/b9py00380k-f10.gif) | ||
| Fig. 10 Formation of supramolecular dendronized polymers (10)n⊂11 through dendritic ammonium compound 10 and dibenzo[24]crown-8-containing polymer 11. | ||
Stoddart and coworkers have recently disclosed the synthesis of a [2]rotaxane dendrimer using click chemistry (Fig. 11).31 As a strong charge-transfer complex could be formed from tetrathiafulvalene 12 and cyclobis(paraquat-p-phenylene) 13 compounds,32 the reported [2]rotaxane dendrimer was synthesized using click chemistry (Cu(I)-catalyzed 1,3-dipolar cycloaddition) in the final coupling step via a threading-followed-by-stoppering approach with a dendritic stopper molecule 14. The product 15 is a [2]rotaxane dendrimer (Type I-A rotaxane dendrimer) which was isolated in 50% yield. The [2]rotaxane dendrimer 15 exhibits bistable electrochemical switching behavior wherein the cyclobis(paraquat-p-phenylene) ring can be encircled to either the tetrathiafulvalene unit or the 1,5-dioxynaphthalene unit upon electrochemical stimuli. Since the [2]rotaxane dendrimer 15 contains six rigid mesogen units (cyclohexylbiphenyl) at the stopper region, furthermore, it forms liquid crystalline self-assemblies in a wide temperature range—from ambient temperature to 150 °C.
![Synthesis of bistable [2]rotaxane dendrimer 15 based on charge-transfer complex and click chemistry.](/image/article/2010/PY/b9py00380k/b9py00380k-f11.gif) | ||
| Fig. 11 Synthesis of bistable [2]rotaxane dendrimer 15 based on charge-transfer complex and click chemistry. | ||
3. Thermodynamic synthesis of rotaxane dendrimers
Preparation of pseudorotaxane dendrimers can be effectively achieved via direct self-assembly as described in the previous section. The synthesis of rotaxane dendrimers, instead of pseudorotaxane dendrimers, relies on kinetically controlled reactions, e.g., click chemistry, nucleophilic, electrophilic and radical reactions. Generally, the synthesis of mechanically interlocked molecular compounds such as rotaxanes and catenanes has relied on kinetically controlled reactions during the past three decades. However, as a result of the irreversible nature of this kinetically controlled synthetic approach, free dumbbell compounds and free macrocycles are inevitably obtained as by-products. Although these by-products are usually separable by chromatographic methods, however, the yield of the desired rotaxane structure would be invariably reduced. Thus, dynamic covalent chemistry (DCC)33 has recently become popular, leading to molecular assemblies that are formed in a thermodynamically controlled manner. In contrast with the kinetic process, the reversible thermodynamic regime allows the undesired or competitive by-products to be recycled to give the most energetically favored product in the presence of a matched template. In polymer chemistry, the reversibility of covalent bonds has been a significant factor in controlled radical polymerizations.34As the understanding of supramolecular template-directed effects11a advances, more and more mechanically interlocked compounds can be constructed by the thermodynamically controlled synthetic methodology.35 Dynamic covalent bonds involve trityl ether, thioether, disulfide, olefin, imine, hydrazone, acetal, etc.33 Some of these dynamic bonds possess relatively high stability but they are all subjected to reversible bond formation/breaking and exchange upon the addition of acids, bases, nucleophiles, metathesis catalysts, etc. Some metal–ligand bonds are also dynamic. In particular, Jeong and Park reported36 a dynamic synthesis of rotaxane dendrimers via Os–N bonds (Fig. 12). Direct seven-component self-assembly can be performed by mixing the dendritic ligand 16, OsO4, 2,3-dimethylbut-2-ene, and the dendritic bisamide dumbbell-shaped thread 17 to afford the dendritic [2]rotaxane 18 (Type I–C rotaxane dendrimer) in high yield. According to NMR studies, the amide groups act as hydrogen bond donor–acceptor templates. The dynamic nature of the [2]rotaxane dendrimer 18 has been proven by mixing preformed Os-based metallocycle 19 and thread 17, which also permits the formation of the same [2]rotaxane dendrimer 18. The mechanism involves the forming and breaking of the dynamic Os–N bonds in the metallocycle, which allows the thread 17 to perform thermodynamically favorable, complementary self-assembly into the metallocycles's cavity. Under these conditions, the formation of the [2]rotaxane dendrimer 18 is readily detected in high yield reminiscent of the “magic” interlocking rings trick.37
![Formation of dynamic [2]rotaxane dendrimer 18 by direct seven-component self-assembly or dynamic threading.](/image/article/2010/PY/b9py00380k/b9py00380k-f12.gif) | ||
| Fig. 12 Formation of dynamic [2]rotaxane dendrimer 18 by direct seven-component self-assembly or dynamic threading. | ||
Dendrimers can be constructed thermodynamically by a slippage approach. Stoddart and coworkers established some years ago that, at elevated temperature (40 °C, CH2Cl2), bis(cyclohexylmethyl)ammonium hexafluorophosphate (50 mM) is converted 98% of the way to a [2]rotaxane in the presence of 150 mM of dibenzo[24]-crown-8.38 In this event, this thermodynamically controlled self-assembly process has been employed to produce a Type I–C [2]rotaxane dendrimer 22 by mixing dendritic (cyclohexylmethyl)ammonium 20 and dendritic crown ether 21 (Fig. 13) via dibenzo[24]crown-8⊃ammonium molecular recognition.39 The cyclohexyl ring of the thread 20 gains sufficient energy at high temperature which can subsequently thread through the crown ether ring. However, this thermodynamically controlled [2]rotaxane dendrimer (Type I–C rotaxane dendrimer) formation lost all its remarkable efficiency from which the best yield of a mechanically interlocked dendrimer in a single slippage experiment was 19% after 90 days of reaction followed by chromatographic separation. The [2]rotaxane dendrimer 22 is stable when it is dissolved in relatively nonpolar solvents, e.g., CH2Cl2 at room temperature; yet dissociates when it is dissolved in hydrogen bond-disfavored solvents, e.g., dimethyl sulfoxide, at room temperature.
![Formation of [2]rotaxane dendrimer 22 by slippage approach.](/image/article/2010/PY/b9py00380k/b9py00380k-f13.gif) | ||
| Fig. 13 Formation of [2]rotaxane dendrimer 22 by slippage approach. | ||
In particular, of the dynamic covalent bonds, imine bonds have been well-studied and widely used to build up complex molecular structures due to their relatively high stability in organic solvents, ease of formation, and the ability of chelating transition metal cations and various cationic templates.40,41 Therefore, various types of imine-containing, linear or hyperbranched polymers have been successfully synthesized; and their intrinsic properties have been explored.34 Moreover, the ability to control imine bond formation and dissociation leads to a versatile tool for solid-phase combinatorial synthesis.42
Imine bonds that have been used as a building block for the construction of dendrimers, exhibit special properties.43 A [G4] polyphenylazomethine dendrimer which possesses a [Ar–NC(Ar)2] group at every branch, reveals to an energy gradient within its spherical dendritic environment. Such energy gradient directs the transfer of charge and energy from the dendrimer surface to its core, leading to a stepwise binding of Sn(II) ions to the [NC] moieties from interior core to exterior surface.43 For other examples, amine-terminated poly(ethylene imine) (PEI) dendrimers which connected with poly(ethylene glycol) (PEG) chains via imine bonds, act as acid labile nanocarriers for drugs.44 The movement of amine-terminated PPI dendrimers can be manipulated by treating them on a glass substrate via gradient-driven multiple imine bond formations.45 Dendrons can be immobilized onto microspheres46via imine bonds and that their stability and rate toward hydrolysis47 have been reported using contact angle measurement.46 Furthermore, imines have been used (1) in the thermodynamic synthesis of mechanically interlocked molecules,35,40b,47,48 (2) as templates for CD-based polyrotaxanes,49 and (3) in controlling the ring movements in rotaxane molecules.50
The versatility and efficiency of DCC that involves imine bonds have been explored in convergent synthesis of Type I–C [4]rotaxane dendrimers.51 Reversible imine bond formation is employed (Fig. 14) to clip two acyclic components, wherein one of them a diformyl pyridine unit bearing a Fréchet-type dendrons from [G0] to [G3] 23a–d, and the other, a complementary dianiline in the shape of the di(o-aminophenyl)ether of tetraethylene glycol 24, around each arm of a tritopic trisammonium ion core 25. Under this thermodynamic synthesis with [24]crown-8⊃ammonium templating strategy, Type I–C [4]rotaxane dendrimers from [G0] to [G3] 26a–d have been obtained in very high yields (>90%) via a seven-component self-assembly. The dynamic dendrimers 26a–d were characterized by NMR spectroscopy and electrospray ionization mass spectrometry (ESI-MS). On account of their six readily hydrolysable imine bonds, the kinetically labile (dynamic) [4]rotaxane dendrimers from [G0] to [G2] 26a–c can be reduced by a boron complex to give kinetically stable [4]rotaxane dendrimers 27a–c in good yields. However, imine reduction of dynamic [G3]-dendrimer 26d did not work perfectly but yielded a mixture of degraded dendrimers. This observation may due to the massive steric hindrance of the [G3]-dendron.
![Formation of (A) imine-containing [4]rotaxane dynamic dendrimers 26a–dvia seven component self-assembly and (B) kinetically stable [4]rotaxane dendrimers 27a–c.](/image/article/2010/PY/b9py00380k/b9py00380k-f14.gif) | ||
| Fig. 14 Formation of (A) imine-containing [4]rotaxane dynamic dendrimers 26a–dvia seven component self-assembly and (B) kinetically stable [4]rotaxane dendrimers 27a–c. | ||
The inherent modularity of the overall process should allow for the rapid and straightforward access to many other analogues of rotaxane dendrimers in which either the branched core, or dendritic periphery can be modified to suit the needs of any given application of these molecules. Indeed, the dynamic nature of the initial thermodynamically driven assembly could be utilized in order to amplify particular products from a potential library as a result of a selective recognition process. The dynamic convergent synthesis of [4]rotaxane dendrimers allows components to be mixed and matched on the basis of a larger system under equilibrium, which would allow the dendrimer to adapt its environment. The dynamic property of these dendrimers was demonstrated by mixing preformed dendrimers and allowing them to come to equilibrium, forming dynamic combinatorial libraries. The ratio of the constituents in the library can be controlled by varying the conditions under which the library is prepared. In particular, two preformed, different generation [4]rotaxane dendrimers 26a(G0/G0/G0) and 26c(G2/G2/G2) (Fig. 15) undergo dendron exchange via reversible imine bonds to convert into two new [4]rotaxane dendrimers 28(G0/G0/G2) and 29(G0/G2/G2). Furthermore, direct mixing of four preformed [4]rotaxane dendrimers 26a–d in one pot gives a mixture of 20 mixed dendron [4]rotaxane dendrimers, as characterized by ESI-MS.51,52 This mix-and-match53 dendron exchange process which affords different dendrimers in one pot, offers a facile method to synthesize rotaxane dendrimers. Eventually, for a small dendritic dynamic library wherein dynamic dendrimers have vastly difference in molecular weight, they can be reduced into their corresponding kinetically stable dendrimers; and can be further separated and isolated by size exclusion chromatographic techniques.
![Two preformed [4]rotaxane dendrimers 26a(G0/G0/G0) and 26c(G2/G2/G2) undergo dendron exchange via reversible imine bonds to convert into two new [4]rotaxane dendrimers 28(G0/G0/G2) and 29(G0/G2/G2) in one pot.](/image/article/2010/PY/b9py00380k/b9py00380k-f15.gif) | ||
| Fig. 15 Two preformed [4]rotaxane dendrimers 26a(G0/G0/G0) and 26c(G2/G2/G2) undergo dendron exchange via reversible imine bonds to convert into two new [4]rotaxane dendrimers 28(G0/G0/G2) and 29(G0/G2/G2) in one pot. | ||
4. Conclusion and perspectives
A wide variety of pseudorotaxane and rotaxane dendrimers has been obtained by direct self-assembly and/or typical rotaxane formation. Thermodynamic synthesis of rotaxane dendrimers sometimes displays enhanced yield and efficiency than using a kinetically controlled synthesis. Dynamic rotaxane dendrimers that contain imine bonds, can be post-modified to become kinetically stable rotaxane dendrimers. In addition, dynamic rotaxane dendrimers can be mixed and matched via reversible bonds to obtain new rotaxane dendrimers that are difficult to be obtained using conventional synthetic methods.Several areas in this topic remain to be explored. First, rapid and efficient rotaxane formation is required to be discovered. When thermodynamic synthesis is used for rotaxane dendrimer formation, other dynamic covalent bonds33,35,37 instead of imine bonds can be employed. The effective transformation from dynamic rotaxane dendrimers into kinetically stable ones is a concern.33 Sanders and coworkers discovered that hydrazone is superior for formation of catenane compounds with controllable dynamic equilibrium.54 Dynamic olefin metathesis using a second generation Grubbs' catalyst is suitable for the effective synthesis of mechanically interlocked compounds.37 Furthermore, the synthesis of well-defined Type III rotaxane dendrimers wherein rotaxane building units grow like a dendrimer, is still a challenge.12 Switchable bistable rotaxanes or other sophisticated mechanically interlocked compounds55 that are responsive to external stimuli such as pH,56 redox,57 temperature, competitive binding,58 light,59etc., can be eventually incorporated into every branch of a dendrimer. Such globular, hyperbranched polyrotaxane dendrimers which act as vehicles for trapping selected substrates/drugs in their dendritic void spaces, may exhibit three-dimensional, reversible size and density control. This control will be based on reversible dendrimer interior contraction and extension behaviors, leading to unprecedented properties in controlled substrate/drug release and surface applications.60
Linear polyrotaxane dendrimers whereas dendrons with a macrocycle unit are mechanically interlocked onto a long linear thread (or polymer), are an interesting class of material. The free movements of the dendritic macrocycle units on the main-chain polymer thread may provide an optimum environment for multivalent interactions between dendron peripheries and biological species.61 The successful preparation of linear polyrotaxane dendrimers may rely on the advances in multiple ring formation on a linear thread62 or supramolecular polymerization14b,14c techniques. The synthesis and properties of another rotaxane dendrimer analogues—catenane dendrimers, polycatenane dendrimers63 and other exotic structures with a significant structural and topological diversity64 remain to be explored.
Acknowledgements
We thank the financial support by a Strategic Investments Scheme from The Chinese University of Hong Kong as well as a General Research Fund (CUHK401808) by The Research Grants Council of Hong Kong. This work is also partially supported by a grant from the University Grants Committee of HKSAR (Area of Excellence Scheme AoE/P-03/08).References and notes
- Reviews on dendrimer chemistry (a) Dendrimers and Other Dendritic Polymers, J. M. J. Fréchet and D. A. Tomalia, Wiley, New York, 2002 Search PubMed; (b) Dendrimers and Dendrons: Concepts, Syntheses, Applications, G. R. Newkome, F. Vögtle and C. N. Moorefield, VCH, New York, 2001 Search PubMed; (c) H.-F. Chow, T. K.-K. Mong, M. F. Nongrum and C.-W. Wan, Tetrahedron, 1998, 54, 8543–8660 CrossRef CAS; (d) O. A. Matthews, A. N. Shipway and J. F. Stoddart, Prog. Polym. Sci., 1998, 23, 1–56 CrossRef; (e) A. W. Bosman, H. M. Janssen and E. W. Meijer, Chem. Rev., 1999, 99, 1665–1688 CrossRef CAS; (f) F. J. Stoddart and T. Welton, Polyhedron, 1999, 18, 3575–3591 CrossRef CAS; (g) F. Vögtle, S. Gestermann, R. Hesse, H. Schwierz and B. Windisch, Prog. Polym. Sci., 2000, 25, 987–1041 CrossRef CAS; (h) D. A. Tomalia, Prog. Polym. Sci., 2005, 30, 294–324 CrossRef CAS; (i) I. Gitsov and C. Lin, Curr. Org. Chem., 2005, 9, 1025–1051 CrossRef CAS; (j) C. C. Lee, J. A. MacKay, J. M. J. Fréchet and F. C. Szoka, Nat. Biotechnol., 2005, 23, 1517–1526 CrossRef CAS.
- E. Buhleier, W. Wehner and F. Vogtle, Synthesis, 1978, 155–158 CrossRef CAS.
- (a) C. J. Hawker and J. M. J. Fréchet, J. Chem. Soc., Chem. Commun., 1990, 1010–1013 RSC; (b) C. J. Hawker and J. M. J. Fréchet, J. Am. Chem. Soc., 1990, 112, 7638–7647 CrossRef CAS.
- (a) T. H. Mourey, S. R. Turner, M. Rubenstein, J. M. J. Fréchet, C. J. Hawker and K. L. Wooley, Macromolecules, 1992, 25, 2401–2406 CrossRef CAS; (b) C. J. Hawker, Adv. Polym. Sci., 1999, 147, 113–160 CAS; (c) S. Hecht and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2001, 40, 74–91 CrossRef CAS.
- (a) P. Milosevic and S. Hecht, Org. Lett., 2005, 7, 5023–5026 CrossRef CAS; (b) U. Boas, J. B. Christensen and P. M. H. Heegaard, J. Mater. Chem., 2006, 16, 3785–3798 RSC.
- (a) F. Zeng and S. C. Zimmerman, Chem. Rev., 1997, 97, 1681–1712 CrossRef CAS; (b) S. C. Zimmerman and L. J. Lawless, Top. Curr. Chem., 2001, 217, 95–120 CAS; (c) D. K. Smith, Chem. Commun., 2006, 34–44 RSC; (d) A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS; (e) K.-N. Lau, H.-F. Chow, M.-C. Chan and K.-W. Wong, Angew. Chem., Int. Ed., 2008, 47, 6912–6916 CrossRef CAS; (f) B. M. Rosen, C. J. Wilson, D. A. Wilson, M. Peterca, M. R. Imam and V. Percec, Chem. Rev., 2009, 109, 6275–6540 CrossRef CAS.
- N. Nishiyama, W.-D. Jang and K. Kataoka, New J. Chem., 2007, 31, 1074–1082 RSC.
- (a) D. Astruc and Françoise Chardac, Chem. Rev., 2001, 101, 2991–3023 CrossRef CAS; (b) P. Arya, G. Panda, N. V. Rao, H. Alper, S. C. Bourque and L. E. Manzer, J. Am. Chem. Soc., 2001, 123, 2889–2890 CrossRef CAS; (c) T. Muraki, K. Fujita and M. Kujime, J. Org. Chem., 2007, 72, 7863–7870 CrossRef CAS.
- W.-S. Li and T. Aida, Chem. Rev., 2009, 109, 6047–6076 CrossRef CAS.
- (a) S. H. Medina and M. E. H. El-Sayed, Chem. Rev., 2009, 109, 3141–3157 CrossRef CAS; (b) A.-M. Caminade, C.-O. Turrin and J.-P. Majoral, Chem.–Eur. J., 2008, 14, 7422–7432 CrossRef CAS.
- (a) F. Aricó, J. D. Badjic, S. J. Cantrill, A. H. Flood, K. C.-F. Leung, Y. Liu and J. F. Stoddart, Top. Curr. Chem., 2005, 249, 203–259 CAS; (b) I. Yoon, M. Narita, T. Shimizu and M. Asakawa, J. Am. Chem. Soc., 2004, 126, 16740–16741 CrossRef CAS; (c) C.-W. Chiu, C.-C. Lai and S.-H. Chiu, J. Am. Chem. Soc., 2007, 129, 3500–3501 CrossRef CAS; (d) E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS.
- J. W. Lee and K. Kim, Top. Curr. Chem., 2003, 228, 111–140 CAS.
- (a) C. Burd and M. Weck, Macromolecules, 2005, 38, 7225–7230 CrossRef CAS; (b) C. R. South, M. N. Higley, K. C.-F. Leung, D. Lanari, A. Nelson, R. H. Grubbs, J. F. Stoddart and M. Weck, Chem.–Eur. J., 2006, 12, 3789–3797 CrossRef CAS; (c) C. R. South, K. C.-F. Leung, D. Lanari, J. F. Stoddart and M. Weck, Macromolecules, 2006, 39, 3738–3744 CrossRef CAS; (d) S. B. Darling, Prog. Polym. Sci., 2007, 32, 1152–1204 CrossRef CAS; (e) F. Wang, B. Zheng, K. L. Zhu, Q. Z. Zhou, C. X. Zhai, S. J. Li, N. Li and F. H. Huang, Chem. Commun., 2009, 4375–4377 RSC.
- (a) S. C. Zimmerman and P. S. Corbin, Struct. Bonding, 2000, 96, 63–94 CAS; (b) J. D. Fox and S. J. Rowan, Macromolecules, 2009, 42, 6823–6835 CrossRef CAS; (c) T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS.
- , W. L. Mock, in Comprehensive Supramolecular Chemistry ed. F. Vögtle, Pergamon, Oxford, 1996, vol. 2, p. 477 Search PubMed.
- (a) H.-J. Kim, J. Heo, W. S. Jeon, E. Lee, J. Kim, S. Sakamoto, K. Yamaguchi and K. Kim, Angew. Chem., Int. Ed., 2001, 40, 1526–1529 CrossRef CAS; (b) K. Kim, Chem. Soc. Rev., 2002, 31, 96–107 RSC; (c) Y. H. Ko, E. Kim, I. Hwang and K. Kim, Chem. Commun., 2007, 1305–1315 RSC.
- J. W. Lee, Y. H. Ko, S.-H. Park, K. Yamaguchi and K. Kim, Angew. Chem., Int. Ed., 2001, 40, 746–749 CrossRef CAS.
- W. Wang and A. E. Kaifer, Angew. Chem., Int. Ed., 2006, 45, 7042–7046 CrossRef CAS.
- J. W. Lee, S. C. Han, J. H. Kim, Y. H. Ko and K. Kim, Bull. Korean Chem. Soc., 2007, 28, 1837–1840 CAS.
- Y. Zeng, Y. Li, M. Li, G. Yang and Y. Li, J. Am. Chem. Soc., 2009, 131, 9100–9106 CrossRef CAS.
- S.-Y. Kim, Y. H. Ko, J. W. Lee, S. Sakamoto, K. Yamaguchi and K. Kim, Chem.–Asian J., 2007, 2, 747–754 CrossRef.
- W. L. Mock, Top. Curr. Chem., 1995, 175, 1–24 CAS.
- (a) K. A. Connors, Chem. Rev., 1997, 97, 1325–1358 CrossRef CAS; (b) S. A. Nepogodiev and J. F. Stoddart, Chem. Rev., 1998, 98, 1959–1976 CrossRef CAS; (c) M. W. P. L. Baars, A. Karlsson, V. Sorokin, B. F. M. de Waal and E. W. Meijer, Angew. Chem., Int. Ed., 2000, 39, 4262–4265 CrossRef CAS; (d) E. Alvarez-Parrilla, P. R. Cabrer, W. Al-Soufi, F. Meijide, E. R. Núñez and J. V. Tato, Angew. Chem., Int. Ed., 2000, 39, 2856–2858 CrossRef CAS; (e) J. J. Michels, M. W. P. L. Baars, E. W. Meijer, J. Huskens and D. N. Reinhoudt, J. Chem. Soc., Perkin Trans. 2, 2000, 1914–1918 RSC; (f) K. J. C. van Bommel, G. A. Metselaar, W. Verboom and D. N. Reinhoudt, J. Org. Chem., 2001, 66, 5405–5412 CrossRef CAS; (g) J. J. Michels, J. Huskens and D. N. Reinhoudt, J. Chem. Soc., Perkin Trans. 2, 2002, 102–105 RSC; (h) A. Harada, A. Hashidzume, H. Yamaguchi and Y. Takashima, Chem. Rev., 2009, 109, 5974–6023 CrossRef CAS.
- C. A. Nijhuis, K. A. Dolatowska, B. J. Ravoo, J. Huskens and D. N. Reinhoudt, Chem.–Eur. J., 2007, 13, 69–80 CrossRef.
- (a) X. Y. Ling, L. Malaquin, D. N. Reinhoudt, H. Wolf and J. Huskens, Langmuir, 2007, 23, 9990–9999 CrossRef CAS; (b) X. Y. Ling, D. N. Reinhoudt and J. Huskens, Chem. Mater., 2008, 20, 3574–3578 CrossRef CAS.
- C. M. Bruinink, C. A. Nijhuis, M. Péter, B. Dordi, O. Crespo-Biel, T. Auletta, A. Mulder, H. Schönherr, G. J. Vancso, J. Huskens and D. N. Reinhoudt, Chem.–Eur. J., 2005, 11, 3988–3996 CrossRef CAS.
- O. Crespo-Biel, B. Dordi, P. Maury, M. Péter, D. N. Reinhoudt and J. Huskens, Chem. Mater., 2006, 18, 2545–2551 CrossRef CAS.
- C. Park, I. H. Lee, S. Lee, Y. Song, M. Rhue and C. Kim, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1199–1203 CrossRef CAS.
- (a) H. Frey, Angew. Chem., Int. Ed., 1998, 37, 2193–2197 CrossRef CAS; (b) A. D. Schlüter and J. P. Rabe, Angew. Chem., Int. Ed., 2000, 39, 864–883 CrossRef CAS; (c) A. D. Schlüter, Top. Curr. Chem., 2005, 245, 151–191; (d) H. Frauenrath, Prog. Polym. Sci., 2005, 30, 325–384 CrossRef CAS.
- (a) K. C.-F. Leung, P. M. Mendes, S. N. Magonov, B. H. Northrop, S. Kim, K. Patel, A. H. Flood, H.-R. Tseng and J. F. Stoddart, J. Am. Chem. Soc., 2006, 128, 10707–10715 CrossRef CAS; (b) A. Ossenbach, H. Rüegger, A. Zhang, K. Fischer, A. D. Schlüter and M. Schmidt, Macromolecules, 2009, 42, 8781–8793 CrossRef CAS.
- I. Aprahamian, T. Yasuda, T. Ikeda, S. Saha, W. R. Dichtel, K. Isoda, T. Kato and J. F. Stoddart, Angew. Chem., Int. Ed., 2007, 46, 4675–4679 CrossRef CAS.
- J. F. Stoddart and H.-R. Tseng, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4797–4800 CrossRef CAS.
- (a) P. A. Brady, R. P. Bonar-Law, S. J. Rowan, C. J. Suckling and J. K. M. Sanders, Chem. Commun., 1996, 319–320 RSC; (b) P. A. Brady and J. K. M. Sanders, Chem. Soc. Rev., 1997, 26, 327–336 RSC; (c) R. L. E. Furlan, S. Otto and J. K. M. Sanders, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4801–4804 CrossRef CAS; (d) J. K. M. Sanders, Pure Appl. Chem., 2000, 72, 2265–2274 CrossRef CAS; (e) P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS; (f) J.-M. Lehn, Chem.–Eur. J., 1999, 5, 2455–2463 CrossRef CAS; (g) J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC; (h) S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef; (i) L. M. Greig and D. Philp, Chem. Soc. Rev., 2001, 30, 287–302 RSC; (j) A. Herrmann, Org. Biomol. Chem., 2009, 7, 3195–3204 RSC.
- T. Maeda, H. Otsuka and A. Takahara, Prog. Polym. Sci., 2009, 34, 581–604 CrossRef CAS.
- P. C. Haussmann and J. F. Stoddart, Chem. Rec., 2009, 9, 136–154 CrossRef CAS.
- K.-S. Jeong and E. J. Park, J. Org. Chem., 2004, 69, 2618–2621 CrossRef CAS.
- It is possible, under the appropriate conditions, to reproduce the conjurer's “magic rings” trick, wherein two apparently “closed” rings can be linked together, one through the other, to form a [2]catenane; wherein one “closed” ring can be linked with a dumbbell thread to form a [2]rotaxane. For selected examples, see: (a) A. F. M. Kilbinger, S. J. Cantrill, A. W. Waltman, M. W. Day and R. H. Grubbs, Angew. Chem., Int. Ed., 2003, 42, 3281–3285 CrossRef CAS; (b) E. N. Guidry, S. J. Cantrill, J. F. Stoddart and R. H. Grubbs, Org. Lett., 2005, 7, 2129–2132 CrossRef CAS.
- P. R. Ashton, I. Baxter, M. C. T. Fyfe, F. M. Raymo, N. Spencer, J. F. Stoddart, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 1998, 120, 2297–2307 CrossRef CAS.
- A. M. Elizarov, T. Chang, S.-H. Chiu and J. F. Stoddart, Org. Lett., 2002, 4, 3565–3568 CrossRef CAS.
- (a) M. Grigoras and C. O. Catanescu, J. Macromol. Sci., Polym. Rev., 2004, 44, 131–173; (b) C. D. Meyer, C. S. Joiner and J. F. Stoddart, Chem. Soc. Rev., 2007, 36, 1705–1723 RSC; (c) A. Herrmann, Org. Biomol. Chem., 2009, 7, 3195–3204 RSC.
- For selected examples, see (a) P. Cerrada, L. Oriol, M. Piñol and J. L. Serrano, J. Polym. Sci., Part A: Polym. Chem., 1996, 34, 2603–2611 CrossRef CAS; (b) T. Nishinaga, A. Tanatani, K. Oh and J. S. Moore, J. Am. Chem. Soc., 2002, 124, 5934–5935 CrossRef CAS; (c) D. Zhao and J. S. Moore, J. Org. Chem., 2002, 67, 3548–3554 CrossRef CAS; (d) F. Kopp, C. Mahlert, J. Grünewald and M. A. Marahiel, J. Am. Chem. Soc., 2006, 128, 16478–16479 CrossRef CAS; (e) H. Kanazawa, M. Higuchi and K. Yamamoto, Macromolecules, 2006, 39, 138–144 CrossRef CAS; (f) V. del Amo, A. M. Z. Slawin and D. Philp, Org. Lett., 2008, 10, 4589–4592 CrossRef CAS.
- For selected examples, see (a) C. Castro, J. Ramos, A. Millán, J. González-Calbet and F. Palacio, Chem. Mater., 2000, 12, 3681–3688 CrossRef CAS; (b) D. E. Robinson and M. W. Holladay, Org. Lett., 2000, 2, 2777–2779 CrossRef CAS; (c) B. B.-N. Ben-Aroya and M. Portnoy, J. Comb. Chem., 2001, 3, 524–527 CrossRef CAS; (d) J. C. Pelletier, A. Khan and Z. Tang, Org. Lett., 2002, 4, 4611–4613 CrossRef CAS; (e) C. Patteux, V. Levacher and G. Dupas, Org. Lett., 2003, 5, 3061–3063 CrossRef CAS.
- (a) K. Yamamoto, M. Higuchi, S. Shiki, M. Tsuruta and H. Chiba, Nature, 2002, 415, 509–511 CrossRef CAS; (b) A. Kimoto, J.-S. Cho, M. Higuchi and K. Yamamoto, Macromolecules, 2004, 37, 5531–5537 CrossRef CAS.
- S. Xu, M. Krämer and R. Haag, J. Drug Targeting, 2006, 14, 367–374 Search PubMed.
- T. Chang, D. I. Rozkiewicz, B. J. Ravoo, E. W. Meijer and D. N. Reinhoudt, Nano Lett., 2007, 7, 978–980 CrossRef CAS.
- K. C.-F. Leung, S. Xuan and C.-M. Lo, ACS Appl. Mater. Interfaces, 2009, 1, 2005–2012 Search PubMed.
- (a) K. C.-F. Leung, W.-Y. Wong, F. Aricó, P. C. Haussmann and J. F. Stoddart, Org. Biomol. Chem., 2010, 8, 83–89 RSC; (b) W.-Y. Wong, K. C.-F. Leung and J. F. Stoddart, Org. Biomol. Chem., 2010, 8, 2332–2343 RSC.
- L. M. Klivansky, G. Koshkakaryan, D. Cao and Y. Liu, Angew. Chem., Int. Ed., 2009, 48, 4185–4189 CrossRef CAS.
- C. I. Simionescu, M. Grigoras, A. Farcas and A. Stoleru, Macromol. Chem. Phys., 1998, 199, 1301–1306 CrossRef CAS.
- (a) H. Kawai, T. Umehara, K. Fujiwara, T. Tsuji and T. Suzuki, Angew. Chem., Int. Ed., 2006, 45, 4281–4286 CrossRef CAS; (b) T. Umehara, H. Kawai, K. Fujiwara and T. Suzuki, J. Am. Chem. Soc., 2008, 130, 13981–13988 CrossRef CAS.
- (a) K. C.-F. Leung, F. Aricó, S. J. Cantrill and J. F. Stoddart, J. Am. Chem. Soc., 2005, 127, 5808–5810 CrossRef CAS; (b) K. C.-F. Leung, F. Aricó, S. J. Cantrill and J. F. Stoddart, Macromolecules, 2007, 40, 3951–3959 CrossRef CAS.
- K. C.-F. Leung, Macromol. Theory Simul., 2009, 18, 328–335 CrossRef CAS.
- (a) K. S. Chichak, S. J. Cantrill and J. F. Stoddart, Chem. Commun., 2005, 3391–3393 RSC; (b) R. Cacciapaglia, S. D. Stefano, G. Ercolani and L. Mansolini, Macromolecules, 2009, 42, 4077–4083 CrossRef CAS.
- T. S. Lam, A. Belenguer, S. L. Roberts, C. Naumann, T. Jarrosson, S. Otto and J. K. M. Sanders, Science, 2005, 308, 667–669 CrossRef CAS.
- (a) Y. Liu, A. H. Flood, P. A. Bonvallet, S. A. Vignon, B. H. Northrop, H.-R. Tseng, J. O. Jeppesen, T. J. Huang, B. Brough, M. Baller, S. Magonov, S. D. Solares, W. A. Goddard, C.-M. Ho and J. F. Stoddart, J. Am. Chem. Soc., 2005, 127, 9745–9759 CrossRef CAS; (b) J. Wu, K. C.-F. Leung, D. Benítez, J.-Y. Han, S. J. Cantrill, L. Fang and J. F. Stoddart, Angew. Chem., Int. Ed., 2008, 47, 7470–7474 CrossRef CAS.
- (a) K. C.-F. Leung, C.-P. Chak, C.-M. Lo, W.-Y. Wong, S. Xuan and C. H. K. Cheng, Chem.–Asian J., 2009, 4, 364–381 CrossRef CAS; (b) C.-P. Chak, S. Xuan, P. M. Mendes, J. C. Yu, C. H. K. Cheng and K. C.-F. Leung, ACS Nano, 2009, 3, 2129–2138 CrossRef CAS.
- (a) C. A. Nijhuis, B. J. Ravoo, J. Huskens and D. N. Reinhoudt, Coord. Chem. Rev., 2007, 251, 1761–1780 CrossRef CAS; (b) S. Nygaard, K. C.-F. Leung, I. Aprahamian, T. Ikeda, S. Saha, B. W. Laursen, S.-Y. Kim, S. W. Hansen, P. C. Stein, A. H. Flood, J. F. Stoddart and J. O. Jeppesen, J. Am. Chem. Soc., 2007, 129, 960–970 CrossRef CAS.
- (a) G. M. Dykes, D. K. Smith and G. J. Seeley, Angew. Chem., Int. Ed., 2002, 41, 3254–3257 CrossRef CAS; (b) K. C.-F. Leung, T. D. Nguyen, J. F. Stoddart and J. I. Zink, Chem. Mater., 2006, 18, 5919–5928 CrossRef CAS.
- T. D. Nguyen, K. C.-F. Leung, M. Liong, Y. Liu, J. F. Stoddart and J. I. Zink, Adv. Funct. Mater., 2007, 17, 2101–2110 CrossRef CAS.
- (a) M. Yoshida and J. Lahann, ACS Nano, 2008, 2, 1101–1107 CrossRef CAS; (b) A. Khan, A. E. Daugaard, A. Bayles, S. Koga, Y. Miki, K. Sato, J. Enda, S. Hvilsted, G. D. Stucky and C. J. Hawker, Chem. Commun., 2009, 425–427 RSC; (c) R. K. Tekade, P. V. Kumar and N. K. Jain, Chem. Rev., 2009, 109, 49–87 CrossRef CAS.
- (a) A. Nelson, J. M. Belisky, S. Vidal, C. S. Joiner, L. G. Baum and J. F. Stoddart, J. Am. Chem. Soc., 2004, 126, 11914–11922 CrossRef CAS; (b) J. D. Badjic, A. Nelson, S. J. Cantrill, W. B. Turnbull and J. F. Stoddart, Acc. Chem. Res., 2005, 38, 723–732 CrossRef CAS; (c) M. A. Kostiainen, G. R. Szilvay, D. K. Smith, M. B. Linder and O. Ikkala, Angew. Chem., Int. Ed., 2006, 45, 3538–3542 CrossRef CAS.
- J. Wu, K. C.-F. Leung and J. F. Stoddart, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17266–17271 CrossRef CAS.
- (a) D. B. Amabilino, P. R. Ashton, V. Balzani, S. E. Boyd, A. Credi, J. Y. Lee, S. Menzer, J. F. Stoddart, M. Venturi and D. J. Williams, J. Am. Chem. Soc., 1998, 120, 4295–4307 CrossRef CAS; (b) Z. Niu and H. W. Gibson, Chem. Rev., 2009, 109, 6024–6046 CrossRef CAS.
- (a) F. M. Raymo and J. F. Stoddart, Chem. Rev., 1999, 99, 1643–1663 CrossRef CAS; (b) L. Fang, M. A. Olson, D. Benítez, E. Tkatchouk, W. A. Goddard III and J. F. Stoddart, Chem. Soc. Rev., 2010, 39, 17–29 RSC.
| This journal is © The Royal Society of Chemistry 2010 |