DOI:
10.1039/C0PY00101E
(Communication)
Polym. Chem., 2010,
1, 827-830
Received
28th March 2010
, Accepted 20th April 2010
First published on
17th May 2010
Abstract
PEG based hyperbranched copolymers have been synthesised using a high concentration of ethylene glycol dimethacrylate (EGDMA) as the branching agent (up to 10% molar ratio of total feed monomers) via a new in situ deactivation enhanced atom transfer radical polymerisation (DE-ATRP). These hyperbranched copolymers exhibited a lower critical solution temperature (LCST) close to body temperature and photo-crosslinkable behavior.
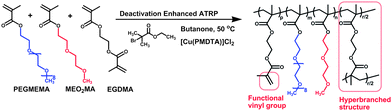
Stimuli-responsive polymers are defined as those polymers which respond with dramatic physical or chemical alterations to small external changes in their environment. Such stimuli include temperature, pH, ionic factors, electric or magnetic fields, chemical or biological agents and mechanical stress.1–4 Thermoresponsive polymers are considered as one of the most widely utilised stimuli-responsive polymers, as they are easy to apply both in vitro and in vivo.5–10 A classic and widely studied thermoresponsive polymer is poly(N-isopropylacrylamide) (PNIPAM) which exhibits a rapid coil-to-globule conversion in aqueous solution around 32 °C.11 However, there are concerns over its safety for in vivo applications.12,13 In the past few years, several thermoresponsive linear and star-shaped copolymers with PNIPAM, poly(ethylene glycol) (PEG), oligo(ethylene glycol) methacrylate (PEGMA), poly(propylene oxide) (PPO), poly(vinyl ether)s (PVEs), poly(lactide) (PLA), poly(D,L-lactide-co-glycolide) (PLGA), poly(caprolactone) (PCL) and 2-(2-methoxyethoxy) ethyl methacrylate (MEO2MA) have been developed for tissue engineering and drug delivery applications.14–18 Among these copolymers, poly(MEO2MA-co-OEGMA) was firstly reported by Lutz and his colleagues.16 This linear copolymer with LCST around 37 °C was prepared via atom transfer radical polymerisation (ATRP).16,19 Compared to linear polymers, hyperbranched or dendritic polymers display a number of unique advantages, such as low solution and melt viscosity, and high functionality.20–22 In Lutz's study, they attempted to introduce the multifunctional vinyl monomer of ethylene glycol dimethacrylate (EGDMA) to achieve hyperbranched polymer structures, however, only one percent of EGDMA caused the polymer macrogelation.19 In contrast, in the present work, a higher degree of EGDMA (up to 10% molar ratio of total feed monomers) was introduced as a multifunctional vinyl monomer. Instead of causing macrogleation, we sucessfully achieved a hyperbranched copolymer of poly(ethylene glycol) methyl ether methacrylate-co-2-(2-methoxyethoxy) ethyl methacrylate-co-ethylene glycol dimethacrylate (PEGMEMA-MEO2MA-EGDMA) via a one-step in situ DE-ATRP approach (Scheme 1). As a result, the vinyl functional groups contributed by the EGDMA component enable the copolymer the capability of easy tailoring and photo-crosslinkable properities. Meanwhile, by adjusting the “long” and “short” PEG chain monomer composition, we can sensitively alter the polymer hydrophilicity and control the LCST value of the copolymers around body temperature. Furthermore, the PEG based structure which is often considered to be nontoxic, nonimmunogenic and have a bio-compatible composition23–27 undoubtedly offers this copolymer great potentials in tissue engineering and biomedical applications.
We previously reported the DE-ATRP approach for the synthesis of dendritic copolymers,28,29 in which an excessive amount of CuII species was added into the system to enhance the deactivation of the polymerisation. In contrast, halogen-CuI/Ligand is used as the catalyst in a conventional ATRP. In activator generated by electron transfer (AGET) ATRP, the reaction is started with CuII/Ligand, however, excessive reducing agent (e.g.L-ascorbic acid) is required to transfer CuII to CuI completely in the reaction.30,31 In this study, an in situ DE-ATRP approach was applied by using CuII/Ligand and a small amount of reducing agent L-ascorbic acid (L-AA) (Scheme 2) at the start of the reaction. This is to ensure the extra amount of CuII species remain in the reaction thus lead to the slow chain growth and delayed gelation. It has been demonstrated that the activation/deactivation equilibrium of ATRP could be easily controlled by adjusting the amount of reducing agent.
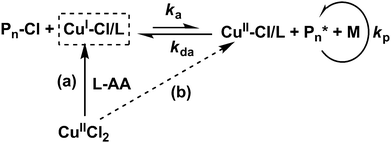 |
| | Scheme 2 Mechanism of in situ deactivation enhanced atom transfer radical polymerisation. (a) In situ produced CuI species from CuII species by reducing agent L-ascorbic acid (L-AA). (b) A small amount of L-AA addition ensures the excessive CuII species in the reaction system further enhance the deactivation reaction, suppress the growth rate and delay the gelation. | |
Using this in situ DE-ATRP approach a hyperbranched PEGMEMA-MEO2MA-EGDMA copolymer was prepared, where ethyl 2-bromoisobutyrate was used as the initiator and CuCl2/bis(2-dimethylaminoethyl)methylamine (PMDTA) were adopted as the catalyst. L-AA was utilised to reduce CuII to CuI. The reaction was monitored by gel permeation chromatography (GPC) analysis. A final yield of up to 57% of water-soluble copolymer was obtained after 26 h (entry 2 in Table 1). Polymer chain growth over time was confirmed via GPC (Fig. 1). The low polydispersity index (PDI) value demonstrated the controlled chain growth and the broader PDI with the polymerisation progressing was commonly considered as a result of the hyperbranched chain growth (Fig. S1 in ESI†).32,33 Moreover, a parallel study which increased the reducing agent (L-AA) from 15 to 20 mol% of [CuCl2] was carried out, and the reaction was clearly accelerated (66% after 16 h). This result indicates that the slight change in the equilibrium between CuII and CuI could sensitively affect the activation/deactivation process of ATRP as aforementioned.
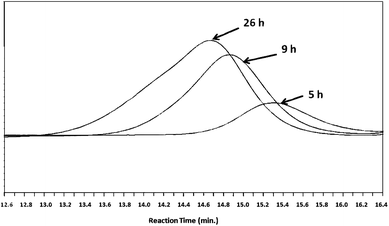 |
| | Fig. 1 GPC traces from RI detector for the samples of entry 2 in Table 1. Increases in molecular weights and polydispersities with monomer conversion are observed. | |
The structure of the copolymers were further characterised by 1H NMR analysis (Fig. 2). The characteristic peaks at chemical shifts of 6.1 and 5.6 ppm were attributed to the vinyl functional groups in the copolymer. The copolymer composition (m, n, r and p) can be calculated from the integral data C, D, E and H assigned as indicated in Fig. 2. The following equations (eqn (1)–(4)) demonstrate the calculation process:
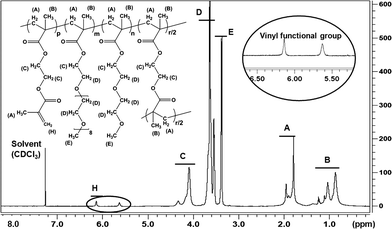 |
| | Fig. 2
1H NMR for the PEGMEMA-MEO2MA-EGDMA copolymer (S3 in Table 2) in CDCl3. Note: the spectrum clearly shows the double bonds within the structure at the chemical shifts of 6.1 and 5.6 ppm. | |
The double bond content and the branching degree of the copolymers were calculated from eqn (5) and (6), respectively:
| | | Double bond content (mol%) = p/ (m + n + r + p) × 100 | (5) |
| | | Branching degree (mol%) = r/ (m + n + r + p) × 100 | (6) |
The composition, double bond content and branching degree of the copolymers are shown in Table 2. For example, the double bond and the branching structure are at 6 and 10 mol % in polymer S3. In addition, the chemical structure of the copolymer was also determined by Fourier transform infrared spectroscopy (FTIR) (Fig. S2 in ESI†).
Table 2 Properties of PEGMEMA-MEO2MA-EGDMA copolymers
| Samples |
f
([PEGMEMA]/[MEO2MA]/[EGDMA])
|
F b([PEGMEMA]/[MEO2MA]/[EGDMA]) |
Double bond content c |
Branching degree d |
LCST/°C e |
|
Monomer feed ratio.
Polymer composition; determined by 1H NMR.
Calculated with eqn (1)–(6) (Unit: mol).
Calculated with eqn (1)–(6) (Unit: mol).
Determined by UV-vis spectrophotometer.
|
| S2 |
10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80:10 :80:10 |
9:77:14 |
4 |
10 |
24 |
| S3 |
15:75:10 |
12:72:16 |
6 |
10 |
28 |
| S4 |
25:65:10 |
22:63:15 |
6 |
9 |
38 |
LCST values of the copolymers were determined by UV-vis spectrophotometery (Fig. 3a). The copolymers (S2, S3 and S4 in Table 2) dissolved in deionized water were quantified by measuring their absorbance of 550 nm at temperatures from 20 to 55 °C with a UV-vis spectrophotometer. Compared with the linear poly(OEGMA-co-MEO2MA) copolymer as reported by Lutz et al., our hyperbranched copolymer showed a lower phase transition temperature. For example they showed LCSTs of 39 and 49 °C with 10 and 20% of PEGMEMA respectively,19 whereas our polymer exhibited 28 and 38 °C with 12 and 22% of PEGMEMA respectively. Two reasons might be responsible: (1) the 3D hyperbranched structure and subsequently the compact space configuration of the copolymer may have resulted in stronger polymer-polymer interactions than polymer-water hydrogen bonding interaction; (2) the hydrophobic EGDMA content of the copolymer also reduces the LCST. Moreover, the high concentration of the copolymer water solution could reversibly form hydrogel above the LCST (Fig. 3b).
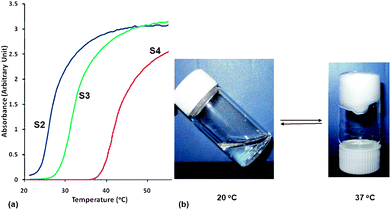 |
| | Fig. 3 The thermoresponsive properties of PEGMEMA-MEO2MA-EGDMA copolymers (S2, S3 and S4 in Table 2). (a) LCST behavior of the copolymer in 0.03% w/v deionized water determined by UV-vis spectrophotometer. (b) Polymer solution (S3 in Table 2, 40% w/v) became thermal gel when the temperature was raised above its LCST from 20 °C to 37 °C. | |
In addition, dynamic light scattering (DLS) indicated an significant change in particle size of the copolymer in deionized water when varying the temperature. At 20 °C, the hydrodynamic radius (Rh) of polymer particles were ca. 7.5 nm, and at 37 °C, it was found to be 125 nm (Fig. S3 in ESI†). This is due to the aggregation occurring at temperatures above the LCST. Furthermore, after exposing the copolymer solution samples (with 0.1% w/v of photoinitiator) to UV light (details in the ESI†), the photo-crosslinking of the polymers occurred to form cosslinked gels. The SEM images (Fig. S4†) show that the polymer at a lower concentration (20% w/v) formed a photo-crosslinked gel with a more porous structure than at a higher concentration (40% w/v). More detailed rheological and mechanical studies both on thermally induced and photo-crosslinked gelling copolymers are under way and will be reported in due course.
3T3 mouse fibroblast cell line was utilised for the cytotoxicity assessment of the copolymer. Cells and the polymer/DMEM solution (S3 in Table 2, dissolved in Dulbecco's Modified Eagle's Medium, 0.5 and 1 mg ml−1) were added into each well of a 48-well tissue culture plate. The alamarBlue® reduction method was used to assess changes in cell viability after one and four days. Percentage of reduced alamarBlue® and percentage of cell viability were calculated from eqn S7–S9 (in ESI†). The results showed no significant different of cells viability between the control (cells alone) and polymer samples after four days, indicating the copolymer is not affecting the cellular metabolism at such concentrations (Fig. 4).
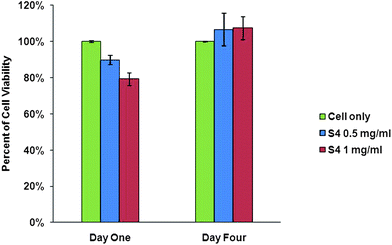 |
| | Fig. 4 Cellular metabolism viability assessment of 3T3 cells after one and four days treatment with PEGMEMA-MEO2MA-EGDMA polymer (S3 in Table 2) using alamarBlue® assay. Cells alone as the control; the polymer concentrations were at 0.5 and 1 mg ml−1. Data plotted shows mean ± standard deviation (n = 3, P < 0.005). | |
In conclusion, we successfully prepared a thermal and photo sensitive hyperbranched copolymer PEGMEMA-MEO2MA-EGDMA via an in situ one-step deactivation enhanced ATRP approach. These copolymers exhibited the LCST around body temperature, which could be specifically adjusted by varying the polymer composition. The unique hyperbranched structure with a high degree of vinyl functional groups enable this dual stimuli responsive polymer to be easily modified further, thus allowing great potential for wide applications, including tissue engineering and drug delivery, polymer composites, UV crosslinkable polymer coatings, prototype mouldings, and micro patterning.
Acknowledgements
Dystrophic Epidermolysis Bullosa Research Association (DEBRA) of Ireland and Austria, Heath Research Board (HRB) of Ireland and Science Fundation Ireland (SFI) are gratefully acknowledged for funding. HT is a EPSRC postdoctoral research fellow at life science interface. Moreover, YD thanks Mr. Yu Zheng at the University of Nottingham for his help with NMR analysis.
References
- Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2001, 53, 321–339 CrossRef CAS.
- E. S. Gil and S. A. Hudson, Prog. Polym. Sci., 2004, 29, 1173–1222 CrossRef CAS.
-
Biorelated Polymers and Gels,, ed. T. Okano, Academic Press, San Diago, CA, 1998 Search PubMed.
- B. Jeong and A. Gutowska, Trends Biotechnol., 2002, 20, 305–311 CrossRef CAS.
- S. Freiberg and X. Zhu, Int. J. Pharm., 2004, 282, 1–18 CrossRef CAS.
- R. X. Liu, M. Fraylich and B. R. Saunders, Colloid Polym. Sci., 2009, 287, 627–643 CrossRef CAS.
- E. Kharlampieva, V. Kozlovskaya, J. Tyutina and S. A. Sukhishvili, Macromolecules, 2005, 38, 10523–10531 CrossRef CAS.
- C. A. Kavanagh, Y. A. Rochev, W. A. Gallagher, K. A. Dawson and A. K. Keenan, Pharmacol. Ther., 2004, 102, 1–15 CrossRef CAS.
- F. Eeckman, A. J. Moes and K. Amighi, Int. J. Pharm., 2002, 241, 113–125 CrossRef CAS.
- M. Hruby, J. Kucka, H. Mackova, O. Lebeda and K. Ulbrich, Chem Listy, 2008, 102, 21–27 CAS.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- A. Chilkoti, M. R. Dreher, D. E. Meyer and D. Raucher, Adv. Drug Delivery Rev., 2002, 54, 613–630 CrossRef CAS.
- H. Vihola, A. Laukkanen, L. Valtola, H. Tenhu and J. Hirvonen, Biomaterials, 2005, 26, 3055–3064 CrossRef CAS.
- I. K. Kwon and T. Matsuda, Biomaterials, 2006, 27, 986–995 CrossRef CAS.
- C. Li, N. J. Buurma, I. Haq, C. Turner, S. P. Armes, V. Castelletto, I. W. Hamley and A. L. Lewis, Langmuir, 2005, 21, 11026–11033 CrossRef CAS.
- J.-F. Lutz, O. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS.
- S. Y. Park, D. K. Han and S. C. Kim, Macromolecules, 2001, 34, 8821–8824 CrossRef CAS.
- W. S. Shim, J. S. Yoo, Y. H. Bae and D. S. Lee, Biomacromolecules, 2005, 6, 2930–2934 CrossRef CAS.
- J.-F. Lutz, K. Weichenhan, O. Akdemir and A. Hoth, Macromolecules, 2007, 40, 2503–2508 CrossRef CAS.
-
A. Hult, M. Johansson and E. Malmstrom, in Advances in Polymer Science: Branched Polymers II, ed. J. Roovers, Springer-Verlag Berlin, Berlin, 1999, vol. 143, pp. 1–34 Search PubMed.
- B. Voit, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2505–2525 CrossRef CAS.
- M. Seiler, Fluid Phase Equilib., 2006, 241, 155–174 CrossRef CAS.
- S. Zalipsky, Adv. Drug Delivery Rev., 1995, 16, 157–182 CrossRef CAS.
- F. M. Veronese, Biomaterials, 2001, 22, 405–417 CrossRef CAS.
- K. T. Nguyen and J. L. West, Biomaterials, 2002, 23, 4307–4314 CrossRef CAS.
- H. Kobayashi, S. Kawamoto, T. Saga, N. Sato, A. Hiraga, T. Ishimori, J. Konishi, K. Togashi and M. W. Brechbiel, Magn. Reson. Med., 2001, 46, 781–788 CrossRef CAS.
- R. B. Greenwald, Y. H. Choe, J. McGuire and C. D. Conover, Adv. Drug Delivery Rev., 2003, 55, 217–250 CrossRef CAS.
- H. Tai, W. Wang, T. Vermonden, F. Heath, W. E. Hennink, C. Alexander, K. M. Shakesheff and S. M. Howdle, Biomacromolecules, 2009, 10, 822–828 CrossRef CAS.
- H. Tai, D. Howard, S. Takae, W. Wang, T. Vermonden, W. E. Hennink, P. S. Stayton, A. S. Hoffman, A. Endruweit, C. Alexander, S. M. Howdle and K. M. Shakesheff, Biomacromolecules, 2009, 10, 2895–2903 CrossRef CAS.
- M. Li, K. Min and K. Matyjaszewski, Macromolecules, 2004, 37, 2106–2112 CrossRef CAS.
- K. Min, W. Jakubowski and K. Matyjaszewski, Macromol. Rapid Commun., 2006, 27, 594–598 CrossRef CAS.
- K. Matyjaszewski, S. G. Gaynor, A. Kulfan and M. Podwika, Macromolecules, 1997, 30, 5192–5194 CrossRef CAS.
- F. Isaure, P. A. G. Cormack, S. Graham, D. C. Sherrington, S. P. Armes and V. Butun, Chem. Commun., 2004, 1138–1139 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and Fig. S1–S4. See DOI: 10.1039/c0py00101e |
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.