Facile polyisobutylene functionalization via thiol–ene click chemistry†‡
Andrew J. D.
Magenau
,
Justin W.
Chan
,
Charles E.
Hoyle
and
Robson F.
Storey
*
School of Polymers and High Performance Materials, The University of Southern Mississippi, 118 College Drive #10076. Hattiesburg, MS 39406-10076, USA. E-mail: robson.storey@usm.com
First published on 10th May 2010
Abstract
Thiol–ene click chemistry was adapted to easily and rapidly modify exo-olefin polyisobutylene with an array of thiol compounds bearing useful functionalities, including primary halogen, primary amine, primary hydroxyl, and carboxylic acid.
Polyisobutylene (PIB) has commercial utility as a stabilizing fuel and motor oil additive, packaging elastomer, adhesive and sealant, and more recently, as a biomaterial. This completely saturated hydrocarbon elastomer has excellent thermal and oxidative stability, gas-barrier properties, and biocompatibility. Due to these and other unique characteristics, PIB remains the subject of continued research, e.g. as a self-separating homogeneous catalyst support,1 a matrix for quantum dot-polymer composites for inks with enhanced photoluminensce,2 and a biostable/biocompatible thermoplastic elastomer (TPU) for the drug-eluting coating on coronary stents.3
Efficient methods for the synthesis of PIB carrying functional end groups have long been sought to facilitate the creation of new PIB-based materials. In the past, post-polymerization modification procedures were employed, which were often laborious, time-consuming, and multistep.4 To circumvent these difficult procedures, in situ quenching has recently been developed to conveniently functionalize PIB through direct reaction of quasiliving PIB with a nucleophilic quenching or capping agent. Various classes of compounds with demonstrated effectiveness include olefins such as alkenylsilanes5 and butadiene,6 hindered nucleophiles,7 and activated aromatic compounds such as 2-alkylfurans, thiophene, N-substituted pyrroles, and alkoxy benzenes.8 Although in situ quenching is attractive due to its simplicity, the quenching agents are often expensive or unavailable commercially. Also, in situ quenching is generally limited to soft nucleophilic (π) quenching agents, since hard nucleophiles (σ) inevitably react with the Lewis acid.9
Since adoption of the thiol–ene reaction into the class of click chemistries,10 numerous researchers have utilized this powerful synthetic tool for polymer modification,11 polymer–protein conjugate synthesis,12 network formation,13 and creation of complex polymeric architectures.10a,14 Thiol–ene chemistry offers many advantages including mild reaction conditions, tolerance to oxygen and water, simple purification, high reaction rates, modularity, and quantitative conversion in the absence of metal catalyst. Moreover, the vast array of commercially available thiol and alkene functionalities provides an incredibly versatile selection of reagents.
Monofunctional PIB possessing a high proportion (up to 85–90%) of exo-olefin (methyl vinylidene) end groups is available commercially (e.g., Glissopal, BASF; Ultravis, BP Chemicals) and both monofunctional and difunctional PIBs with ∼100% exo-olefin termini are readily produced viain situ quenching of quasiliving PIB.7 Application of thiol–ene chemistry to exo-olefin PIB would thus provide a practical means to a variety of functional PIBs. However, only a few literature reports have appeared on this topic, and reaction conversions were typically low and/or reaction times were long. For example, Blackborow et al.15 reported the radical addition of thiols carrying a number of functional groups including hydroxyl, carboxylic acid, and alkoxysilane to Ultravis (52 or 84% exo-olefin end groups), using either photo- or thermal initiation. High conversions were qualitatively claimed but reaction times were long (12–17 h for photoinitiation, 20–50 h for thermal initiation). Gorski et al. reported the radical addition of alkyl and hydroxyl functional thiols to Glissopal; although reaction kinetics with Glissopal were not reported, a model compound for exo-olefin PIB, 2,4,4-trimethyl-1-pentene, required long reaction times even when reacted neat in the presence of a ten-fold excess of thiol (p ≈ 85% at 6 h).16 Later, based partly on their earlier patent,15 Blackborow et al. reported the radical addition to Ultravis of thiols with various functional groups including hydroxyl, methoxyethoxyethyl, carboxylic acid, and organosilane.17 Conversions determined using FTIR, ranged from 35 to 80%, with reaction times of 5 h or more; in the few examples where high conversions were reached (p ≥ 90%), reaction times between 44 and 88 h were necessary.17
The PIB modifications described in these previous literature reports do not meet the qualifications of a click reaction. Herein, we show that the thiol–ene reaction, under appropriate conditions, can be used to simply, rapidly, and quantitatively functionalize exo-olefin PIB with an array of functional thiols (Scheme 1). These reactions achieve near-quantitative conversion of the exo-olefin in less than 10 min, are applicable to both mono and difunctional PIBs, and require minimal equipment, and the products can be purified by either a simple precipitation or wash.
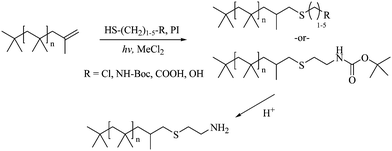 | ||
| Scheme 1 Photo-initiated radical addition of functional thiols (halo, NH–Boc, carboxylic acid, hydroxyl) to exo-olefin-terminated PIB and subsequent Boc deprotection. PI = dimethoxy-2-phenylacetophenone. | ||
Telechelic mono- and difunctional exo-olefin PIBs were synthesized via quasiliving cationic polymerization followed by quenching with the hindered amine, 1,2,2,6,6-pentamethylpiperidine.7 Low molecular weight polymers were targeted to facilitate end group characterization by NMR spectroscopy. Molecular weight, polydispersity index (PDI), and functionality of the exo-olefin PIBs are summarized in Table 1. Both polymers exhibited very low PDIs with unimodal, symmetrical elution peaks in GPC (Fig. S1 and S2, ESI†) and high chain end functionality (≥97%), calculated as previously reported in literature.7 Excellent agreement between GPC and 1H NMR molecular weights was observed for both PIB precursors.
A variety of photo-initiated thiol–ene reactions were conducted, and the results are summarized in Table 2. Particular emphasis was placed on primary halogen and amine functionalities due to their novelty and utility. The clear, transparent nature and inert hydrocarbon backbone of PIB facilitated efficient photochemical reactions. Typical synthetic procedures involved charging a scintillation vial with an appropriate mass of polymer, thiol, and 1 wt% photoinitiator, and then dissolving the mixture in a suitable solvent. Solvent selection (CH2Cl2 or CHCl3) and reagent concentrations were crucial parameters, and were chosen to ensure a homogenous reaction mixture. Reaction conversions were improved by reducing reaction temperature, in agreement with prior literature for thiol–ene functionalization of PIB.17 In the absence of external cooling, internal reaction temperatures were found to rise as high as 40 °C, whereas by placing the reactor in an ice bath, reaction temperatures were maintained below 15 °C.
| Exp. | Thiol | [ene]/M | [SH]:[ene] | t/min | p ene (%) |
|---|---|---|---|---|---|
| a Ice bath. b Difunctional PIB. c Sunlight. d One-pot deprotection. e NMR. | |||||
| 1 | HS(CH2)3Cl | 0.80 | 1.50 | 3.5 | 97 |
| 2a | HS(CH2)3Cl | 0.80 | 1.50 | 3.5 | >99 |
| 3a,b | HS(CH2)3Cl | 0.80 | 1.50 | 3.5 | >99 |
| 4 | HS(CH2)2NH2 | 0.80 | 1.50 | 3.5 | 0 |
| 5 | HS(CH2)2NH2·HCl | 0.80 | 1.50 | 3.5 | 0 |
| 6 | HS(CH2)2NH–Boc | 0.80 | 1.50 | 3.5 | 93 |
| 7a | HS(CH2)2NH–Boc | 0.80 | 1.50 | 3.5 | >99 |
| 8a,b | HS(CH2)2NH–Boc | 0.80 | 1.50 | 3.5 | >99 |
| 9a,c | HS(CH2)2NH–Boc | 0.80 | 1.50 | 180 | >99 |
| 10a,d | HS(CH2)2NH–Boc | 0.80 | 1.50 | 3.5 | >99 |
| 11a | HSCH2COOH | 0.40 | 3.00 | 8 | >99 |
| 12a | HS(CH2)5OH | 0.38 | 4.00 | 6 | 98 |
Thiol–ene reactions involving 3-chloropropane thiol in 50% excess (Exp. 1–3) achieved high conversions in less than 5 min, with essentially quantitative conversion achieved with cooling. Fig. 1 depicts 1H NMR spectra of the monofunctional exo-olefin PIB precursor and resulting 3-chloropropane thioether functionalized PIB (Exp. 2). Resonances associated with the exo-olefin terminus of the precursor, consisting of methylene (2.00 ppm), methyl (1.78 ppm), and olefinic protons (4.64 and 4.85 ppm), were all absent in the final product. New resonances in the product associated with the 3-chloropropane thioether moiety were located at 3.66 (triplet), 2.66 (triplet), 2.04 (quintet), and 1.74 ppm. In addition, diastereotopic protons, d1 and d2, were observed at 2.52 and 2.35 ppm, each appearing as a doublet of doublets due to the adjacent chiral center.
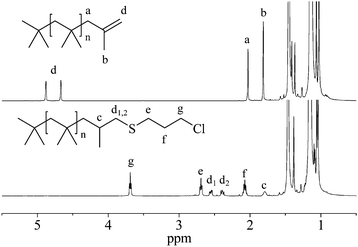 | ||
| Fig. 1 1H NMR spectra of exo-olefin PIB (top) and PIB–Cl (bottom). | ||
Thiol–ene reactions with cysteamine and cysteamine hydrochloride, Exp. 4 and 5, resulted in no thioether formation. These results were theorized to be a consequence of thiolate formation, due to amine basicity, rendering the thiol group incapable of thiyl radical formation. Furthermore, incompatibility of these reagents in a common reaction medium, creating a heterogeneous reaction mixture, impeded their ability to react. Typical solvents for cysteamine, including water, methanol, and DMF, were incompatible with the hydrophobic PIB; THF and THF/H2O solvent systems were explored with thermal and photoinitiators, respectively, with limited success.
Conversion of the olefin increased dramatically when the amine group of cysteamine was protected with a tert-butoxycarbonyl (Boc) group (Exp. 6). The Boc group produced a completely homogenous reaction mixture and considerably reduced the basicity of the amine, diminishing thiolate formation. Although the Boc-protected cysteamine yielded the desired product, complete conversion was still not achieved in Exp. 6. However, near-quantitative conversion was achieved for both the mono (Exp. 7) and difunctional (Exp. 8) exo-olefin polymers by cooling the reaction with an ice-bath. Thiol–ene reaction kinetics by FTIR for cysteamine–Boc are shown in Fig. S3 in ESI†.
GPC analysis of the PIB–Boc and PIB–Cl products of Table 2 revealed symmetrical traces with no high molecular weight coupled species. Slight decreases in elution volume were observed, indicative of increased molecular weight, with respect to the exo-olefin PIB. Fig. S4 in ESI† shows GPC traces of PIB-exo, PIB–Boc, and PIB–Cl.
Sunlight-activated radical generation was also attempted (Exp. 9); however, extended reaction times were required to achieve complete olefin conversion and substantial by-products were detected. Prolonged exposure to sunlight (3 h) may have caused unwanted photo-oxidization to occur. The susceptibility of thiols and various sulfur derivatives to photo-oxidation is well known in literature.18
In an effort to obtain amine functionality directly from Boc-protected cysteamine, a procedure was developed in which the thiol–ene reaction and subsequent deblocking were carried out in one pot. Deblocking was accomplished by simply charging a 50 vol% trifluoroacetic acid (TFA) solution in CH2Cl2 to the reactor after the thiol–ene reaction was complete (Exp. 10). No by-products were detected, and near quantitative conversion and simple purification were maintained. 1H NMR spectra of Boc-protected and deprotected amine functionalized PIB (PIB–Boc and PIB–NH2) are shown in Fig. 2. Peaks due to residual exo-olefin were not observed. PIB–Boc resonances appeared at 4.92 (g′), 3.31 (f), 2.62 (e), 1.72 (c), 1.45 (h′), and at 2.51 (d1) and 2.37 (d2) ppm (diastereotopic protons, doublet of doublets). As expected, after Boc deprotection disappearance of the secondary amine (g′) and tert-butyl (h′) proton peaks occurred. Furthermore, the ultimate methylene protons adjacent to nitrogen (f) shifted slightly upfield; whereas the penultimate methylene protons (e) shifted downfield. Further confirmation was provided by comparison of FTIR spectra of PIB-exo, PIB–Boc, and PIB–NH2 as shown in Fig. S5 in ESI†.
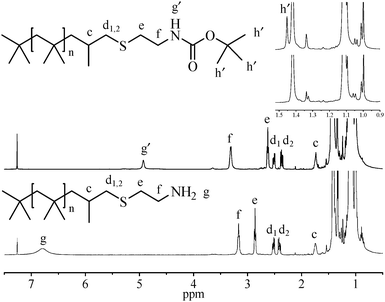 | ||
| Fig. 2 1H NMR spectra of PIB–Boc (top) and PIB–NH2 (bottom). | ||
To further demonstrate the versatility and modularity of the thiol–ene approach, two additional functionalities were selected, carboxylic acid (Exp. 11) and hydroxyl (Exp. 12). Both reactions achieved high conversions (≥98) in minutes (<10 min). 1H NMR, FTIR, and GPC data are provided in the ESI† for the carboxylic acid (Fig. S6–8) and hydroxyl (Fig. S9–11) functionalized PIBs.
To conclude, the above described is a simple, rapid synthesis of primary halogen, amine, carboxylic acid, and hydroxyl-functional PIBs (mono- and difunctional) via thiol–ene click chemistry. The methods produce near-quantitative functionalization within 10 min without difficult purification or reaction conditions. Reduced reaction temperature (ice bath) facilitates increased conversion. Functionalization using cysteamine requires Boc protection; however, the Boc protecting group may be easily removed in one pot.
Notes and references
- (a) D. E. Bergbreiter, P. N. Hamilton and N. M. Koshti, J. Am. Chem. Soc., 2007, 129, 10666–10667 CrossRef CAS.
- V. Wood, M. J. Panzer, J. Chen, M. S. Bradley, J. E. Halpert, M. G. Bawendi and V. Bulović, Adv. Mater., 2009, 21, 2151–2155 CrossRef CAS.
- L. Pinchuk, G. J. Wilson, J. J. Barry, R. T. Schoephoerster, J.-M. Parel and J. P. Kennedy, Biomaterials, 2008, 29, 448–460 CrossRef CAS.
- J. P. Kennedy and B. Ivan, Designed Polymers by Carbocationic Macromolecular Engineering: Theory and Practice, Hanser, New York, 1992 Search PubMed.
- (a) L. Wilczek and J. P. Kennedy, J. Polym. Sci., Part A: Polym. Chem., 1987, 25, 3255–3265 CrossRef CAS; (b) L. V. Nielsen, R. R. Nielsen, B. Gao, J. Kops and B. Iván, Polymer, 1997, 38, 2529–2534 CrossRef; (c) M. Roth and H. Mayr, Macromolecules, 1996, 29, 6104–6109 CrossRef CAS.
-
(a)
K. Knoll, K. Bronstert and D. Bender, US Pat., 5
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 212248, 1993;
(b) P. De and R. Faust, Macromolecules, 2006, 39, 6861–6870 CrossRef CAS.
212248, 1993;
(b) P. De and R. Faust, Macromolecules, 2006, 39, 6861–6870 CrossRef CAS. - (a) K. L. Simison, C. D. Stokes, J. J. Harrison and R. F. Storey, Macromolecules, 2006, 39, 2481–2487 CrossRef CAS; (b) A. J. D. Magenau, N. Martinez-Castro and R. F. Storey, Macromolecules, 2009, 42, 2353–2359 CrossRef CAS.
- (a) S. Hadjikyriacou and R. Faust, Macromolecules, 1999, 32, 6393–6399 CrossRef CAS; (b) N. Martinez-Castro, M. G. Lanzendörfer, A. H. E. Müller, J. C. Cho, M. H. Acar and R. Faust, Macromolecules, 2003, 36, 6985–6994 CrossRef CAS; (c) N. Martinez-Castro, D. L. Morgan and R. F. Storey, Macromolecules, 2009, 42, 4963–4971 CrossRef CAS; (d) D. L. Morgan and R. F. Storey, Macromolecules, 2009, 42, 6844–6847 CrossRef CAS; (e) A. J. D. Magenau, N. Martinez-Castro, D. A. Savin and R. F. Storey, Macromolecules, 2009, 42, 8044–8051 CrossRef CAS.
- (a) R. Faust and J. P. Kennedy, J. Macromol. Sci., Chem., 1990, A27, 649–667 CAS; (b) B. Iván and J. P. Kennedy, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 89–104 CrossRef CAS.
- (a) K. L. Killops, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 5062–5064 CrossRef CAS; (b) C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900–4908 CrossRef CAS; (c) T. P. Lodge, Macromolecules, 2009, 42, 3827–3829 CrossRef CAS; (d) A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC; (e) R. K. Iha, K. L. Wooley, A. M. Nyström, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS.
- (a) Z. Hordyjewicz-Baran, L. You, B. Smarsly, R. Sigel and H. Schlaad, Macromolecules, 2007, 40, 3901–3903 CrossRef CAS; (b) B. Yu, J. W. Chan, C. E. Hoyle and A. B. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3544–3557 CrossRef CAS; (c) M. Li, P. De, S. R. Gondi and B. S. Sumerlin, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5093–5100 CrossRef CAS.
- (a) A. A. Aimetti, A. J. Machen and K. S. Anseth, Biomaterials, 2009, 30, 6048–6054 CrossRef CAS; (b) M. W. Jones, G. Mantovani, S. M. Ryan, X. Wang, D. J. Brayden and D. M. Haddleton, Chem. Commun., 2009, 5272–5274 RSC.
- (a) C. E. Hoyle, T. Y. Lee and T. Roper, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5301–5559 CrossRef CAS; (b) J. Shin, S. Nazarenko and C. E. Hoyle, Macromolecules, 2009, 42, 6549–6557 CrossRef CAS; (c) V. S. Khire, Y. Yi, N. A. Clark and C. N. Bowman, Adv. Mater., 2008, 20, 3308–3311 CrossRef CAS.
- J. W. Chan, B. Yu, C. E. Hoyle and A. B. Lowe, Chem. Commun., 2008, 4959–4961 RSC.
-
J. R. Blackborow, S. Boileau and B. Mazeaud, Eur. Pat., EP 0342792 A1, 1989.
- U. Gorski, K. Maenz and D. Stadermann, Angew. Makromol. Chem., 1997, 253, 51–64 CrossRef CAS.
- S. Boileau, B. Mazeaud-Henri and R. Blackborow, Eur. Polym. J., 2003, 39, 1395–1404 CrossRef CAS.
- G. Capozzi and G. Modena, in The Chemistry of the Thiol Group, ed. S. Patai, John Wiley & Sons, Bristol, 1974, vol. 2, ch. 17, pp. 832–833 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental, instrumentation, and characterization. See DOI: 10.1039/c0py00094a |
| ‡ This paper is dedicated to the late Prof. Charles E. Hoyle (1948–2009). |
| This journal is © The Royal Society of Chemistry 2010 |