Metal catalysts for ε-caprolactone polymerisation†
Abdessamad
Arbaoui
and
Carl
Redshaw
*
Energy Materials Laboratory, School of Chemistry, University of East Anglia, Norwich, UK NR4 7TJ. E-mail: carl.redshaw@uea.ac.uk; Fax: +44 (0) 1603 592003; Tel: +44 (0) 1603 593137
First published on 13th January 2010
Abstract
Economic pressure, environmental issues and ever increasing demand are driving the shift from oil-based polymers to those available from renewable resources. Ring opening polymerisation of cyclic esters is currently a topical field with metal complex-induced coordination/insertion type polymerisation leading the way. Such a polymerisation method offers a wide range of advantages from control over the polymer structure to kinetic enhancement; however industrially available catalysts based on tin suffer from inherent toxicity and as a result a policy change from the Food and Drug Administration (FDA) is not unrealistic. This review underlines the efforts made in the past five years or so, and shows how low toxicity metals are attracting increasing attention in the field of ε-caprolactone polymerisation.
 Abdessamad Arbaoui | Abdessamad Arbaoui was born in 1980 in Tayerza, Morocco. He received his Masters Degree from the University of Lille (2005) in Villeneuve d'Ascq (France) under the supervision of André Mortreux. He also graduated from the National Graduate School of Engineering Chemistry of Lille (2005) before moving to England. He recently received his PhD (2009) under the supervision of Carl Redshaw from the University of East Anglia, and was a postdoctoral research associate in the same group. He has now joined AkzoNobel as a Graduate in Research and Development. |
 Carl Redshaw | Carl Redshaw received his PhD (with John Errington) from Newcastle University. He was a Robert A. Welch Fellow at the University of Texas, Austin (1989–90), and a postdoctoral fellow with the late Professor Sir G. Wilkinson at Imperial College, London (1990–92), and the IRC Durham (1993–95). Following a further stint at Imperial College, he was awarded a Leverhulme Fellowship and moved to the University of East Anglia in 1999, where he was appointed as a lecturer in Inorganic Chemistry, and promoted to Senior Lecturer (2007) and Reader (2009). His research interests include macrocyclic chemistry, catalysis and the use of metal reagents in the battle against Cancer. |
1. Introduction
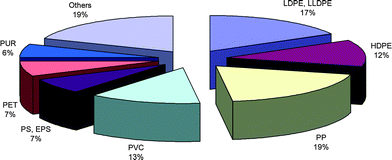
Plastic materials are essential in our every day life and find use in almost all domestic and industrial appliances. Low density and low cost combined with high versatility have made such materials ideal substitutes for ferrous and non-ferrous metallic materials. Commodity polymers are used in packaging, clothing, car manufacturing, etc… Speciality polymers find widespread use typically in electronics, bullet-proof vests and modern aircrafts. The consumption of plastics is mainly driven by the food and packaging sector which, for the UK, represents 38% of all annual plastic consumption.1 In Western Europe, plastic production is estimated at over 60 million tons per year and globally well above 240 million tons per year.2 Indeed, Fig. 1 shows how plastic consumption is now expected to rise by over 5% per year, with experts setting the annual plastic consumption in 2016 at over 400 million tons.3 It is clear that such a high demand will result in increased industrial pressure both in terms of competitiveness and process efficiency, which in turn will lead to demand for new high activity and/or new innovative polymers, all at minimal cost. Polymer production is still widely dominated by poly(olefin)s, with poly(ethylene) (LDPE, LLDPE and HDPE) and poly(propylene) (PP) representing over 47% of Western Europe's annual plastic consumption (Fig. 2).2 These poly(olefin)s raise two issues: (i) they are obtained primarily from limited, price-rising fossil fuels which can contribute to ca. 80% of their total cost, (ii) their inertness makes them environmentally unfriendly as they suffer from a severe lack of biodegradability. For example, poly(ethylene) plastic bags can take up to 400 years to degrade in landfill sites.4 This environmental problem has been overcome by introducing degrading agents such as photo-sensitizers5 that allow for the degradation of the long polymeric chains into shorter oligomeric chains which can in turn undergo degradation by micro-organisms and/or by mixing poly(olefin)s with biodegradable polymers.4 However, petroleum resources are becoming increasingly expensive and the risk of shortage is now seen as inevitable; the only viable way to keep producing commodity polymers in such tonnage, and avoiding releasing CO2 from fossil fuels, is to turn to polymers obtained from renewable resources. It is for these reasons that there is immense current interest in polymers such as poly(lactide). However, it is also worthy of note here that polyethylene can now be produced from bioethanol obtained from sugar cane, as recently patented by Braskem (Brazil).6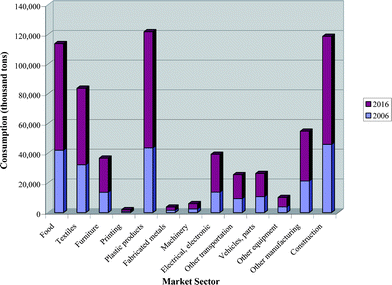 | ||
| Fig. 1 Global annual plastic consumption by market sector in 2006 and 2016 (estimated).3 | ||
Poly(lactide), poly(ε-caprolactone) and copolymers thereof are amongst the most studied polymers, and their biocompatibility makes them ideal for use in numerous biomedical and pharmaceutical applications.7–9 Most of the data available concerning the biocompatibility of poly(ε-caprolactone) involves the development of Capronor™, an implantable contraceptive delivery system. No toxic effects were observed when testing this implant9,10 and the oral LD50 of poly(caprolactone) for rats is expected to be over 2,000 mg kg−1.11
Another attractive aspect of poly(lactide) and poly(ε-caprolactone), besides their inherent biodegradability, is their availability from renewable resources. Whilst lactide is already available from corn starch12,13 and poly(lactide) already produced by companies such as Nature Works (a joint venture between Cargill and Teijin Limited with a 140,000 tons annual production capacity),14 ε-caprolactone is still mainly produced through the Bayer Williger oxidation of cyclohexanone with peracids or hydrogen peroxide (Scheme 1a)15 by companies such as Perstorp (through a subsidiary previously owned by Solvay), BASF and Daicel. However, reports in the patent literature make mention of the possible availability of ε-caprolactone from starch (Scheme 1b),16 making poly(ε-caprolactone) a polymer with high industrial potential.
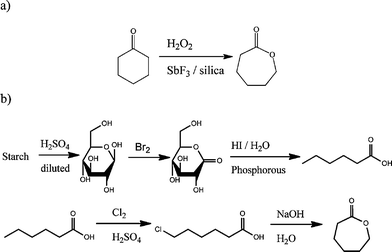 | ||
| Scheme 1 Synthesis of ε-caprolactone monomer (a) via the Bayer Williger oxidation process and (b) from renewable resources. | ||
The main characteristics of poly(ε-caprolactone) (its biodegradability and potential availability from renewable resources) have made this polymer and its copolymers a prefered choice for potential environmentally friendly commodity plastic.17
Poly(ε-caprolactone) is now produced industrially by companies such as Solvay Interox (CAPA®), which was recently acquired by the Perstorp group of Sweden, Dow Union Carbide (Tone®) and Daicel (Placcel®), and the demand for this polymer is ever increasing.18
An upcoming challenge is to produce biodegradable polymers or copolymers based on poly(lactide) or poly(ε-caprolactone) at low cost and with physical properties that would make them industrial competitors to non-biodegradable, well established commodity polymers such as poly(ethylene).
The industrial synthesis of polyesters such as poly(lactide) and poly(ε-caprolactone) is based on more than half a century of research. These polyesters can be formed in a variety of ways: by polycondensation,19 enzymatic polymerisation,20–23 cationic polymerisation,24,25 anionic polymerisation,9,26–29 or coordination/insertion polymerisation.30,31 Complexes based on metals with empty p, d or f orbitals react as coordination catalysts and not as anionic initiators. The use of such catalysts enables the production of high molecular weight polymers via the coordination/insertion mechanism (Scheme 2).
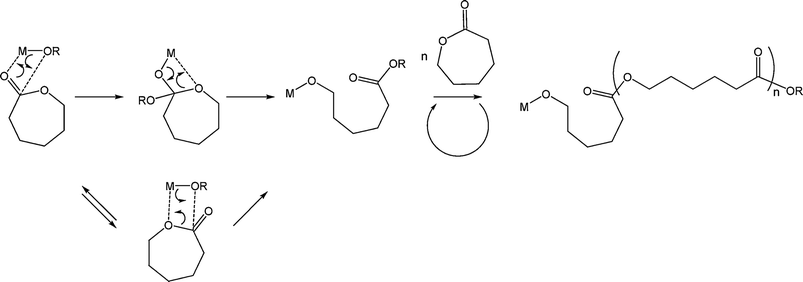
This is often a living process which allows for good control over the polymer molecular weight and affords polymers with very narrow polydispersities and well defined end groups.30,31 Furthermore, this process allows the production of stereoregular polymers (e.g. poly(lactide) or polymers of functionalised ε-caprolactone), whereas epimerisation regularly occurs during the often more active anionic polymerisation; this process also limits side reactions that hamper the anionic polymerisation process.32 The polymerisation mechanism involves the coordination of the monomer to the Lewis acidic metal centre through the exocyclic oxygen. The carbonyl group of the lactone is therefore rendered more susceptible to nucleophilic attack (Scheme 2). At high temperature, or for long reaction times, intermolecular and intramolecular (i.e. back-biting) transesterification reactions become important.
A wide range of metals have been studied for the ring opening polymerisation of ε-caprolactone through the coordination/insertion mechanism; typically magnesium, calcium, aluminium, titanium, iron, zinc, tin, lanthanides and rare earth metals.30
When considering the literature for references on poly(lactide) or poly(ε-caprolactone) synthesis, two problems arise: (i) the lack of a standardised measurement of initiator/catalyst activity and (ii) the deviation between the actual average molecular weight and the one obtained by GPC with reference to polystyrene standards. Wherever possible, these problems have been overcome in this review by (i) quoting the conditions under which the initiators/catalysts have been screened and classifying the activity according to Table 1 and (ii) quoting the corrected values for Mn or by clarifying the measurement method (e.g.1H NMR).
| Monomer/metal ratio | Polymerisation time a | TOF/h−1b | M n(calculated)/g mol−1 | Activity |
|---|---|---|---|---|
| a Polymerisation time at quantitative monomer conversion. b Indicative values only. | ||||
| < 100 | days | < 4 | < 11,400 | low |
| hours | 0.04 to 100 | poor | ||
| minutes | > 1 | moderate | ||
| 100–500 | days | < 21 | 11,400–57,000 | poor |
| hours | 4 to 500 | moderate | ||
| minutes | > 100 | good | ||
| 500–1,000 | days | < 42 | 57,000–114,100 | moderate |
| hours | 21 to 1,000 | good | ||
| minutes | > 500 | high | ||
| 1,000–5,000 | days | < 208 | 114,100–570,700 | good |
| hours | 42 to 5,000 | high | ||
| minutes | > 1,000 | very high | ||
| >5,000 | days | > 30 | >570,700 | high |
| hours | > 205 | very high | ||
| minutes | > 5,000 | exceptional |
A number of excellent reviews that cover different aspects of lactone polymerisation, including the use of enzyme catalysts,13,31 of metal catalysts,13,14,32–35 and of organocatalysts36 have previously appeared in the literature and provide the reader with useful further reading. Herein, we will focus on discrete metal based catalytic systems for the polymerisation of ε-caprolactone that have been developed over the past five years or so.
The review is organised such that for each group of the periodic table, catalytic systems are discussed in terms of ligand classification. After first reviewing metallocene-based systems (wherever applicable), post metallocene complexes bearing ligands bound through oxygen only are discussed, followed by those combining a mixed nitrogen/oxygen donor set, and then those utilising only nitrogen. Finally, any ligand systems not covered by the four preceding sections are discussed.
2. Alkali and alkali earth metal complexes
2.1. Complexes based on phenolate ligands
Alkali metal based compounds are often associated with anionic polymerisation. The anionic ring opening polymerisation often leads to very active systems, but with poor side reaction control and therefore broad polydispersities.32Nevertheless, Chang and Liang reported the synthesis and catalytic screening of alkali metal complexes supported by a bis(phenolate) phosphine ligand (1–3, Chart 1).37 Compound 1 led to 91% conversion after 12 h, at 80 °C and for a monomer : metal ratio of 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Despite the reaction yielding polymers with a higher molecular weight than expected for a living polymerisation, the low polydispersity observed (< 1.6) suggested a controlled mechanism with low occurrence of transesterification reactions.
:1. Despite the reaction yielding polymers with a higher molecular weight than expected for a living polymerisation, the low polydispersity observed (< 1.6) suggested a controlled mechanism with low occurrence of transesterification reactions.
 | ||
| Chart 1 Alkali metal complexes M = Li (1), Na (2), K (3). | ||
2.2. Ligands containing mixed nitrogen/oxygen donor sets
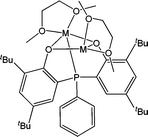
Using a bis(phenolate) amine ligand bearing a pendant nitrogen donor arm, Bochmann and co-workers have investigated the synthesis and catalytic behaviour of the complexes 4 and 5 (Chart 2).38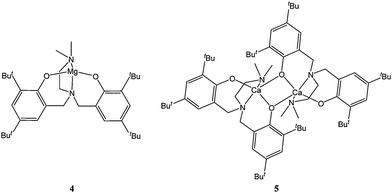 | ||
| Chart 2 Alkali earth metal amine bis(phenolate) complexes 4 and 5. | ||
At 60 °C and for a monomer : metal ratio of 200:1, the magnesium complex 4 proved to be poorly active, reaching only 5% conversion after 24 h. The calcium analogue 5 was somewhat more active, reaching 40% conversion after 12 h. The low polydispersity (< 1.4) suggested once again a polymerisation with limited side reactions. However, analysis of GPC traces showed in this case the broadening of the main peak during the polymerisation and the formation of a shoulder indicative of transesterification reactions which occur generally for long polymerisation times and at high temperature.39
A current trend in ring opening polymerisation catalyst design involves the use of dinuclear complexes driven by attempts to mimic the mechanistic behaviour of some hydrolases.40 Ligand frameworks suited to supporting such dinuclear complexation often include diimino or diamino phenolate structural motifs (see also section 7.2). Hillmyer, Tolman and co-workers reported the synthesis of complex 6 (Fig. 3).41 The dinuclear complex 6 displayed poor activity, leading to quantitative conversion after ca 3 h, at room temperature and for a monomer : metal ratio of 100:1.
 | ||
| Fig. 3 Di-nuclear magnesium complex 6. | ||
Ketiminate ligands have also been used to support magnesium complexes. Five coordinate magnesium complexes 7 and 8 (Chart 3) were synthesised by reaction of dibutyl magnesium with two equivalents of the ketimine ligand followed by treatment with one equivalent of ketimine ligand (7) or excess pyridine (8).42
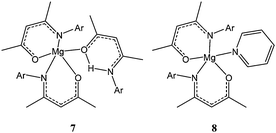
Interestingly, both compounds were found to be inactive towards ε-caprolactone polymerisation at room temperature and were only moderately active at 70 °C, complex 8 leading to 98% conversion after 3 h and for a monomer : metal ratio of 200:1. The polydispersity indexes (PDI) obtained for these two catalysts (up to 3.2) combined with an observed polymer molecular weight significantly higher than expected (e.g. Mn(measured) 32,310 vs Mn(calculated) 22,370 g mol−1) showed poor control over the polymerisation process, possibly due to ligand exchange at this temperature.
2.2. Ligands with nitrogen donor sets only
The use of bulky heteroscorpionate ligands to support alkyl magnesium complexes such as 9–14 (Chart 4) 43 led to moderate to good polymerisation activities. For instance, mononuclear complex 14 led to 98% conversion at room temperature with a monomer : metal ratio of 500:1.
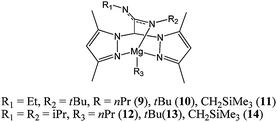 | ||
| Chart 4 Heteroscorpionate magnesium complexes 9–14. | ||
However, the polydispersity (PDI up to 1.5) and the lack of control over the polymer molecular weight (Mn(corrected) 22,790 vs Mn(calculated) 55,930 g mol−1) suggested that the polymerisation was not as controlled as the lactide polymerisation reported for these complexes. Lowering the polymerisation temperature, however, led to a decrease in polydispersity index to 1.1, consistent with suppression of back-biting and transesterification reactions. The β-diketiminate magnesium complex 15 (Chart 5), reported by the group of Bochmann, displayed good activity at room temperature, whilst the dimeric magnesium hydroxo analogue 16 was inactive under the same conditions.44
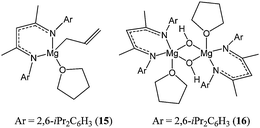 | ||
| Chart 5 β-diketiminate magnesium complexes 15 and 16. | ||
Some magnesium complexes have also been shown to be active at very low catalyst loadings. This is of great importance, and allows access to very high molecular weight polymers.45 Complex 17 (Fig. 4) afforded 33% monomer conversion after 30 min at 110 °C and for a monomer : metal ratio of 1,000 : 1. However, the recorded molecular weight was quite low (Mn(corrected) 3,360 g mol−1) compared with the one expected for this component ratio and at this conversion (Mn(calculated) 37,210); the polydispersity index (PDI 1.6) also suggested a lack of control over the polymerisation process.
 | ||
| Fig. 4 Magnesium complex 17. | ||
Despite their high activities (quantitative conversions in minutes in some cases), alkali and alkali earth metal complexes suffer from a lack of control over the polymerisation process (large polydispersity index and significant disparity with the predicted molecular weight). Employing bulkier ligands is one option to bring some control to these catalysts, however possibly an easier alternative is to move to the use of other metal systems.
3. Rare earth metal complexes
Rare earth metal alkoxides are generally very active for the ring opening polymerisation of cyclic esters; the polymerisation rate obtained is comparable to that of an anionic polymerisation.32 The use of bulky ligands reduces transesterification reactions and usually affords polymers with narrow polydispersities.31,463.1. Metallocene-based catalytic systems
In several reports, Wang and co-workers reported the synthesis and catalytic evaluation of organolathanide(II) complexes bearing indenyl or fluorenyl ligands functionalised with nitrogen or oxygen donor arms.Compounds where Ln = Yb and Eu (i.e.18–29, Chart 6) were synthesised by the reaction of the Ln(III) starting material ([Me3Si]2N)3Ln(μ-Cl)Li(THF)3 with the corresponding neutral indene derivatives. The mechanism of formation was thought to involve a reductive elimination step that is not observed when reacting a Ln(III) starting material with an unsubstituted indenyl ligand.47,48 When Ln = Sm or Nd, the Ln(III) complexes 30 and 31 (Chart 7) were obtained from the same reaction rather than Ln(II) complexes; their formation pathway involved a silylamine elimination followed by a ligand redistribution.49,50
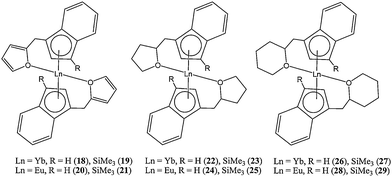 | ||
| Chart 6 Organolanthanide(II) complexes (18–29) bearing an oxygen donor arm | ||
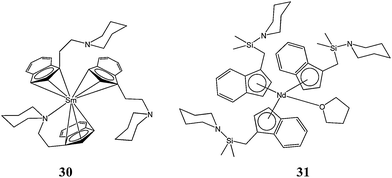 | ||
| Chart 7 Organolanthanide(III) complexes 30 and 31. | ||
The catalytic activity of these organolanthanide complexes depended strongly on the nature of the substituents on the indenyl moiety. The trimethylsilylated complexes (19, 21, 23, 25, 27 and 29) displayed good to high activity for the ring opening polymerisation of ε-caprolactone, whereas their non-silylated counterparts (18, 20, 22, 24, 26 and 28) were completely inactive.47,51
The silyl group, crucial for polymerisation activity, can be incorporated into the bridge between the indenyl ring and the heteroatom donor and still yield active catalysts (32–35, Chart 8). For instance, complex 32 was found to be moderately active at 30 °C, yielding 57% monomer conversion after 20 min and for a monomer : metal ratio of 500:1.49
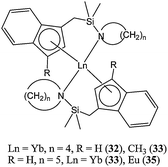 | ||
| Chart 8 Organolanthanide(II) complexes (32–35) bearing a nitrogen donor pendant arm. | ||
In search of possible improvement in terms of solubility in non-polar organic solvents, the same group recently synthesised lanthanide(II) complexes (36–39, Chart 9) supported by functionalised fluorenyl ligands.52 These catalysts showed good activity, with complex 36 reaching 99% conversion after 30 s at 30 °C in toluene and for a monomer : metal ratio of 500:1. The polydispersity index remained very low in this case (PDI = 1.08) indicative of a controlled polymerisation process.
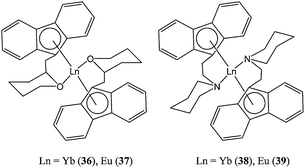 | ||
| Chart 9 Organolanthanide(II) complexes (36–39) supported by fluorenyl ligands. | ||
The polymerisation pathway for all Ln(II) catalysts was thought to involve oxidation of the Ln(II) metal centre by the first monomer unit, followed by a coordination/insertion mechanism as described in Scheme 3.
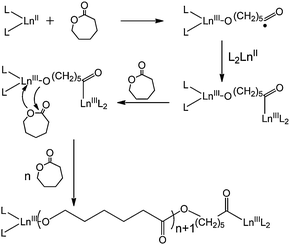 | ||
| Scheme 3 Polymerisation mechanism proposed by Wang and co-workers.48 | ||
The samarium(III) and neodymium(III) complexes 30 and 31 were also active for ε-caprolactone polymerisation, although the polymerisation mechanism would be expected to be different to the one for the Ln(II) species.53 Rare earth metal silylamido complexes supported by bridge indenyl ligands 40 and 41 (Chart 10) displayed good activity, leading to quantitative conversion after 1 min at 30 °C and for a monomer : metal ratio of 500:1.48
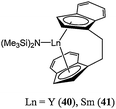 | ||
| Chart 10 Ethylenebis(indenyl) rare earth complexes 40 and 41. | ||
Lanthanide(III) complexes supported by cyclopentadienyl or methylcyclopentadienyl ligands, Cp3Ln (Ln = Y (42), Er (43), Sm (44)) or (MeC5H4)3Ln (Ln = Er (45), Sm (46)), were moderately active for the ring opening polymerisation of ε-caprolactone.
Complex 42 yielded 98% conversion after 1 h at room temperature and for a monomer : metal ratio of 500:1. The broad polymer molecular weight distribution (PDI up to 2.51) and the difference between calculated and observed polymer molecular weight (e.g. Mn(corrected) 14,170 vs Mn(calculated) 57,070) suggested poor control over the polymerisation process and the occurrence of side reactions.54 Roesky and co-workers also investigated heterobimetallic complexes containing a Ln(III)–O–Al unit (47–50, Chart 11).55
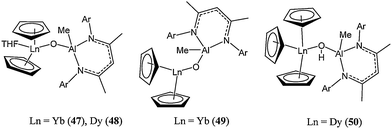 | ||
| Chart 11 Organolanthanide(III) complexes (47–50) containing and Ln–O–Al motif (Ar = 2,6-iPr2C6H3). | ||
The cyclopentadienyl based complexes showed moderate activity for the ring opening polymerisation of ε-caprolactone; complex 47 reaching 95% conversion after 30 min, at 70 °C in toluene and for a monomer : metal ratio of 100:1.
3.2. Lanthanide alkoxides and related compounds
Given the form of the proposed propagating species for organolanthanocene(II) complexes (see Scheme 3), simple systems based on alkoxide or aryloxide ligands are of interest for ε-caprolactone polymerisation.Zhang and co-workers investigated the use of bulky aryloxide ligands such as 2,4,6-tri-tert-butylphenolate.56 The complex La(2,4,6-tri-tert-butylphenolate)3 (51, Chart 12) was found to be a very highly active single component system for the ring opening polymerisation of ε-caprolactone, affording 100% conversion after 20 min, at 20 °C and for a monomer : metal ratio of 1,500:1. However, the complexes Ln(III)(2,4,6-tri-tert-butylphenolate)351–56 led to polymers with a large molecular weight distribution (PDI = 2.10 at 20 °C and 3.53 at 0 °C). Furthermore, the molecular weights obtained differed from those expected even when more than one active aryloxide per metal was considered. The polymerisation seemed to be hampered by unavoidable side reactions when using such monodentate ligands.
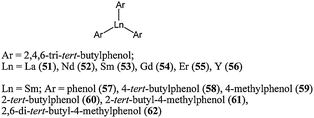 | ||
| Chart 12 Aryloxide lanthanide complexes 51–62. | ||
In order to understand whether the bulk of the ligand had an effect on the catalytic performance of these Ln(III) aryloxide complexes, Shen and co-workers synthesised a series of samarium complexes (57–62, Chart 12) bearing substituted phenolate ligands.57 At 60 °C, after 60 min and for a monomer : metal ratio of 800:1, the activity trend (61 ≈ 62 > 60 > 59 > 58 ≈ 57) revealed the importance of bulky ortho substituents on the aryl rings. The absence of ortho substituent led to moderately active (59) or even inactive complexes (57 and 58). Surprisingly, the polydispersity index followed almost the opposite trend : increasing PDI values were in the order 59 < 60 ≈ 61 < 62. Ortho substituted tert-butylphenolate afforded more active species but the repulsion between ligands around the metal centre tended to favour transesterification reactions. A cationic analogue of complex 62 was synthesised and screened for ε-caprolactone polymerisation.58 The samarium(III) complex (63, Fig. 5) was obtained via oxidation of Sm(OAr)2(THF)3 (Ar = 2,6-di-tert-butyl-4-methylphenol) by AgBPh4. The cationic complex 63 was found to be highly active, reaching quantitative conversion after 3 min, at 20 °C and for a monomer/metal ratio of 1000:1. This catalyst was as active as its neutral Sm(II) analogue, and much more active than its Sm(III) analogue Sm(OAr)3(THF)2 (Ar = 2,6-di-tert-butyl-4-methylphenol). This difference in activity was explained by the improved electrophilicity for the samarium, leading to enhanced coordination to the monomer. It is noteworthy here that a single crystal X-ray diffraction study showed an absence of an interaction between the cation and the B(Ph)4− anion in the solid state.
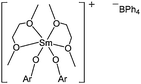 | ||
| Fig. 5 Complex 63 (Ar = 2,6-tBu2-4-Me-C6H2). | ||
Rare earth metal trifluoromethanesulfonate complexes have also been investigated by the groups of Kunioka and N. Nomura.59,60 The air and moisture stable yttrium(III) trifluoromethanesulfonate complex, Y(OSO2CF3)3, was tested as a polymerisation catalyst using unpurified monomer and in air. Under these conditions, Y(OSO2CF3)3 was inactive as a single component at 60 °C. However, the introduction of initiators such as alcohols led to active, albeit poorly, systems. Hydrated Y(OSO2CF3)3, in the presence of 5 equivalents of 1,3-propanediol, yielded 70% monomer conversion after 48 h, at 60 °C and for a monomer : metal ratio of 500:1.59 Under dry conditions, the complex Sc(OSO2CF3)3 displayed poor activity reaching quantitative conversion after 2 h at room temperature, for a monomer : metal ratio of 50:1, in the presence of 1 equivalent of benzyl alcohol.60
Recycling of Ce(OSO2CF3)3 was achieved through a biphasic polymerisation process, using an ionic liquid and toluene. The recyclable catalyst was poorly active, giving only low molecular weight polymer; however, its catalytic properties were retained for up to three polymerisation cycles.60 Gauvin, Mortreux and co-workers also investigated immobilized lanthanide complexes.61 Preliminary studies showed lanthanum amides grafted on silica formed highly active catalysts in the presence of 600 equivalents of monomer.61
Lanthanide(III) thiolate complexes such as [Sm(SC6H5)3(OP(NMe2)3)3] (64, Fig. 6) were investigated for their catalytic properties. Complex 64 showed good activity and yielded 90% conversion after 20 min, at 35 °C and for a monomer : metal ratio of 500:1.62 The polydispersity indexes of the polymers obtained were large and indicative of transesterification side reactions.
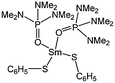 | ||
| Fig. 6 Samarium thiolate complex 64. | ||
Bis(phenolate) ligands have been used by Shen and co-workers to support lanthanide alkoxide, amide or chloride complexes (65–74, Chart 13)63,64
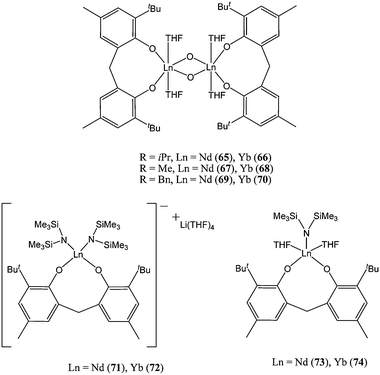 | ||
| Chart 13 Anionic and neutral lanthanide complexes 65–74 supported by bis(phenolate) ligands. | ||
When comparing complexes 65–74, the neodymium based catalysts appeared to be more active than their ytterbium counterparts. At 50 °C in toluene and for a monomer : metal ratio of 400:1, catalyst 65 reached quantitative conversion after 1 h, whereas the ytterbium catalyst 66 yielded 15% monomer conversion in the same polymerisation time.63 This activity trend was consistent with previous observations.49 Furthermore, the anionic complexes were more active than their neutral counterparts: i.e.71 > 73 and 72 > 74. Although, for the anionic complexes, the polymer molecular weight distribution was found to be wider than that obtained from the neutral catalysts (e.g. PDI = 1.85 for 72vs. 1.32 for 74). The difference in activity may well stem from an enhanced nucleophilicity of the amido group and the coordination of the lithium counter ion to the caprolactone monomer, rendering the latter more susceptible to nucleophilic attack.14
With these last observations in mind, the same group has investigated the synthesis of heterobimetallic complexes such as 75–83 (Chart 14).65–67 The complexes 75–83 proved to be exceptionally active polymerisation catalysts. At room temperature, in toluene and for a monomer : metal ratio of 15,000 : 1, catalyst 75 achieved quantitative conversion after 1 min.65 The polydispersity indexes obtained were slightly lower than those for the lithium salt 72. The polymer molecular weight obtained suggested, unsurprisingly, the involvement of more than one alkoxide group per lanthanide in the polymerisation process and the occurrence of transesterification reactions. The activity trend, 75 > 76 > 78, was in accordance with results for related neutral lanthanide alkoxides (vide supra). Fluorinated alkoxide and tert-butoxide bridged mixed lanthanide/sodium cage complexes were also synthesised. The fluorinated alkoxide derivatives 81–83 showed very high activity for ε-caprolactone polymerisation, although lower than the activity described for 75–80, possibly due to a lower nucleophilicity of the alkoxide groups.66,68
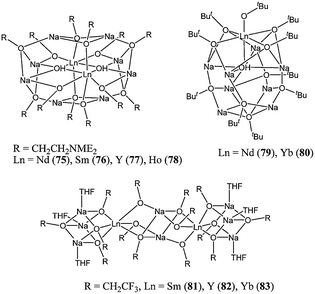 | ||
| Chart 14 Heterobimetallic cage structures of complexes 75–83. | ||
3.3. Ligands with mixed nitrogen/oxygen donor sets
Along with the carbon bridged bis(phenolate) ligands, amine bis(phenolate) ligands are amongst the most utilised ancillary ligands for metal involved in caprolactone polymerisation. The effect of extra coordination by the nitrogen bridge and by potential pendant donor arms on catalysts activity is discussed below.The lanthanide(II) complexes 84–90 (Chart 15) supported by amino bis(phenolate) ligands were synthesised by reaction of the corresponding [Ln(N(SiMe3)2)2(THF)2] starting material with the neutral ligand.69–71 Among these Ln(II) complexes, the monomeric Yb(II) complex 84 reported by Shen and co-workers was the most active, reaching quantitative conversion after 5 min, at room temperature and for a monomer : metal ratio of 1,000 : 1.69
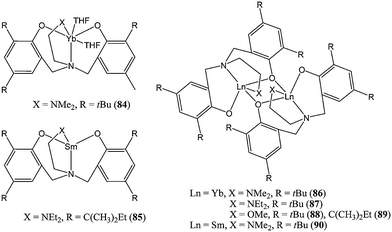 | ||
| Chart 15 Lanthanide(II) complexes (84–90) supported by amine bis(phenolate) ligands. | ||
Lanthanide(III) complexes supported by amino bis(phenolate) ligands have received more attention than their Ln(II) counterparts. Complexes 91–95 (Chart 16) were synthesised by oxidation of compounds 86 or 88 by AgPF6, AgCF3SO3, tert-butanol and phenol, respectively. Complexes 96 and 97 were synthesised from the reaction of [Yb(N(SiMe)3)2)3] and the appropriate neutral ligand.70 Complexes 91–95 were inactive towards ε-caprolactone polymerisation; whereas the amido complexes 96 and 97 displayed good activity. Compound 96 led to quantitative conversion after 10 min, at room temperature and for a monomer : metal ratio of 200:1. The disparity in catalytic activity could once again be attributed to poor nucleophilicity (R = PF6 or CF3SO3) or steric hindrance (R = Ot-Bu) of the initiating Ln–R group. Interestingly, the chloride analogues 98–104 (Chart 17) were catalytically active, although they only displayed low activities.72 The activity order followed the classical trend (vide supra): i.e.101 ≈ 100 > 102 > 103 > 104.
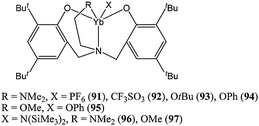 | ||
| Chart 16 Ytterbium(III) complexes 91–97. | ||
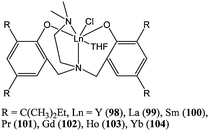 | ||
| Chart 17 Lanthanide(III) chloride complexes 98–104. | ||
The samarium(III) amido complex 105 (Chart 18) showed good activity, yielding 67% conversion after 2 min at room temperature and for a monomer : metal ratio of 275:1. The zwitterionic analogues 106 and 107 (Chart 18) were in the same activity range, but yielded polymers with higher molecular weight (Mn(corrected) = 28,980 and 51,280 for 106 and 107 respectively, vs. 16,830 g mol−1 for 105).73
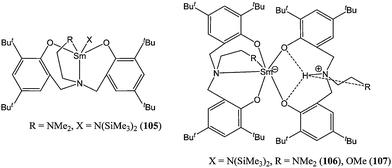 | ||
| Chart 18 Samarium(III) complexes 105–107. | ||
As part of the same study, organolanthanide(III) complexes supported by amine bis(phenolate) ligands (108 and 109, Chart 19) were found to be poorly to moderately active.
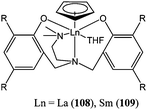 | ||
| Chart 19 Organolanthanide(III) complexes 108 and 109. | ||
The lanthanum(III) complex 108 was much more active than its samarium(III) analogue. At 50 °C, after 4 h and for a monomer : metal ratio of 200:1, the samarium complex 109 reached 2% conversion against 93% conversion for the lanthanum complex 108.74 Highly active amine bis(phenolate) lanthanide(III) complexes were reported by Shen and co-workers. The ytterbium(III) and erbium(III) amido complexes 110 and 111 (Chart 20) afforded 95 to 97% conversion after 1 min at room temperature and for a monomer : metal ratio of 1,000 : 1.75 Unsurprisingly, the methyl complex 112 was less active than its amido analogues, reaching 33% conversion after 30 h under similar polymerisation conditions.
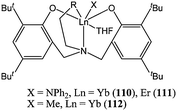 | ||
| Chart 20 Lanthanide(III) complexes 110–112. | ||
Amine bis(phenolate) ligands were also used to support a series of rare earth borohydride complexes (113–115, Chart 21). Complexes 113–115 were synthesised by reacting Ln(BH4)3(THF)3 with the sodium salt of the corresponding ligand.76 These complexes displayed good activity for the ring opening polymerisation of ε-caprolactone with 115 yielding 90% conversion after 30 s, at room temperature and for a monomer : metal ratio of 275:1. Corrected polymer molecular weights were in good agreement with calculated values which is indicative of a controlled polymerisation with limited side reactions.
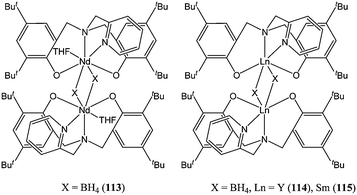 | ||
| Chart 21 Rare earth borohydride complexes 113–115. | ||
It is noteworthy here that other rare earth metal borohydride complexes were used for ε-caprolactone polymerisation such as rare earth metal tris(borohydride) Ln(BH4)3(THF)3 (Ln = La (116), Nd (117), Sm (118)) and the cationic bis(borohydride) complexes [Ln(BH4)2(THF)5]+[BPh4]− (Ln = Y (119), La (120), Nd (121), Sm (122)).77,78 Complex 117 afforded quantitative conversion after 15 min, at room temperature and for a monomer : metal ratio of 664:1. At high catalyst concentration (monomer : metal ratio lower than 250:1), calculated and experimental polymer molecular weights were in good agreement; at lower catalyst concentration however, a deviation was observed between Mn(calculated) and Mn(measured). This deviation was attributed to transfer reactions under these polymerisation conditions.77 Cationic complexes 119–122 showed good polymerisation activity. Although the polydispersity indexes were relatively low (PDI < 1.38), the low polymer molecular weights obtained were again indicative of transfer reactions.78
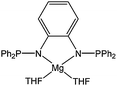
Kerton and co-workers used a high-throughput approach to screen a large number of rare earth metal complexes with the general formula Ln(L)(N(SiMe2R)2) where Ln = Y, La, Pr, Sm, Gd or Yb, R = H or Me and L is an ONNO type ligand.79 This screening confirmed features discussed previously for complexes 57–62: ortho substituted rather than non-substituted phenolate ligands are able to give species active in polymerisation. However, unlike for the lanthanide aryloxide complexes 57–62, the bulky tert-pentyl and tert-butyl ortho substituents seemed to yield polymers with narrower molecular weight distributions. The phenoxytriamine complex 123 (Fig. 7) was obtained by reaction of Y(N(SiMe2H)2)3(THF)2 with the neutral ligand. This ligand contrasts with those reported in this section thus far as it behaves as a tridentate monoanionic ligand set leading to a di-amido yttrium complex.80
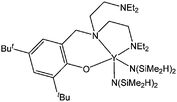 | ||
| Fig. 7 Yttrium phenoxytriamine complex 123. | ||
Complex 123 was highly active at room temperature, affording 98% conversion after 30 min and for a monomer : metal ratio of 1,000 : 1. A similar strategy was adopted by Delbridge and co-workers81 when reacting the potentially tridentate monoanionic ligand L1H (Chart 22) with Ln(N(SiMe3)2)3. Instead of binding in a tridentate fashion as expected, the ancillary ligand bound in a bidentate mode affording the di-phenolate complexes 124–128 (Chart 22).
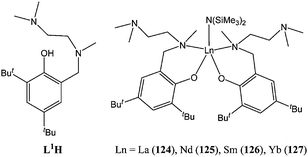 | ||
| Chart 22 Phenolate ligand L1H and its lanthanide complexes 124–127. | ||
Treatment of complex 124 with one equivalent of [Et3NH][BPh4] afforded the cationic complex [L12La][BPh4] (128). Compounds 124–128 were moderately active catalysts, affording quantitative conversions in 1 to 30 min, at room temperature and for a monomer : metal ratio of 100:1. Lactide polymerisation studies monitored by ESI mass spectrometry showed that the phenolate ligands also acted as initiating groups; the lability of the ancillary ligand may stem from its binding mode bidentate instead of the more stationary tridentate fashion. It was envisaged that the use of di-anionic tridentate phenolate and di-anionic tetradentate di-phenolate ligands (L2H2 and L3H2, Chart 23) would prevent the anchoring ligand from initiating the polymerisation process.82 With this in mind, treatment of L2H2 or L3H2 with [Yb(N(SiMe3)2)2(THF)2] afforded the ytterbium(II) complexes [Yb(L2)]2 (129) and [Yb(L3)]2 (130, Chart 23). Compound 129 was further oxidised to give the ytterbium(III) complexes [Yb(L2)(OSO2CF3)] (131) and [Yb(L2)(OPh)] (132). Direct transamination reaction between [Ln(N(SiMe3)2)3] and L2H2 afforded the silylamido lanthanide(III) complexes [Ln(L2)(N(SiMe3)2)(THF)x] (Ln = La, x = 2 (133); Ln = Sm, x = 1 (134); Ln = Yb, x = 1 (135)).
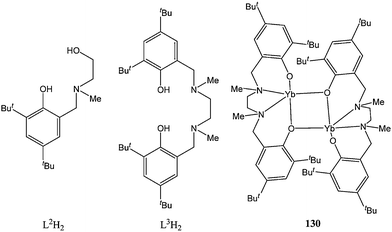 | ||
| Chart 23 Ligands L2H2 and L3H2 and complex 130. | ||
Catalysts 129–135 displayed poor to moderate activity with the exception of complex 131 which was inactive in ring opening polymerisation, consistent with results obtained for Y(OSO2CF3)3.59 ESI mass spectrometry showed that the ancillary ligand remained bound to the metal centre during the polymerisation.
Making use of the labile properties of monoanionic bidentate ancillary ligands, Shen and co-workers reported the homoleptic complexes 136–138 (Chart 24) for which the chelating phenoxyimine was needed to initiate the ring opening polymerisation.83 Complexes 136 and 137 were moderately active for ε-caprolactone polymerisation. Both complexes reached 97 to 98% conversion after 3 h at 70 °C and for a monomer : metal ratio of 500:1. Complex 138 was inactive under these polymerisation conditions, which was first attributed to the bulk of the ligand in the case of 138 preventing monomer coordination to the metal centre. However, in light of the differing behaviour of bidentate and tridentate monoanionic ligands discussed previously, the difference of activity between 136, 137 and 138 was most probably due to the inertness of the chelating tridentate ligand in 138. This was confirmed by the deviation between calculated and measured polymer molecular weights obtained with catalysts 136 and 137. For both complexes, the relationship Mn(calculated) < Mn(measured) indicated that (1) there was at the most only one initiating group per metal centre and that (2) only ca. 80% (for 136) and ca. 89% (for 137) of catalytic species were active in the polymerisation process. Bidentate monoanionic ligands, despite being labile, are not as efficient in the first step (initiation) of polymerisation as monodentate ligands such as an aryloxide. Schafer and co-workers also investigated the initiation properties of the homoleptic rare earth metal complexes 139–141 (Chart 25) bearing labile amidate bidentate ligands.84 Complexes 139–141 showed good catalytic activity at room temperature with 139 yielding 63% conversion after 15 min and for a monomer : metal ratio of 500:1. Again, the deviation between measured and predicted polymer molecular weights (Mn(measured) > Mn(calculated)) suggested that only part of the catalyst was involved in the polymerisation process.
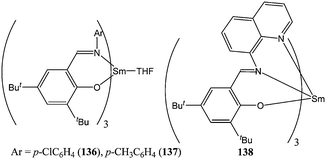 | ||
| Chart 24 Samarium(III) complexes 136–138. | ||
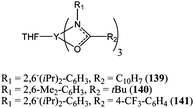 | ||
| Chart 25 Yttrium(III) amidate complexes 139–141. | ||
The main drawback of using homoleptic bidentate ligands seemed to be the lack of control over the polymer molecular weight. The behaviour of such catalysts is intermediate between lanthanide alkoxide complexes (high activity, large polydispersity) and lanthanide complexes supported by bulky spectator ligands such as amino bis(phenolate) (lower activity, narrow polydispersity).
3.4. Ligands with nitrogen donors
In an effort to further explore the reactivity of (L)LnR2 complexes, the attention of several research groups turned to β-diketiminate and related ligands. (L)LnR2 type complexes tend to dimerise (as in 150 and 151) or retain solvent molecules (as in 142–145). Residual solvent molecules can hamper the polymerisation reaction as they may compete with the monomer for coordinating to the metal centre, whereas bridged di-nuclear species may exhibit enhanced activity due to the proximity of the two metal centres.40,85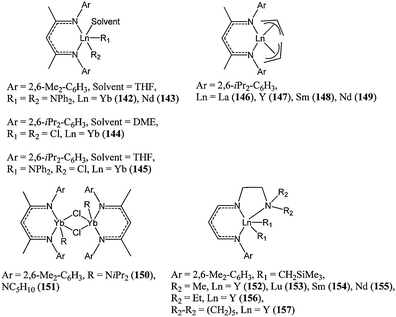 | ||
| Chart 26 β-Diketiminate rare earth metal complexes 142–157. | ||
The diamido complexes 142 and 143 showed very high activity, with 143 reaching 96% conversion after 15 min at room temperature and for a monomer : metal ratio of 1,500 : 1. Consistent with previous observations, the neodymium catalyst was more active than its ytterbium counterpart. The polymer molecular weight obtained suggested that both amido groups were active during the polymerisation.86 Allyl groups are also active initiators, and indeed complexes 146–149 were very active catalysts with 146 displaying very high activity, reaching 94% conversion after 20 min at room temperature and for a monomer : metal ratio of 5,000 : 1.87 Decreasing the polymerisation temperature to −50 °C led to narrower polymer molecular weight distribution concomitant with a slower polymerisation reaction.
Reports have suggested low activity for halide initiating groups when compared to alkoxide, amide and alkyl groups.88 This trend was verified for complexes 144, 145, 150 and 151.89 The mononuclear dichloride complex 144 was moderately active, leading to 16% conversion after 16 h at room temperature and for a monomer : metal ratio of 200:1. Under similar conditions, the mixed chloride/amido catalysts 145, 150 and 151 led to quantitative conversion after 1 min. Although end chain analysis and polymer molecular weight suggest that the β-diketiminate ligand was retained on the metal centre and was not an initiating group during the polymerisation, Chen and co-workers investigated the design of a tridentate monoanionic ligand based on a β-diketiminate moiety.90 Complexes 152–157 showed very high activity for ε-caprolactone polymerisation; however, the large deviation between predicted and measured polymer molecular weights was indicative of side reactions. The relatively bulkier catalysts 156 and 157 showed a slightly better control over the polymerisation process leading to a two-fold increase in polymer molecular weight.
Given the success of metal complexes supported by amine bis(phenolate) ligands bearing a pendant donor arm, Shen and co-workers used another dianionic tetradentate ligand, a bridged bis(amidinate) ligand, to support the di- and mono-nuclear rare earth metal complexes (158–163, Chart 27). Catalysts 158–161 displayed very high activity for the polymerisation of ε-caprolactone with 158 yielding quantitative conversion after 30 min at room temperature and for 2,500 equivalents of monomer.91 However, the high polydispersity indexes (PDI up to 2.80) and the deviation between predicted and measured polymer molecular weights indicated a lack of control over the polymerisation process. Lower polydispersity indexes were obtained using catalysts 162 and 163.92 Interestingly, 163 was only moderately active at room temperature, but became highly active at 40 °C without broadening of the molecular weight distribution. Contrastingly, similar treatment of 162, which was highly active at room temperature, led to a broadening of the polymer molecular weight distribution. It may be that compound 163 retains a di-nuclear structure at room temperature with less active μ-OiPr bridges, whereas at 40 °C, dissociation leads to terminal OiPr groups which are more active for polymerisation.
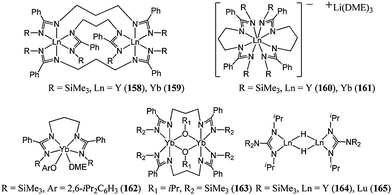 | ||
| Chart 27 Bis(amidinate) and guanidinate rare earth metal complexes 158–165. | ||
Guanidinate rare earth metal complexes 164 and 165 were moderately active at room temperature. For these hydrido dimeric complexes, the relationship Mn(measured) = 2 × Mn(calculated) was observed. Furthermore, 1H NMR spectroscopy did not reveal the expected C(O)H end group. This suggested a more complex initiation mechanism that may involve the reduction of the first caprolactone monomer leading to an initiating species of the form Ln-O-(CH2)6-O–Ln.93
Other nitrogen donor ligands have been studied for their ability to support rare earth metal complexes. The halide complex 166 (Chart 28)94 was found to be inactive for ε-caprolactone polymerisation, despite being a mixed alkali/rare earth metal complex akin to the exceptionally active compounds 75–83.
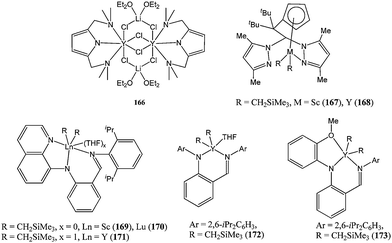 | ||
| Chart 28 Other rare earth complexes bearing nitrogen donor ligands (166–173). | ||
The heteroscorpionate rare earth metal complexes 167 and 168 were moderately active for ε-caprolactone polymerisation, in contrast to the good activity displayed by the magnesium heteroscorpionate complexes 9–14.95 Cui and co-workers reported the use of a tridentate monoanionic ligand to support rare earth metal complexes such as 169–171 (Chart 28) and compared their polymerisation activity with complex 172 bearing a bidentate ligand and complex 173 bearing an NNO tridentate ligand.96 The activity trend 171 ≈ 170 ≈ 173 > 172 > 169 reflected the influence of the nature of the metal centre on the polymerisation activity. Complexes 169–173 were highly active catalysts and exhibited good control over the polymerisation process with Mn(calculated) ≈ Mn(measured).
Roesky and co-workers used bulky phosphiniminomethanide ligands to support rare earth metal complexes in an attempt to produce poly(ε-caprolactone) with narrow polydispersity.97–99 Among complexes 174–182, the most active system was the samarium(II) catalyst 182 (Chart 29), which showed very high activity, producing high molecular weight polymer and yielding 95% monomer conversion at room temperature after 1 h and for a monomer : metal ratio of 10,000:1. However, the polymer molecular weight distribution obtained with catalyst 182 was quite large with PDI up to 2.75.98 Better polydispersity indexes were obtained when using complexes 174–176 activated in situ by addition of iso-propanol. The system 174/iPrOH was moderately active, leading to quantitative conversion after 90 min, in the presence of one equivalent of iso-propanol, at room temperature and for a monomer : metal ratio of 422:1. The polymerisation proceeded here with minimum transesterification reactions as the polymer molecular weights obtained were close to the predicted values and the polydispersity indexes remained low (PDI < 1.20).99
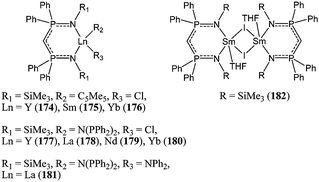 | ||
| Chart 29 Bis(phosphinimino)methanide rare earth metal complexes 174–182. | ||
4. Group IV metal complexes
4.1. Titanocene and zirconocene derivatives
Given the success of group IV metallocenes in α-olefin polymerisation over the past three decades,100–107 the ability of such compounds to polymerise cyclic esters has been studied with interest.Royo and co-workers reported the catalytic screening of complexes 183 and 184 (Chart 30).108 Both di- (183) and mono-nuclear (184) complexes showed poor activities in toluene with conversions of up to 14% at 100 °C for a monomer : metal ratio of 110:1. Increasing the polymerisation temperature to 140 °C led to a dramatic improvement in terms of catalytic activity (76% conversion). Interestingly, the polydispersity index of polymers produced at this temperature remained quite low (1.2 and 1.1 for 183 and 184 respectively).
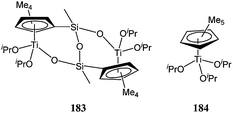 | ||
| Chart 30 Titanocene complexes 183 and 184. | ||
Xie and co-workers studied the zirconium complex 185 (Fig. 8) for its ability to polymerise ε-caprolactone.109 The constrained geometry catalyst 185 was moderately active at room temperature leading to 55% conversion after 2 h and for a monomer : metal ratio of 500:1. As in the case of catalysts 183 and 184 (vide supra), an increase in polymerisation temperature (from 25 to 60 °C) led to a catalytic improvement (from 55 to 95% conversion after 2 h) without significant broadening of the polymer molecular weight distribution (PDI = 1.26 at 25 °C vs. 1.33 at 60 °C), which sometimes was observed in other systems.
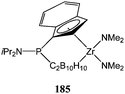 | ||
| Fig. 8 Zirconium constrained geometry complex 185. | ||
The zwitterionic titanocene monohalide complexes 186 and 187 (Chart 31) were screened by Baumann and co-workers for their potential as cationic ring opening polymerisation initiators.110 At low initiator loading and in toluene solution, complexes 186 and 187 displayed good activity, giving 36 and 22% yield, respectively, after 8 days for a monomer : metal ratio of 5,000:1 and at 75 °C. The significant difference between the measured polymer molecular weight and the one predicted by calculation (61,090 measured vs. 205,450 g mol−1 calculated for 186 and 56,000 measured vs. 125,550 g mol−1 calculated for 187), along with polydispersity indexes of 1.5, suggested a departure from living processes for these systems.
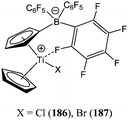 | ||
| Chart 31 Zwitterionic complexes 186 and 187. | ||
The same group reported the synthesis and use of the heterobimetallic complex 188 (Fig. 9).111 Preliminary screening revealed a somewhat higher activity than that of complexes 186 and 187, with 188 yielding 22% conversion after 1 h and for a monomer : metal ratio of 5,000:1 at 60 °C. It was unclear here whether only the zirconium centre or a combination of zirconium/aluminium were involved in the polymerisation process.
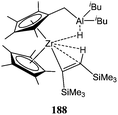 | ||
| Fig. 9 Heterobimetallic complex 188. | ||
4.2. Titanium tetraalkoxide and related complexes
Looking for structural alternatives to metallocenes, several research groups investigated the catalytic behaviour of oxygen and/or nitrogen bound ligands. Monoanionic alkoxide and aryloxide ligands are attractive substitutes for cyclopentadienyl ligands.Bounor-Legaré and co-workers studied the influence of the alkoxo or aryloxo group on the polymerisation process, in particular titanium tetra-n-propoxide and titanium tetra-phenoxide complexes.1121H NMR spectra of polymers obtained at low conversion or at low monomer : metal ratio confirmed the coordination/insertion mechanism with acyl-oxygen bond cleavage in ε-caprolactone. In the bulk, and for a monomer : metal ratio of 300:1, Ti(OnPr)4 was found to be more active than Ti(OPh)4 reaching quantitative conversion after 3 min, compared with 8 min for Ti(OPh)4. This difference in activity can be related to the disparity in polymer molecular weight between the two systems. Ti(OnPr)4 yielded higher molecular weight polymers than Ti(OPh)4: Mn(corrected) = 5,800 for Ti(OnPr)4 and 12,070 g mol−1 for Ti(OPh)4. These disparities in activity and polymer molecular weight find their explanation in a larger number of active alkoxide/aryloxide ligands per metal centre for Ti(OnPr)4 over Ti(OPh)4.
Titanium(IV) complexes of the type Ti(OR)2(OiPr)2 and Ti(OR)2Cl2 (189–196, Chart 32) were reported by del Hierro and co-workers.113 For a monomer : metal ratio of 100:1 and at room temperature, these catalysts displayed low activity. The dichloride complexes were less active than their alkoxide counterparts. Complex 189 for instance afforded 77% monomer conversion under these conditions after 24 h, whereas complex 194 only reached 65% conversion. This was consistent with the activity trend often observed: alkoxide > alkyl > halide (vide supra).88 The polydispersity indexes for polymers obtained with catalysts 189–196 were large (up to 3.1) and showed a lack of control similar to that exhibited by titanium alkoxide or aryloxide complexes.
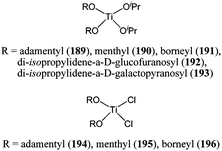 | ||
| Chart 32 Titanium complexes 189–196. | ||
The group of Davidson investigated the synthesis and catalytic behaviour of metal organic frameworks such as compound 197 the dimeric building block of which is shown in Fig. 10.114 In toluene, for a monomer : metal ratio of 100:1, at room temperature and after 48 h, a monomer conversion of 95% was obtained, making complex 197 a slower catalyst than the other titanium alkoxide complexes reported above. However, the polydispersity index in the case of complex 197 was much more favourable (PDI = 1.24) and the polymer molecular weight close to the predicted value (Mn(corrected) = 10,840 vs.Mn(calculated) = 12,260 g mol−1), indicative of a much more controlled polymerisation process than for the other titanium alkoxides.
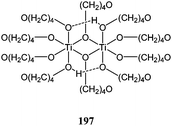 | ||
| Fig. 10 Dimeric building block of complex 197. | ||
Use of bulky ancillary ligands has been shown to improve the control over the polymerisation process and to yield polymers with a narrow polydispersity index and controlled molecular weight. The bis(amido) titanium complexes 198 and 199 (Chart 33) showed poor to moderate activity for ε-caprolactone polymerisation. Interestingly, the tetracoordinate complex 198 was more active than the pentacoordinate complex 199; the former affording 91% conversion after 8 h at 100 °C for 200 equivalents of monomer, whereas a 90% conversion took 32 h for complex 199 under the same conditions.115
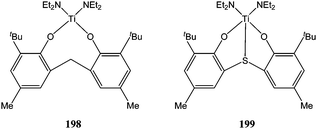 | ||
| Chart 33 Bis(amido) titanium complexes 198 and 199. | ||
Using catechol as a chelating ligand, Davidson et al. reported the synthesis and catalytic behaviour of compounds 200–206 (Chart 34).116 Typically, complex 200 displayed moderate activity affording 79% conversion at room temperature and for a monomer : metal ratio of 100:1. The polydispersity index reported for catalysts 200–206 were relatively low (1.13–1.27), the polymer molecular weights however differed dramatically from the expected values (Mn(measured) (NMR) = 2,400 vs Mn(calculated) = 9,020 g mol−1) indicating poor control over the polymerisation process.
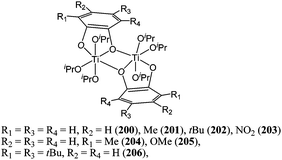 | ||
| Chart 34 Titanium complexes 200–206. | ||
Substituted acetylacetonato ligands have also been used to support titanium dialkoxide and zirconium dichloride complexes such as 207–210 (Chart 35).117 The zirconium(IV) dichloride complex 210 displayed good activity for the ring opening polymerisation of ε-caprolactone in the bulk; reaching a conversion of 99% after 6 h at 110 °C and for a monomer : metal ratio of 600:1. Complex 210 was even more active than any of the dialkoxide titanium complexes 207–209, which was surprising considering the general activity trend already mentioned (vide supra). Furthermore, as seen for complex 210, both halide groups seemed to be active in the polymerisation as evidenced by the polymer molecular weight obtained: Mn(corrected) = 34,940 and Mn(calculated) = 67,590 for one active chlorine group and 33,800 g mol−1 for two active chlorine groups.
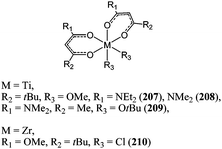 | ||
| Chart 35 Titanium and zirconium complexes 207–210. | ||
4.3. Complexes supported by mixed nitrogen/oxygen donor ligands
The aminobisphenolate complex 211 (Chart 36) was synthesised and structurally characterised by Davidson and co-workers. ε-Caprolactone polymerisation screening revealed a low activity for this complex with quantitative conversion after 24 h at room temperature for 100 equivalents of monomer. The bulky ligand framework here was of dramatic importance as the use of the less hindered N,N-bis(3,5-dimethyl-2-hydroxybenzyl)methylamine led to the formation of the disubstituted compound 212.118 Interestingly, catalyst 211 was less active than complex 213 featuring a tetrahedral titanium centre (Fig. 11). Low coordinate tetrahedral titanium complexes (e.g.213 and 198) seemed to be more active than the more hindered trigonal bipyramidal complexes (e.g.199 and 211).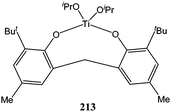 | ||
| Fig. 11 Bisphenolate titanium complex 213. | ||
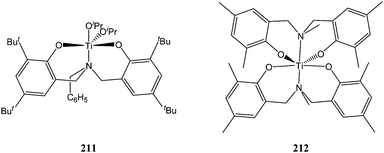 | ||
| Chart 36 Amino bis(phenolate) titanium complexes 211 and 212. | ||
The use of amino bis(phenolate) ligands with pendant donor arm was also investigated.119 The combination of such a ligand with titanium or zirconium yielded the complexes 214–218 (Chart 37). The titanium complex 214 was shown to be inactive for the polymerisation of ε-caprolactone and complex 216 showed low activity, yielding quantitative conversion after 24 h, at room temperature and for a monomer : metal ratio of 100:1. In the latter case, the polydispersity index was quite large (PDI = 2.60) owing to poor control over the polymerisation process and the occurrence of transesterification reactions. The dialkoxide zirconium species 215 and 218 were poorly active, reaching 50% conversion after 2 and 6 h, respectively, for a monomer : metal ratio of 100:1 and at room temperature.
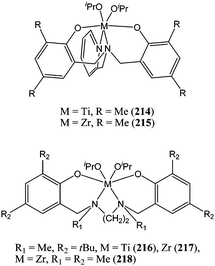 | ||
| Chart 37 Titanium and zirconium complexes 214–218. | ||
A similar amino bis(phenolate) ligand with a pendant nitrogen donor arm was used by Bochmann and co-workers to support titanium complexes of the form 219–221 (Chart 38).38 Interestingly, the titanium(IV) complexes 219 and 220 were found to be inactive for the ring opening polymerisation of ε-caprolactone at 60 °C, whereas the titanium(III) zwitterionic complex 221 was moderately active at the same temperature giving 95% conversion after 2 h and for a monomer : metal ratio of 200:1. Here, the presence of the sodium ion was of dramatic importance. It was thought to enhance the polymerisation process by coordinating to the exocyclic oxygen of the monomer, rendering it more susceptible to nucleophilic attack as reported for other sodium/iron and sodium/lanthanide mixed systems.14,120
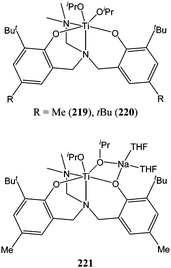 | ||
| Chart 38 Amino bis(phenolate) titanium complexes 219–221. | ||
Lee and co-workers investigated the synthesis and catalytic behaviour of titanium(IV) complexes supported by N-alkoxy-β-ketoiminate ligands (Chart 39).121 Complexes 222–225 displayed poor catalytic activities with complex 222 yielding 70% conversion after 5.3 h at 70 °C and for a monomer : metal ratio of 100:1. These complexes also displayed poor control over the polymerisation process as evidenced by the bimodal polymer molecular weight distribution.
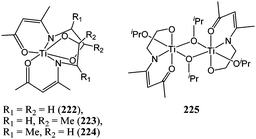 | ||
| Chart 39 Titanium complexes 222–225. | ||
The titanium complex 226 supported by a tetradentate imino-2-2′-2′′-triethanolate ligand and bearing a 2,6-di-tert-butylphenoxy group was synthesised by Kim and co-workers (Fig. 12).122 The bulky tert-butyl subsituents were of key importance here for the production of a monomeric complex. At 110 °C, complex 226 displayed low activity yielding 87% conversion after 24 h and for a monomer : metal ratio of 100:1. Again, a bimodal polymer molecular weight distribution and a large polydispersity index (PDI = 2.12) indicated a lack of control over the polymerisation process.
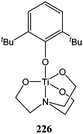 | ||
| Fig. 12 Titanium complex 226. | ||
4.4. Complexes supported by nitrogen donor ligands
Given the success of halide and alkoxide zirconium catalysts, Huang and co-workers synthesised and screened zirconium and hafnium complexes supported by substituted pyrrolyl ligands (227–232, Chart 40).123 These complexes were active for the ring opening polymerisation of ε-caprolactone. More recently, Martins and co-workers reported the use of dichloride zirconium complexes supported by tetradentate diamido/diamino macrocyclic ligands (233–235, Chart 40).124 Preliminary screening showed complex 233 to be the most active, reaching quantitative conversion after ca. 10 h at 80 °C for a monomer : metal ratio of 100:1.125
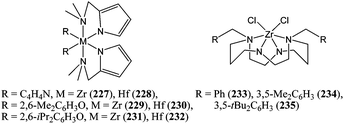 | ||
| Chart 40 Zirconium and hafnium complexes 227–235. | ||
5. Vanadium complexes
Mirroring the difference in academic interest between group IV and group V complexes for α-olefin polymerisation,126 vanadium and other group V metals have received relatively little attention for the ring opening polymersation of cyclic esters.Atlamsani, Brégault and co-workers reported the use of vanadium complexes, including heteropolyacids based on vanadium and molybdenum, VOSO4 and VO(acac)2 for the oligomerisation of ε-caprolactone under an atmosphere of dioxygen.127 Both vanadium heteropolyacid and VOSO4 were catalytically active, yielding oligo(ε-caprolactone) with a low molecular weight (Mw = 4,900 g mol−1), whereas VO(acac)2 displayed low activity under similar conditions.
Interestingly, when the polymerisations were conducted under an atmosphere of dry nitrogen, the heteropolyacids (containing vanadium(V)) displayed low activity and the vanadium(IV) species (i.e. VO(acac)2 and VOSO4) were completely inactive. This could be related to the observation of an intense green colour when using Mo(VI)/V(V) based heteropolyacids in polymerisations conducted under dry nitrogen: the intense colour was associated with the conversion of the heteropolyacid to reduced Mo(V)/V(IV) species. In the first step of the polymerisation mechanism proposed by Atlamsani and Brégeault, the dioxygen introduced in the reaction medium (re-)oxidised the inactive vanadium(IV) species to a dioxovanadium(V) cationic species (Scheme 4).
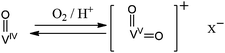 | ||
| Scheme 4 Simplified redox activation mechanism of the vanadium complex as proposed by Atlamsani and Brégeault (some of the ligands on the vanadium have been omitted for clarity). | ||
Efforts in our group were directed towards the application of chloride and alkoxide vanadium compounds in the ring opening polymerisation of ε-caprolactone.68 Organoimido vanadium(V) complexes 236–246 (Chart 41) were synthesised from the readily available starting material [V(NAr)(OR)3].68
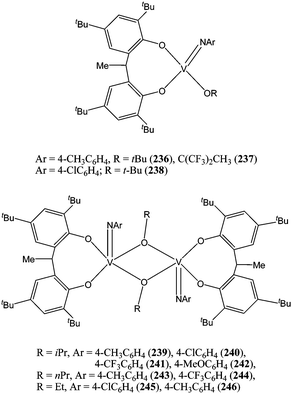 | ||
| Chart 41 Vanadium complexes 236–246. | ||
Single crystal X-ray structural determination revealed complexes 236–238 for which R = C(CH3)3 or C(CH3)(CF3)2 to be monomeric, whereas complexes 239–246 for which R = Et, n-Pr, i-Pr are dimeric, bridged by the μ-alkoxide ligand. At 40 °C and for a monomer : metal ratio of 500:1, the dimeric species 239–246 were found to be more active than their monomeric counterparts 236–238. The propagating species was likely to be monomeric as a polymeric chain is bulkier than a propoxide group; the catalytic advantage must therefore be in the first stage of the polymerisation process during the coordination of the first molecule of ε-caprolactone and its subsequent insertion into the V–OR bond. Interestingly, use of the fluorinated alkoxide group OC(CH3)(CF3)2 led to the inactive catalyst 237; in this case, the improvement in Lewis acidity of the metal centre was counterbalanced by the loss of nucleophilicity of the alkoxide group.
Yamada and K. Nomura reported the organoimido vanadium(V) complex 247 (Fig. 13), reminiscent of complexes 236–246.128 Complex 238 displayed poor activity with 57% conversion after 48 h at 100 °C and for a monomer : metal ratio of 250:1. 1H NMR study revealed the presence of C(O)O–2,6-Me2C6H3 end group suggesting a coordination/insertion mechanism and the inertness of the methyl substituent during the polymerisation process.
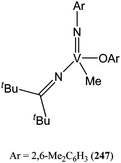 | ||
| Fig. 13 Organoimido vanadium(V) complex 247. | ||
6. Group VIII and IX metal complexes
Iron is a metal centre that has emerged in recent years with promising catalytic performances,129 and as a result, it has received increased attention in relation to biodegradable polymer production.Hillmyer, Tolman and co-workers studied the synthesis and catalytic behaviour of complex 248 (Fig. 14), the dinuclear cobalt analogue of compound 6.41 The cobalt complex was found to be less active than its magnesium counterpart.
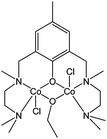 | ||
| Fig. 14 Di-nuclear cobalt complex 248. | ||
Shen and co-workers synthesised the N-heterocyclic carbene based iron(II) complex 249 (Fig. 15), and were able to screen this catalyst for the ring opening polymerisation of ε-caprolactone. Complex 249 was found to be moderately active, leading to quantitative conversion after 12 h at 80 °C for 300 equivalents of monomer.130 However, a decrease of polymer molecular weight with increasing monomer conversion and polydispersity indexes in the range 1.9–3.1 indicated transesterification reactions and poor control over the polymerisation process.
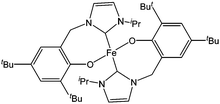 | ||
| Fig. 15 N-heterocyclic carbene iron complex 249. | ||
Commercially available metal halides are economically attractive alternatives for the ring opening polymerisation of cyclic esters. With this in mind, Gowda and Chakraborty investigated the catalytic behaviour of iron(II), iron(III) and ruthenium(III) chloride.131 In the bulk, FeCl3·6H2O afforded quantitative monomer conversion after 27 min at room temperature and for a monomer : metal ratio of 200:1. The same polymerisation required 1 h in toluene but afforded polymers with lower polydispersity index (PDI = 2.22 in bulk vs. 1.27 in toluene). The polymerisation mechanism envisaged for these Lewis acid catalysts follows an activated monomer pathway rather than the coordination/insertion mechanism for related alkoxide complexes. Surprisingly though, the polymerisation process seemed to be well controlled with bulk ε-caprolactone polymerisations at room temperature affording polymers of predictable molecular weight (after complete conversion).
The iron(III) complexes 250–257 (Charts 42 and 43) were studied within our group.132 They invariably display the widely reported Fe(III)–O–Fe(III) unit.133 Their anionic nature, the presence of alkali metals along with the complexation ability of the calixarene cavity were expected to favour the polymerisation process. Surprisingly, complexes 250–257 only showed poor to moderate activity. In contrast to vanadium based ethylene polymerisation catalysts,134 the use of an oxacalixarene moiety here (255–257) did not improve the catalytic behaviour of the complex.
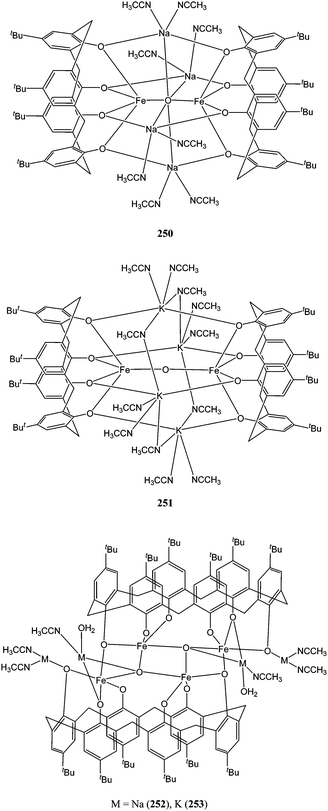 | ||
| Chart 42 Heterobimetallic alkali metal/iron calixarene and oxacalixarene complexes 250–253. | ||
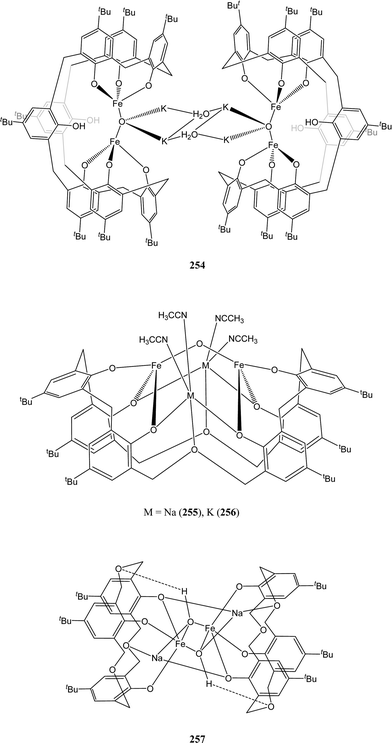 | ||
| Chart 43 Heterobimetallic alkali metal/iron calixarene and oxacalixarene complexes 254–257. | ||
7. Groups XII–XV metal complexes
7.1. Tin, bismuth, aluminium and zinc supported by oxygen donor ligands
For both industrial production and academic research, the most commonly used catalyst remains tin(II) 2-ethylhexanoate (or tin(II) octanoate). It is therefore natural that despite their high toxicity, some mechanistic and innovative process studies are still based on tin complexes. Stassin and Jérôme investigated the potential use of supercritical carbon dioxide as a reaction medium for the ring opening polymerisation of ε-caprolactone.135 When Bu2Sn(OCH3)2 was used, the polymerisation process was hampered by a carbonation reaction. The proposed reaction mechanism involved the dissociation of carbon dioxide to allow for the incorporation of a new monomer. Sobczak and Kolodziejski introduced the use of the amino acid L-carnitine for the production of low molecular weight polyesters in the presence of tin(II) octanoate.136Kricheldorf and co-workers envisaged bismuth(III) complexes as alternatives for the more toxic tin(II) octanoate.137–139 Bismuth(III) n-hexanoate (258), bismuth(III) subsalicylate (259, Fig. 16) and diphenyl bismuth(III) ethoxide (260) were screened for their polymerisation ability and their activities were compared to that of the well established tin(II) octanoate catalyst.
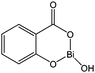 | ||
| Fig. 16 Bismuth subsalicylate complex 259. | ||
In bulk, at 120 °C, compounds 258 and 259 were found to be less reactive than tin(II) octanoate; however 258 was also less active as a transesterification catalyst than tin(II) octanoate, yielding polymers with narrower polydispersities and less cyclic oligomers (resulting from back-biting reactions) when used with tetra(ethylene) glycol.137 Considering the activity trend already discussed above (metal alkoxide > metal carboxylate), the alkoxide bismuth complex 260 was synthesised by the same group. Although less active than tin(II) octanoate (with 1 equivalent of ethanol) at 120 °C, in bulk, complex 260 proved to be more active at lower temperatures (i.e. at temperature ≤ 90 °C). MALDI-TOF mass spectrometry showed ethyl ester end groups indicative of a coordination/insertin mechanism involving only the Bi–OEt group, the phenyl groups acting as spectator ligands.139
Early studies by Penczek, Duda et al. demonstrated an alkoxide intermediate, formed via alkoxide/carboxylate ligand exchange, in the mechanism of polymerisation using tin(II) octanoate (Scheme 5).140 The same group undertook a similar study focusing on zinc 2-ethylhexanoate and aluminium tris(acetylacetonate) catalysts.141 A similar polymerisation mechanism was demonstrated for these two pro-catalysts.
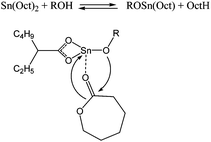 | ||
| Scheme 5 General mechanism for tin(II) octanoate based polymerisation. | ||
Aluminium and zinc alkoxide species are attractive as they often lead to high polymerisation activity and good control over the polymerisation process. However, the dialkyl aluminium complexes 261 and 262 (Chart 44) were poorly active for ε-caprolactone polymerisation. MALDI-TOF mass spectrometry indicated that, as it was the case for 260, the polymerisation took place at the Al–O bond and that the alkyl groups act as spectator ligands. It is noteworthy that a lactide insertion product (263, Chart 44) was isolated and structurally characterised. Complex 263 was inactive for further lactide or ε-caprolactone polymerisation at 40 °C, but became active upon heating at 70 °C.142
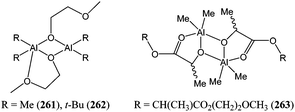 | ||
| Chart 44 Complexes 261, 262 and intermediate compound 263. | ||
This behaviour was explained by the coordination of the ester group to the metal centre preventing the approach of another monomer. The same structural feature could explained the difference in lactide polymerisation behaviour of 261 and 262, with 261 inactive at 40 °C, whereas 262 was active at that temperature. The bulk of the tert-butyl groups may prevent the ester from coordinating to the metal centre. The diaryloxide aluminium complex 264 (Fig. 17) was moderately active for ε-caprolactone polymerisation, reaching 13% conversion after 24 h at room temperature and for a monomer : metal ratio of 1,000:1.143
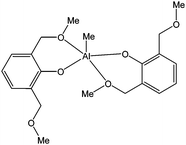 | ||
| Fig. 17 Diaryloxide aluminium complex 264. | ||
Following our early reports on the catalytic improvement in ethylene polymerisation when shifting from a calixarene to an oxacalixarene supported complex,134 our group investigated the use of the mixed alkyl/alkoxide zinc complex 265 (Fig. 18). The hexa-nuclear complex 265 was poorly active for the ring opening polymerisation of ε-caprolactone with 43% conversion after 24 h at 60 °C, for a monomer : metal ratio of 300:1 and with 1 equivalent of benzyl alcohol. The polydispersity index remained low (PDI ≤ 1.3), except when the polymerisation was performed without benzyl alcohol co-initiator (PDI = 1.7). Such a phenomenon has already been described for other metals.144
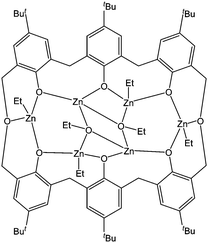 | ||
| Fig. 18 Hexa-nuclear zinc complex 265. | ||
7.2. Ligands with mixed nitrogen/oxygen donor sets
Recently, Fujita and co-workers have shown that phenoxy-imine supported group IV and group V complexes could achieve very high activities in the polymerisation of α-olefins.145 This and the readily availability of such phenoxy-imine ligands have prompted several groups as well as ourselves to investigate the catalytic behaviour of aluminium and, to a lesser extent, zinc complexes supported by such ligands.N. Nomura and co-workers were first interested in the in situ formation of ethyl aluminium complexes supported by phenoxy-imine ligands.146 The simplest of the phenoxy-imine di-methyl aluminium complexes (complex 266, Chart 45) was found by us and others to be inactive for ε-caprolactone polymerisation.85,147 However, the mixture of ligand L4H (Chart 45) and Et3Al gave rise to an active catalyst, although it displayed poor activities. In general, greater activities were obtained for catalysts supported by ligands possesing bulky ortho substituents on the phenolic residue such as L5H. A mixture of L5H with Et3Al gave rise to a very active species yielding 98% conversion after 1 h at room temperature and for a monomer : metal ratio of 300:1. The polymerisation required one equivalent of benzyl alcohol; this complex showed good control over the polymerisation process (PDI < 1.2). Following this study, K. Nomura and co-workers isolated a series of well defined complexes (267–282, Chart 45) in an attempt to identify steric and electronic effects of the ligands on polymerisation activity.148–152
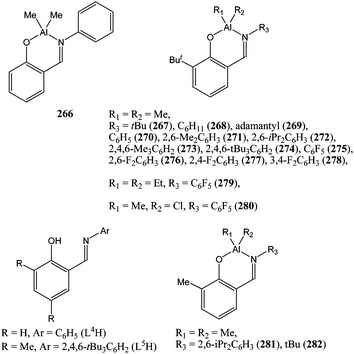 | ||
| Chart 45 Aluminium and zinc complexes (266–282) supported by phenoxy-imine ligands. | ||
In accordance with findings from N. Nomura and co-workers,146 a bulky ortho phenolic substituent appeared to be crucial for polymerisation activity. However, the exact nature of this group (i.e. Me or tBu) had little effect on the polymerisation process. On the other hand, the nature of the imino substituent had a strong influence on the catalytic behaviour of the complexes 267–282. Among these complexes, the most active catalyst was compound 275 bearing a pentafluoro phenyl substituent. Complex 275, in the presence of one equivalent of benzyl alcohol was found to be very active for ε-caprolactone polymerisation, reaching 94% conversion in 45 min at 50 °C and for a monomer : metal ratio of 250:1. In comparison, complex 267 bearing a tert-butyl ligand on the imine moiety was only moderately active. The difference in activity could not be explained only by steric factors. Complex 276 bearing a 2,6-F2C6H3 substituent was more active than its methylated counterpart, complex 271 bearing a 2,6-Me2C6H3 substituent. Here, the polymerisation activity seemed to be enhanced by improved Lewis acidity of the aluminium metal centre bearing electron withdrawing groups.
The same electronic effect was observed by Carpentier and co-workers when using dialkoxy/diamino or dialkoxy/diimino fluorinated ligands. The resulting aluminium complexes 283–285 (Chart 46) demonstrated moderate to good activity.45,153 Catalyst 285 was also found to be active at low concentration. For a monomer/aluminium ratio of 1,000:1, a conversion of 98% was reached after 12 h. Although the polymerisation at this concentration required the use of extra-pure ε-caprolactone monomer due to catalyst deactivation. It is noteworthy here that a similar improvement in activity was observed in the polymerisation of lactide by methyl aluminium complexes supported by chlorinated phenoxy-imine type ligands.154
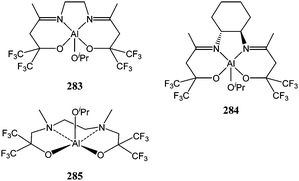 | ||
| Chart 46 Aluminium complexes 283–285. | ||
Other aluminium and zinc phenoxy-imine complexes have been screened for their catalytic behaviour. The aluminium (286–290 and 296–298, Chart 47) and zinc complexes (291–295, Chart 47) were studied by the group of Wang.155 The activity order for the aluminium complexes, i.e.290 > 289 > 288 > 287 > 286, reflected the need for ortho substituted phenolate moiety (vide supra). The zinc complexes were found to be moderately active with 294 yielding quantitative conversion after 75 min at 80 °C and for a monomer : metal ratio of 200:1; they were also more active than their aluminium counterparts. The zinc complexes 299–301 were synthesised from the templated reaction of 2-hydroxy-5-tert-butyl-1,3-benzene dicarboxaldehyde with various amino acids. These anionic di-nuclear zinc complexes were surprisingly inactive for the ring opening polymerisation of ε-caprolactone.156
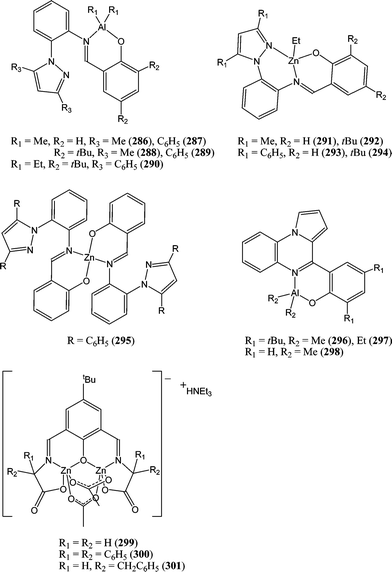 | ||
| Chart 47 Aluminium and zinc complexes 286–301 supported by phenoxy-imine ligands. | ||
The di-nuclear zinc complexes 299–301 are reminiscent of the magnesium complex 6, inspired by hydrolase enzymes (vide supra). In an attempt to pursue this route and to study possible cooperative effect between metal centres, our group turned to the use of macrocyclic Schiff base ligands157 to support di- and tetra-nuclear aluminium complexes (302–305, Chart 48). The catalytic behaviour of complexes 302–305 was compared to that of their acyclic mono and di-nuclear counterparts (306 and 307, Chart 48).85
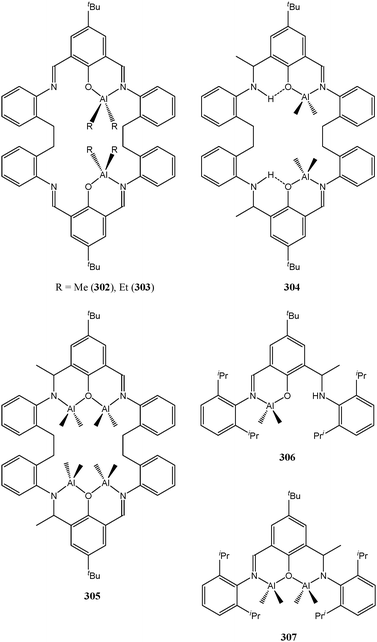 | ||
| Chart 48 Aluminium complexes 302–307 supported by macrocyclic and acyclic Schiff base ligands. | ||
Complex 304 was found to be moderately active, with quantitative conversion after 12 h at room temperature and for a monomer : metal ratio of 500:1. The activity increased in the order 304 > 305 > 306 > 307 > 302 ≈ 303, which suggested that there was a beneficial effect in having metal centres in a close proximity (304 > 306 and 305 > 307) so long as these aluminium metals are not linked in an aluminoxane (Al–O–Al) fashion (304 > 305). The Al⋯Al distance in complex 304 (5.7818(10) Å) was believed to favour the coordination of a single monomer to both catalytic centres from the same complex; one being used as a Lewis acid and the other one using its Al–R functionality to attack the carbonyl group, such a mechanism has been suggested for propylene oxide polymerisation. In the case of 305, catalytic activity was monitored by in situ IR, and the activity was found to peak at ca 1 h, thereafter decreasing slowly with monomer conversion up to 20 h. 158
The zinc analogue of complex 6 was reported by Hillmyer, Tollman and co-workers. According to kinetic studies, complex 308 (Fig. 19) was ten times slower than its magnesium analogue.41
 | ||
| Fig. 19 Dinuclear zinc complex 308. | ||
N,O ligand systems other than phenoxy-imine ligands have also attracted a great deal of attention. Several groups reported complexes of the type 309–321 (Chart 49) supported by amino phenolate, amine bis(phenolate) or heteroscorpionate ligands.
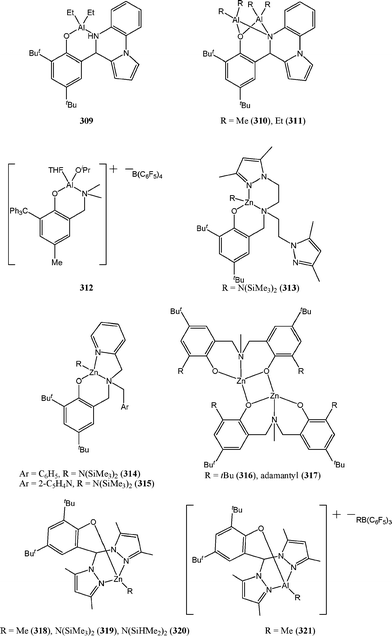 | ||
| Chart 49 Aluminium and zinc complexes, 309–321, supported by amino phenolate, amine bis(phenolate) or heteroscorpionate ligands. | ||
The cationic aluminium complexes 312 and 321 were found to be more active than their neutral counter parts 309 and 310. Furthermore, the alkoxy complex 312 was noticeably more active than the alkyl complex 321. The activity for the zinc complexes decreased in the order 313 ≈ 314 > 316 ≈ 317 > 319 > 320 with complex 313 and 314 exhibiting high activies. The monoanionic tridentate amine phenolate ligand in these two complexes seems to promote highly active catalytic systems. Despite a strong structural similarity, complexes 313 and 314 were more active than the imino phenolate complexes 291–294; this activity improvement could result from the substitution of the imino for an amino group or more likely by the amido functional group instead of the less reactive alkyl group in 291–294.
7.3. Ligands with nitrogen donors only
In light of the dramatic influence of the ortho substituent in amine phenolate ligands, the anilido-imine based complexes 322–329 (Chart 50) have been screened in order to explore the steric effect of the substituent on the coordinating atoms of the ligand.30,159,160 Complexes 322–329 displayed moderate to good activities comparable to those of the phenoxy-imine complexes 266–282. The zinc complexes showed higher activities than the aluminium analogues, with the activity decreasing in the order 328 > 329 > 326 > 327. Interestingly, the heterobimetallic complex 325 was more active than the mono-nuclear equivalent 324.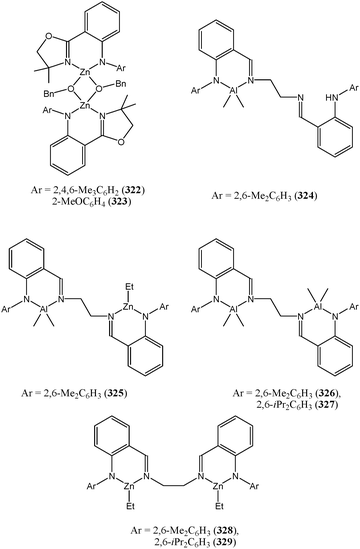
Huang and co-workers screened indium and aluminium complexes (330–336, Chart 51) supported by amino or amido pyrrolyl ligands.161,162 The amino pyrrolyl indium complexes 330–332 displayed poor activities, with complex 332 affording 97% conversion after 1 h at 30 °C and for a monomer : metal ratio of 100:1. The di-nuclear aluminium amido pyrrolyl complexes also displayed poor activities in the order 334 > 335 ≫ 336 > 333 suggesting the lack of reactivity of hydride complexes. The mixed lithium/aluminium complex 336 failed to show any catalytic improvement despite the presence of alkali metals capable of coordinating to the caprolactone monomer.
 | ||
| Chart 51 Amino and amido pyrrolyl complexes 330–336. | ||
The widely used β-diketiminate ligands have also been recently used to support aluminium and zinc complexes such as 337–351 (Chart 52).
 | ||
| Chart 52 β-Diketiminate aluminium and zinc complexes 337–351. | ||
The complexes 337–350 were screened at 80 °C and for a monomer : metal ratio of 100:1.163 Under these conditions, they displayed low to poor activities. Complexes 337–344 were more active than the complexes 345–350 bearing a bulky ortho substituent. The iso-propyl groups here may hamper the coordination of the monomer to the aluminium centre. The electronic influence of the ligand framework on the activity was marked. Compounds 338 and 344 containing a CF3 substituent were found to be the most active and complex 341 containing the electron donating group OCH3 displayed the lowest activity among complexes 337–344. However, halogen atoms in para or meta position of the aryl groups led to species less active than expected. Halogenated complexes 342 and 343 were less active than complex 340. A proposed explanation for this behaviour lies in the electron-donating conjugated effect via p–π bonding from the halogen atom.163 The tri-nuclear zinc complex 351 (Chart 52) showed moderate polymerisation activity.164 Compound 351 led to 92% conversion after 105 min at room temperature and for a monomer : metal ratio of 170:1. Surprisingly, catalyst 351 was found to be more active for lactide than for ε-caprolactone polymerisation.
Other zinc and aluminium complexes based on nitrogen donor ligands include compounds 352–366 (Chart 53). The heteroscorpionate complexes 352–354 were based on the same ligand framework as the magnesium complexes 9–14. Complexes 352–354 displayed moderate to good activity, with complex 354 affording 97% conversion after 40 min at 85 °C and for a monomer : metal ratio of 500:1.165 The zinc complexes 352–354 were much less active than the magnesium analogues 9–14. The iminophosphorano organometallic zinc complexes 355–358 showed moderate activity at 60 °C.166 Low polydispersity indexes and good agreement between calculated and measured polymer molecular weights indicated that the polymerisation occurred in a living fashion. Pyridine based ligands have also been used to support aluminium and zinc complexes such as compounds 359–362.167 These catalysts were moderately active at 60 °C, with complex 361 yielding 81% conversion after 480 min and for a monomer : metal ratio of 200:1.
 | ||
| Chart 53 Aluminium and zinc complexes 352–366. | ||
Zhu and Chen reported the catalytic screening of the aluminium complexes 363 and 364 supported by tripodal triamine ligands.168 These complexes were found to be moderately active at room temperature with and without the addition of benzyl alcohol. The polydispersity indexes recorded here were high (PDI up to 2.59) and suggested a non living polymerisation process.
Zinc and aluminium diamine and amino/imino complexes 365 and 366 were screened for their catalytic properties.17 They displayed moderate to good activity. Complex 365 afforded quantitative conversion after 24 h at 110 °C in the bulk and for a monomer : metal ratio of 2,000:1.
7.4. Other ligand systems
Phosphine donor ligands have been scarcely used to support ε-caprolactone polymerisation catalysts. Recently, Dagorne and co-workers reported the phosphinophenolate aluminium complexes 367–370 (Chart 54).169 The cationic aluminium catalysts displayed poor activity at 75 °C, with complex 370 affording 95% conversion after 2 h and for a monomer : metal ratio of 100:1.
 | ||
| Chart 54 Aluminium and tin complexes 367–373. | ||
Bis(2,4,6-tri-iso-propoxyphenyl)tin(IV) derivatives (371–373, Chart 54) were screened for ε-caprolactone polymerisation.170,171 They were active polymerisation catalysts and complex 373 displayed poor activity, affording 86% conversion after 24 h at 75 °C and for a monomer : metal ratio of 120:1.
Similarly to the work undertaken for lanthanide complexes, the group of Biesemans has studied the possibility of supporting organotin catalysts on polystyrene supports.172–174 The heterogeneous catalysts obtained therefrom were hoped to be easily isolated from the polymer mixture, therefore removing any measurable trace of tin from the polymer and solving the toxicity issue of such complexes. For instance, the undecyltin trichloride graphted to crosslinked polystyrene displayed good polymerisation activity even after ten recycling cycles and with low metal leaching.
8. Summary
Metal complexes with various ancillary ligands represent very attractive initiators/catalysts for ε-caprolactone polymerisation. One of the key factors that dictates the selection of a particular system is its observed catalytic activity. However, in the literature the ways in which this is reported varies considerably. In this review, we have introduced a new means of classifying initiator/catalyst activity to enable easier comparisons between reports in the literature. Chart 55 uses this scale to assemble the activity rating of the different ligand/metal combinations discussed herein; the colour coding relating to the donor atoms present at the ligand. Several generalisations can be drawn from this chart when these combinations are considered in concert. First of all, mirroring the well-established academic research on α-olefin polymerisation catalysts, efforts here have clearly focused not only on the many of the same ligand systems (e.g. β-diketiminate or diphenolate ligands), but also similar choice of metal centre (group IV, iron etc.). Moreover, the omissions from each grouping tend to be the same, for example sulfur-based ligands are rarely employed, whilst little is known about the catalytic activity of group VII metals.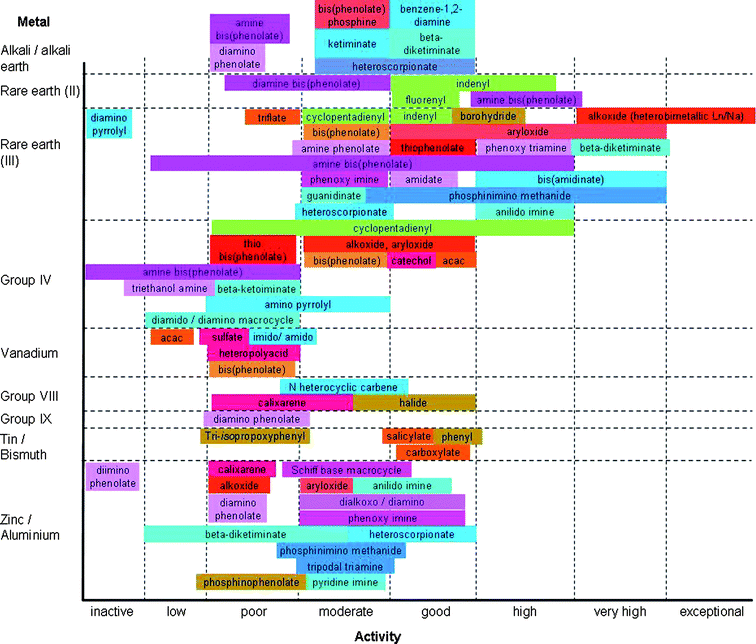 | ||
| Chart 55 Polymerisation activity observed for the different ligand/metal combination discussed in this review. Colour code: green: metallocene; red/orange: O donor; blue: N donor; pink/purple: N/O donor. | ||
It is clear that ligand systems which are inert and possess bulky ligand frameworks are beneficial to the polymerisation process, and act by (i) impeding the approach of the polymer in formation to the metal centre, therefore reducing the occurrence of back-biting and (ii) hampering the aggregation of complexes that may lead to dormant species. Interestingly, homo and hetero-multinuclear species seem to be the key to very high and exceptional activities through a cooperative polymerisation process. Despite the excellent developments to-date, there still remain large gaps in the emerging coordination chemistry of these systems. Indeed, it is hoped that the diversity of new initiators/catalysts presented in Chart 55 will stimulate others to predict useful combinations when looking to design highly active metal-based systems for ε-caprolactone polymerisation.
Notes and references
- British Plastics Federation, Annual Review, 2007 Search PubMed.
- Plastics Europe, The Compelling Facts About Plastics, 2008 Search PubMed.
- P. Bjacek, Oil Gas J., 2008, 106, 40 Search PubMed.
- A. Guttag, US patent 1994/5346929, 1994.
- B. Freedman and M. J. Diamond, US patent 1977/4017668, 1977.
- S. A. Braskem (for US only de-C.R.L.A. Morschbacker) WO patent 2009/070858 A1, 2009.
- E. Chiellini and R. Solaro, Adv. Mater., 1996, 8, 305 CAS.
- E. Mathiowitz, J. S. Jacob, Y. S. Jong, G. P. Carino, D. E. Chickering, P. Chaturvedi, C. A. Santos, K. Vijayaraghavan, S. Montgomery, M. Basset and C. Morrell, Nature, 1997, 386, 410 CrossRef.
- S. Dumitriu, Polymeric Biomaterials, Marcel Dekker, New York, 2002 Search PubMed.
- M. Chasin and R. S. Langer, Biodegradable Polymers as Drug Delivery Systems, Informa Health Care, 1990 Search PubMed.
- Based on safety data available for the Tone™ products from Dow Chemicals.
- A. P. Gupta and V. Kumar, Eur. Polym. J., 2007, 43, 4053 CrossRef CAS.
- C. K. Williams, Chem. Soc. Rev., 2007, 36, 1573 RSC.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147 CrossRef.
- M. C. Rocca, G. Carr, A. B. Lambert, D. J. MacQuarrie and J. H. Clark, S. A. Solvay, US patent 2003/6531615 B2, 2003.
- M. Minami and S. Kozaki, US patent 2003/0023026 A1, 2003.
- F. Majoumo-Mbe, E. Smolensky, P. Lönnecke, D. Shpasser, M. S. Eisen and E. Hey-Hawkins, J. Mol. Catal. A: Chem., 2005, 240, 91 CAS.
- O. Coulembier, P. Degée, J. L. Hedrick and P. Dubois, Prog. Polym. Sci., 2006, 31, 723 CrossRef CAS.
- W. H. Carothers, Chem. Rev., 1931, 8, 353 CrossRef.
- R. A. Gross, A. Kumar and B. Kalra, Chem. Rev., 2001, 101, 2097 CrossRef CAS.
- R. T. MacDonald, S. K. Pulapura, Y. Y. Svirkin, R. A. Gross, D. L. Kaplan, J. Akkara, G. Swift and S. Wolk, Macromolecules, 1995, 28, 73 CrossRef CAS.
- A. Córdova, T. Iversen, K. Hult and M. Martinelle, Polymer, 1998, 39, 6519 CrossRef CAS.
- S. Kobayashi, K. Takeya, S. Suda and H. Uyama, Macromol. Chem. Phys., 1998, 199, 1729 CrossRef CAS.
- P. Kubisa and S. Penczek, Prog. Polym. Sci., 1999, 24, 1409 CrossRef CAS.
- Y. Shibasaki, H. Sanada, M. Yokoi, F. Sanda and T. Endo, Macromolecules, 2000, 33, 4316 CrossRef CAS.
- B. A. Rozenberg, Pure Appl. Chem., 1981, 53, 1715 CrossRef CAS.
- K. Ito, Y. Hashizuka and Y. Yamashita, Macromolecules, 1977, 10, 821 CrossRef CAS.
- K. Ito and Y. Yamashita, Macromolecules, 1978, 11, 68 CrossRef CAS.
- M. Bero, G. Adamus, J. Kasperczyk and H. Janeczek, Polym. Bull., 1993, 31, 9 CrossRef CAS.
- W. Yao, Y. Mu, A. Gao, W. Gao and L. Ye, Dalton Trans., 2008, 3199 RSC.
- A.-C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466 CrossRef CAS.
- S. Penczek, M. Cypryk, A. Duda, P. Kubisa and S. Słomkowski, Prog. Polym. Sci., 2007, 32, 247 CrossRef CAS.
- B. J. O'Keefe, M. A. Hillmyer and W. B. Tolman, J. Chem. Soc., Dalton Trans., 2001, 2215 RSC.
- J. Wu, T.-L. Yu, C.-T. Chen and C.-C. Lin, Coord. Chem. Rev., 2006, 250, 602 CrossRef CAS.
- M. H. Chisholm, Inorg. Chim. Acta, 2009, 362, 4284 CrossRef CAS.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813 CrossRef CAS.
- Y.-N. Chang and L.-C. Liang, Inorg. Chim. Acta, 2007, 360, 136 CrossRef CAS.
- Y. Sarazin, R. H. Howard, D. L. Hughes, S. M. Humphrey and M. Bochmann, Dalton Trans., 2006, 340 RSC.
- D. Mecerreyes, R. Jérôme and P. Dubois, Adv. Polym. Sci., 1999, 147, 1 CAS.
- C. K. Williams, N. R. Brooks, M. A. Hillmyer and W. B. Tolman, Chem. Commun., 2002, 2132 RSC.
- L. E. Breyfogle, C. K. Williams, V. G. Young, M. A. Hillmyer and W. B. Tolman, Dalton Trans., 2006, 928 RSC.
- W.-Y. Lee, H.-H. Hsieh, C.-C. Hsieh, H. M. Lee, G.-H. Lee, J.-H. Huang, T.-C. Wu and S.-H. Chuang, J. Organomet. Chem., 2007, 692, 1131 CrossRef CAS.
- L. F. Sánchez-Barba, A. Garcés, M. Fajardo, C. Alonso-Moreno, J. Fernández-Baeza, A. Otero, A. Antiñolo, J. Tejeda, A. Lara-Sánchez and M. I. López-Solera, Organometallics, 2007, 26, 6403 CrossRef CAS.
- L. F. Sánchez-Barba, D. L. Hughes, S. M. Humphrey and M. Bochmann, Organometallics, 2006, 25, 1012 CrossRef CAS.
- A. Amgoune, L. Lavanant, C. M. Thomas, Y. Chi, R. Welter, S. Dagorne and J.-F. Carpentier, Organometallics, 2005, 24, 6279 CrossRef CAS.
- Y. Shen, Z. Shen, Y. Zhang and K. Yao, Macromolecules, 1996, 29, 8289 CrossRef CAS.
- Y. Wu, S. Wang, C. Qian, E. Sheng, M. Xie, G. Yang, Q. Feng, L. Zhang and X. Tang, J. Organomet. Chem., 2005, 690, 4139 CrossRef CAS.
- S. Zhou, S. Wang, G. Yang, Q. Li, L. Zhang, Z. Yao, Z. Zhou and H.-B. Song, Organometallics, 2007, 26, 3755 CrossRef CAS.
- S. L. Zhou, S. W. Wang, E. H. Sheng, L. J. Zhang, Z. Y. Yu, X. B. Xi, G. D. Chen, W. Luo and Y. Li, Eur. J. Inorg. Chem., 2007, 1519 CrossRef CAS.
- S. W. Wang, X. L. Tang, A. Vega, J. Y. Saillard, S. L. Zhou, G. S. Yang, W. Yao and Y. Wei, Organometallics, 2007, 26, 1512 CrossRef CAS.
- S. W. Wang, S. Y. Wang, S. L. Zhou, G. S. Yang, W. Luo, N. Hu, Z. H. Zhou and H. B. Song, J. Organomet. Chem., 2007, 692, 2099 CrossRef CAS.
- Y. Wei, Z. Y. Yu, S. W. Wang, S. L. Zhou, G. S. Yang, L. J. Zhang, G. D. Chen, H. M. Qian and J. X. Fan, J. Organomet. Chem., 2008, 693, 2263 CrossRef CAS.
- X. M. Deng, M. L. Yuan, C. D. Xiong and X. H. Li, J. Appl. Polym. Sci., 1999, 73, 1401 CrossRef CAS.
- H. M. Sun, S. Chen, Y. M. Yao, Q. Shen and K. B. Yu, Appl. Organomet. Chem., 2006, 20, 310 CrossRef CAS.
- J. F. Chai, V. Jancik, S. Singh, H. P. Zhu, C. He, H. W. Roesky, H. G. Schmidt, M. Noltemeyer and N. S. Hosmane, J. Am. Chem. Soc., 2005, 127, 7521 CrossRef CAS.
- L. F. Zhang, Y. H. Niu, Y. Wang, P. Wang and L. J. Shen, J. Mol. Catal. A: Chem., 2008, 287, 1 CrossRef CAS.
- F. Peng, J. Ling, Z. Q. Shen and W. W. Zhu, J. Mol. Catal. A: Chem., 2005, 230, 135 CrossRef CAS.
- H. T. Sheng, H. Zhou, H. D. Guo, H. M. Sun, Y. M. Yao, J. F. Wang, Y. Zhang and Q. Shen, J. Organomet. Chem., 2007, 692, 1118 CrossRef CAS.
- Y. Wang, S. Onozawa and M. Kunioka, Green Chem., 2003, 5, 571 RSC.
- N. Nomura, A. Taira, A. Nakase, T. Tomioka and M. Okada, Tetrahedron, 2007, 63, 8478 CrossRef CAS.
- R. M. Gauvin, T. Chenal, R. A. Hassan, A. Addad and A. Mortreux, J. Mol. Catal. A: Chem., 2006, 257, 31 CrossRef CAS.
- H. M. Sun, H. R. Li, C. S. Yao, Y. M. Yao, H. T. Sheng and Q. Shen, Chin. J. Chem., 2005, 23, 1541 CrossRef CAS.
- Y. M. Yao, X. P. Xu, B. Liu, Y. Zhang, Q. Shen and W. T. Wong, Inorg. Chem., 2005, 44, 5133 CrossRef CAS.
- X. P. Xu, Z. J. Zhang, Y. M. Yao, Y. Zhang and Q. Shen, Inorg. Chem., 2007, 46, 9379 CrossRef CAS.
- H. T. Sheng, F. Xu, Y. M. Yao, Y. Zhang and Q. Shen, Inorg. Chem., 2007, 46, 7722 CrossRef CAS.
- H. T. Sheng, J. M. Li, Y. Zhang, Y. M. Yao and Q. Shen, Polyhedron, 2008, 27, 1665 CrossRef CAS.
- H. T. Sheng, J. M. Li, Y. Zhang, Y. M. Yao and Q. Shen, J. Appl. Polym. Sci., 2009, 112, 454 CrossRef CAS.
- A. Arbaoui, C. Redshaw, D. M. Homden, J. A. Wright and M. R. J. Elsegood, Dalton Trans., 2009, 8911–8922 RSC.
- H. Zhou, H. D. Guo, Y. M. Yao, L. Y. Zhou, H. M. Sun, H. T. Sheng, Y. Zhang and Q. Shen, Inorg. Chem., 2007, 46, 958–964 CrossRef CAS.
- E. E. Delbridge, D. T. Dugah, C. R. Nelson, B. W. Skelton and A. H. White, Dalton Trans., 2007, 143 RSC.
- D. T. Dugah, B. W. Skelton and E. E. Delbridge, Dalton Trans., 2009, 1436 RSC.
- C. E. Willans, M. A. Sinenkov, G. K. Fukin, K. Sheridan, J. M. Lynam, A. A. Trifonov and F. M. Kerton, Dalton Trans., 2008, 3592 RSC.
- H. E. Dyer, S. Huijser, A. D. Schwarz, C. Wang, R. Duchateau and P. Mountford, Dalton Trans., 2008, 32 RSC.
- L. Z. Zhou, Y. M. Wang, Y. M. Yao, Y. Zharig and Q. Shen, J. Rare Earths, 2007, 25, 544 CrossRef.
- Y. M. Yao, M. T. Ma, X. P. Xu, Y. Zhang, Q. Shen and W. T. Wong, Organometallics, 2005, 24, 4014 CrossRef CAS.
- F. Bonnet, A. R. Cowley and P. Mountford, Inorg. Chem., 2005, 44, 9046 CrossRef CAS.
- I. Palard, A. Soum and S. M. Guillaume, Macromolecules, 2005, 38, 6888 CrossRef CAS.
- D. Robert, M. Kondracka and J. Okuda, Dalton Trans., 2008, 2667 RSC.
- F. M. Kerton, A. C. Whitwood and C. E. Willans, Dalton Trans., 2004, 2237 RSC.
- I. Westmoreland and J. Arnold, Dalton Trans., 2006, 4155 RSC.
- P. I. Binda and E. E. Delbridge, Dalton Trans., 2007, 4685 RSC.
- P. I. Binda, E. E. Delbridge, H. B. Abrahamson and B. W. Skelton, Dalton Trans., 2009, 2777 RSC.
- B. Li, Y. Wang, Y. Yao, Y. Zhang and Q. Shen, J. Organomet. Chem., 2009, 694, 2409 CrossRef CAS.
- L. J. E. Stanlake, J. D. Beard and L. L. Schafer, Inorg. Chem., 2008, 47, 8062 CrossRef CAS.
- A. Arbaoui, C. Redshaw and D. L. Hughes, Chem. Commun., 2008, 4717 RSC.
- M. Xue, Y. Yao, Q. Shen and Y. Zhang, J. Organomet. Chem., 2005, 690, 4685 CrossRef CAS.
- L. F. Sánchez-Barba, D. L. Hughes, S. M. Humphrey and M. Bochmann, Organometallics, 2005, 24, 3792 CrossRef CAS.
- M. Endo, T. Aida and S. Inoue, Macromolecules, 1987, 20, 2982 CrossRef CAS.
- Y. Yao, Z. Zhang, H. Peng, Y. Zhang, Q. Shen and J. Lin, Inorg. Chem., 2006, 45, 2175 CrossRef CAS.
- X. Xu, X. Xu, Y. Chen and J. Sun, Organometallics, 2008, 27, 758 CrossRef CAS.
- J. Wang, H. Sun, Y. Yao, Y. Zhang and Q. Shen, Polyhedron, 2008, 27, 1977 CrossRef CAS.
- J. Wang, Y. Yao, Y. Zhang and Q. Shen, Inorg. Chem., 2009, 48, 744 CrossRef CAS.
- D. M. Lyubov, A. M. Bubnov, G. K. Fukin, F. M. Dolgushin, M. Y. Antipin, O. Pelce, M. Schappacher, S. M. Guillaume and A. A. Trifonov, Eur. J. Inorg. Chem., 2008, 2090 CrossRef CAS.
- P. C. Kuo, J. C. Chang, W. Y. Lee, H. M. Lee and J. H. Huang, J. Organomet. Chem., 2005, 690, 4168 CrossRef CAS.
- A. Otero, J. Fernandez-Baeza, A. Antinolo, A. Lara-Sanchez, E. Martinez-Caballero, J. Tejeda, L. F. Sanchez-Barba, C. Alonso-Moreno and I. Lopez-Solera, Organometallics, 2008, 27, 976 CrossRef CAS.
- W. Gao, D. M. Cui, X. M. Liu, Y. Zhang and Y. Mu, Organometallics, 2008, 27, 5889 CrossRef CAS.
- M. T. Gamer, M. Rastatter, P. W. Roesky, A. Steffens and M. Glanz, Chem.–Eur. J., 2005, 11, 3165 CrossRef CAS.
- M. Wiecko, P. W. Roesky, V. V. Burlakov and A. Spannenberg, Eur. J. Inorg. Chem., 2007, 876 CrossRef CAS.
- M. T. Gamer, P. W. Roesky, I. Palard, M. Le Hellaye and S. M. Guillaume, Organometallics, 2007, 26, 651 CrossRef CAS.
- H. Sinn, W. Kaminsky, H. J. Vollmer and R. Woldt, Angew. Chem., Int. Ed. Engl., 1980, 19, 390 CrossRef.
- J. Herwig and W. Kaminsky, Polym. Bull., 1983, 9, 464 CAS.
- W. Kaminsky, M. Miri, H. Sinn and R. Woldt, Makromol. Chem. Rapid Commun., 1983, 4, 417 CrossRef CAS.
- P. C. Mohring and N. J. Coville, J. Organomet. Chem., 1994, 479, 1 CrossRef.
- X. M. Yang, C. L. Stern and T. J. Marks, J. Am. Chem. Soc., 1994, 116, 10015 CrossRef CAS.
- H. H. Brintzinger, D. Fischer, R. Mulhaupt, B. Rieger and R. M. Waymouth, Angew. Chem., Int. Ed. Engl., 1995, 34, 1143 CrossRef CAS.
- W. Kaminsky, Macromol. Chem. Phys., 1996, 197, 3907 CrossRef CAS.
- W. Kaminsky, Pure Appl. Chem., 1998, 70, 1229 CrossRef CAS.
- L. Postigo, J. Sánchez-Nieves, P. Royo and M. E. G. Mosquera, Dalton Trans., 2009, 3756 RSC.
- H. Wang, H.-S. Chan, J. Okuda and Z. Xie, Organometallics, 2005, 24, 3118 CrossRef CAS.
- L. I. Strunkina, M. K. Minacheva, K. A. Lyssenko, V. V. Burlakov, W. Baumann, P. Arndt, B. N. Strunin and V. B. Shur, J. Organomet. Chem., 2006, 691, 557 CrossRef CAS.
- V. V. Burlakov, P. Arndt, W. Baumann, A. Spannenberg and U. Rosenthal, Organometallics, 2006, 25, 519 CrossRef CAS.
- J. Cayuela, V. Bounor-Legaré, P. Cassagnau and A. Michel, Macromolecules, 2006, 39, 1338 CrossRef CAS.
- Y. Pérez, I. del Hierro, I. Sierra, P. Gómez-Sal, M. Fajardo and A. Otero, J. Organomet. Chem., 2006, 691, 3053 CrossRef CAS.
- C. J. Chuck, M. G. Davidson, M. D. Jones, G. Kociok-Köhn, M. D. Lunn and S. Wu, Inorg. Chem., 2006, 45, 6595 CrossRef CAS.
- Y. Takashima, Y. Nakayama, T. Hirao, H. Yasuda and A. Harada, J. Organomet. Chem., 2004, 689, 612 CrossRef CAS.
- M. G. Davidson, M. D. Jones, M. D. Lunn and M. F. Mahon, Inorg. Chem., 2006, 45, 2282 CrossRef CAS.
- F. Gornshtein, M. Kapon, M. Botoshansky and M. S. Eisen, Organometallics, 2007, 26, 497 CrossRef CAS.
- A. J. Chmura, M. G. Davidson, M. D. Jones, M. D. Lunn and M. F. Mahon, Dalton Trans., 2006, 887 RSC.
- A. J. Chmura, M. G. Davidson, M. D. Jones, M. D. Lunn, M. F. Mahon, A. F. Johnson, P. Khunkamchoo, S. L. Roberts and S. S. F. Wong, Macromolecules, 2006, 39, 7250 CrossRef CAS.
- D. S. McGuinness, E. L. Marshall, V. C. Gibson and J. W. Steed, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3798 CrossRef CAS.
- M. H. Cho, J. S. Yoon and I.-M. Lee, Bull. Korean Chem. Soc., 2007, 28, 2471 CAS.
- S.-D. Mun, Y. Hong and Y. Kim, Bull. Korean Chem. Soc., 2007, 28, 698 CAS.
- K.-C. Hsieh, W.-Y. Lee, L.-F. Hsueh, H. M. Lee and J.-H. Huang, Eur. J. Inorg. Chem., 2006, 2306 CrossRef CAS.
- R. F. Munhá, L. G. Alves, N. Maulide, M. T. Duarte, I. E. Markó, M. D. Fryzuk and A. M. Martins, Inorg. Chem. Commun., 2008, 11, 1174 CrossRef CAS.
- Value calculated for consistency from the catalytic activity reported.
- G. J. P. Britovsek, V. C. Gibson and D. F. Wass, Angew. Chem., Int. Ed., 1999, 38, 428 CrossRef CAS.
- Y. Mahha, A. Atlamsani, J.-C. Blais, M. Tessier, J.-M. Brégeault and L. Salles, J. Mol. Catal. A: Chem., 2005, 234, 63 CrossRef CAS.
- J. Yamada and K. Nomura, Organometallics, 2005, 24, 3621 CrossRef CAS.
- V. C. Gibson, C. Redshaw and G. A. Solan, Chem. Rev., 2007, 107, 1745 CrossRef CAS.
- M.-Z. Chen, H.-M. Sun, W.-F. Li, Z.-G. Wang, Q. Shen and Y. Zhang, J. Organomet. Chem., 2006, 691, 2489 CrossRef CAS.
- R. R. Gowda and D. Chakraborty, J. Mol. Catal. A: Chem., 2009, 301, 84 CrossRef CAS.
- A. Arbaoui, C. Redshaw, M. R. J. Elsegood, A. Yoshizawa and T. Yamato, Chem.–Asian J. DOI:10.1002/asia.200900514.
- D. M. Kurtz, Chem. Rev., 1990, 90, 585 CrossRef CAS.
- C. Redshaw, M. A. Rowan, L. Warford, D. M. Homden, A. Arbaoui, M. R. J. Elsegood, S. H. Dale, T. Yamato, C. P. Casas, S. Matsui and S. Matsuura, Chem.–Eur. J., 2007, 13, 1090 CrossRef CAS.
- F. Stassin and R. Jérôme, Chem. Commun., 2003, 232 RSC.
- M. Sobczak and W. Kolodziejski, Molecules, 2009, 14, 621 Search PubMed.
- H. R. Kricheldorf, K. Bornhorst and H. Hachmann-Thiessen, Macromolecules, 2005, 38, 5017 CrossRef CAS.
- H. R. Kricheldorf and S. Rost, Biomacromolecules, 2005, 6, 1345 CrossRef CAS.
- H. R. Kricheldorf, G. Behnken, G. Schwarz and J. Kopf, Macromolecules, 2008, 41, 4102 CrossRef CAS.
- S. Penczek, A. Duda, A. Kowalski, J. Libiszowski, K. Majerska and T. Biela, Macromol. Symp., 2000, 157, 61 CrossRef CAS.
- A. Kowalski, J. Libiszowski, K. Majerska, A. Duda and S. Penczek, Polymer, 2007, 48, 3952 CrossRef CAS.
- J. Lewiński, P. Horeglad, K. Wójcik and I. Justyniak, Organometallics, 2005, 24, 4588 CrossRef CAS.
- L. Dostál, R. Jambor, I. Císařová, J. Merna and J. Holeček, Appl. Organomet. Chem., 2007, 21, 688 CrossRef CAS.
- M. Moller, R. Kange and J. L. Hedrick, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2067 CrossRef CAS.
- Y. Nakayama, H. Bando, Y. Sonobe and T. Fujita, J. Mol. Catal. A: Chem., 2004, 213, 141 CrossRef CAS.
- N. Nomura, T. Aoyama, R. Ishii and T. Kondo, Macromolecules, 2005, 38, 5363 CrossRef CAS.
- L. S. Baugh and J. A. Sissano, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 1633 CrossRef CAS.
- N. Iwasa, J. Y. Liu and K. Nomura, Catal. Commun., 2008, 9, 1148 CrossRef CAS.
- J. Liu, N. Iwasaa and K. Nomura, Dalton Trans., 2008, 3978 RSC.
- N. Iwasa, M. Fujiki and K. Nomura, J. Mol. Catal. A: Chem., 2008, 292, 67 CrossRef CAS.
- N. Iwasa, S. Katao, J. Y. Liu, M. Fujiki, Y. Furukawa and K. Nomura, Organometallics, 2009, 28, 2179 CrossRef CAS.
- D. Pappalardo, L. Annunziata and C. Pellecchia, Macromolecules, 2009, 42, 6056 CrossRef CAS.
- M. Bouyahyi, E. Grunova, N. Marquet, E. Kirillov, C. M. Thomas, T. Roisnel and J.-F. Carpentier, Organometallics, 2008, 27, 5815 CrossRef CAS.
- P. A. Cameron, D. Jhurry, V. C. Gibson, A. J. P. White, D. J. Williams and S. Williams, Macromol. Rapid Commun., 1999, 20, 616 CrossRef CAS.
- C. Zhang and Z.-X. Wang, J. Organomet. Chem., 2008, 693, 3151 CrossRef CAS.
- A. Arbaoui, C. Redshaw, N. M. Sanchez, M. R. J. Elsegood and D. L. Hughes, to be published.
- A. Arbaoui, C. Redshaw and D. L. Hughes, Supramol. Chem., 2009, 21, 35 CrossRef CAS.
- W. Braune and J. Okuda, Angew. Chem., Int. Ed., 2003, 42, 64 CrossRef CAS.
- C. T. Chen, C. Y. Chan, C. A. Huang, M. T. Chen and K. F. Peng, Dalton Trans., 2007, 4073 RSC.
- A.-H. Gao, W. Yao, Y. Mu, W. Gao, M.-T. Sun and Q. Su, Polyhedron, 2009, 28, 2605 CrossRef CAS.
- I. P. Hsieh, C. H. Huang, H. M. Lee, P. C. Kuo, J. H. Huang, H. I. Lee, J. T. Cheng and G. H. Lee, Inorg. Chim. Acta, 2006, 359, 497 CAS.
- Y. C. Chen, C. Y. Lin, C. Y. Li, J. H. Huang, L. C. Chang and T. Y. Lee, Chem.–Eur. J., 2008, 14, 9747 CrossRef CAS.
- S. G. Gong and H. Y. Ma, Dalton Trans., 2008, 3345 RSC.
- H.-Y. Chen, B.-H. Huang and C.-C. Lin, Macromolecules, 2005, 38, 5400 CrossRef CAS.
- C. Alonso-Moreno, A. Garcés, L. F. Sánchez-Barba, M. Fajardo, J. Fernández-Baeza, A. Otero, A. Lara-Sánchez, A. Antiñolo, L. Broomfield, M. I. López-Solera and A. M. Rodríguez, Organometallics, 2008, 27, 1310 CrossRef CAS.
- Z.-X. Wang and C.-Y. Qi, Organometallics, 2007, 26, 2243 CrossRef CAS.
- Z.-Y. Chai, C. Zhang and Z.-X. Wang, Organometallics, 2008, 27, 1626 CrossRef CAS.
- H. Zhu and E. Y.-X. Chen, Organometallics, 2007, 26, 5395 CrossRef CAS.
- M. Haddad, M. Laghzaoui, R. Welter and S. Dagorne, Organometallics, 2009, 28, 4584 CrossRef CAS.
- L. Annunziata, D. Pappalardo, C. Tedesco, S. Antinucci and C. Pellecchia, J. Organomet. Chem., 2006, 691, 1505 CrossRef CAS.
- D. Pappalardo, L. Annunziata, C. Pellecchia, M. Biesemans and R. Willem, Macromolecules, 2007, 40, 1886 CrossRef CAS.
- G. Deshayes, K. Poelmans, I. Verbruggen, C. Camacho-Camacho, P. Degée, V. Pinoie, J. C. Martins, M. Piotto, M. Biesemans, R. Willem and P. Dubois, Chem.–Eur. J., 2005, 11, 4552 CrossRef CAS.
- K. Poelmans, V. Pinoie, I. Verbruggen, M. Biesemans, G. Van Assche, G. Deshayes, P. Degée, P. Dubois and R. Willem, Appl. Organomet. Chem., 2007, 21, 504 CrossRef CAS.
- K. Poelmans, V. Pinoie, I. Verbruggen, M. Biesemans, G. Deshayes, E. Dusquesne, C. Delcourt, P. Degée, H. E. Miltner, P. Dubois and R. Willem, Organometallics, 2008, 27, 1841 CrossRef CAS.
- M. Shen, W. Zhang, K. Nomura and W.-H. Sun, Dalton Trans., 2009, 9000 RSC.
- E. Grunova, T. Roisnel and J.-F. Carpentier, Dalton Trans., 2009, 9010 RSC.
- A. J. Chmura, M. G. Davidson, C. J. Frankis, M. D. Jones and M. D. Lunn, Chem. Commun., 2008, 1293 RSC.
- A. J. Chmura, C. J. Chuck, M. G. Davidson, M. D. Jones, M. D. Lunn, S. D. Bull and M. F. Mahon, Angew. Chem., Int. Ed., 2007, 46, 2280 CrossRef CAS.
- E. L. Whitelaw, M. D. Jones, M. F. Mahon and G. Kociok-Kohn, Dalton Trans., 2009, 9020 RSC.
- B. Lian, H. Ma, T. P. Spaniol and J. Okuda, Dalton Trans., 2009, 9033 RSC.
- M.-T. Chen, P.-J. Chang, C.-A. Huang, K.-F. Peng and C.-T. Chen, Dalton Trans., 2009, 9068 RSC.
Footnote |
| † Since this review was prepared, a number of relevant publications have appeared in the ‘Metal Catalysed Polymerisation’ themed issue of Dalton Transactions. Sun et al. have prepared a series of dialkyl aluminium complexes bearing 2-methyl-N-arylquinolin-8-amine type ligands. Such dialkyl species were shown to be highly active towards ROP of ε-caprolactone affording high molecular weight polymer (2.1–21.9 × 104) with Mw/Mn in the range 1.19–3.57. Interestingly, related monoalkyl complexes were inactive, both in the presence or absence of nBuOH. 175 Carpentier et al. have shown that zinc complexes, prepared from fluorous imino-alcohols and either Zn[N(SiMe3)2]2 or ZnEt2, when combined with BnOH are active for ROP of racemic lactide (and β-butyrolactone) at 20–50 °C. The product is atactic polylactide (or poly(3-hydroxybutyrate) with molecular weight (Mn) upto 20,700 and Mw/Mn 1.06–1.57. Use of excess benzyl alcohol(acts as a transfer agent) versus zinc, allows for the immortal ROP of lactide (or β-butyrolactone). 176 Amine tris(phenolate) complexes of titanium(IV) and zirconium(IV) have been screened for ROP of rac-lactide, and in general there is a better degree of control when using the zirconium-based systems (Mw/Mn 1.26–1.34). Only a slight degree of heterotacticity was observed (via1H homonuclear decoupled NMR) in the resultant polymers, which is in stark contrast to observations using related C3 complexes of zirconium, hafnium and germanium.177,178 ROP of 1,3-dioxan-2-one afforded polycarbonate in conversions of between 24 and 99% with Mw/Mn 1.60–2.72. 179 Neutral methyl aluminium complexes bearing chiral (OSSO)-type bis(phenolate) ligands were shown, in the presence of isopropanol, to polymerise rac-lactide affording, in a living fashion, atactic polymer. 180 A series of magnesium bis(amido-oxazolinate) complexes have demonstrated efficient activities for ROP of L-lactide in the presence of benzyl alcohol. However, for ligand backbones bearing pendant phenyl-containing functionality, there is poor conversion. 181 |
| This journal is © The Royal Society of Chemistry 2010 |