Synthesis of thermoresponsive phenyl- and naphthyl-terminated poly(NIPAM) derivatives using RAFT and their complexation with cyclobis(paraquat-p-phenylene) derivatives in water†
Julien
Bigot
ab,
David
Fournier
ab,
Joël
Lyskawa
ab,
Thomas
Marmin
ab,
Frédéric
Cazaux
abc,
Graeme
Cooke
*d and
Patrice
Woisel
*abc
aUniv Lille Nord de France, F-59000, Lille, France
bUSTL, Unité des Matériaux Et Transformations (UMET, UMR 8207), Ingénierie des Systèmes polymères (ISP) Team, F-59650, Villeneuve d'Ascq Cedex, France. E-mail: patrice.woisel@ensc-lille.fr
cENSCL, F-59652, Villeneuve d'Ascq, France
dGlasgow Centre for Physical Organic Chemistry, WestCHEM, Department of Chemistry, University of Glasgow, Joseph Black Building, Glasgow, UK G12 8QQ. E-mail: graemec@chem.gla.ac.uk
First published on 21st May 2010
Abstract
A series of poly(N-isopropylacrylamide)s (poly(NIPAM)s) have been synthesized via reversible addition-fragmentation chain transfer (RAFT) polymerization from a functionalized chain transfer agent (CTA) bearing either dialkoxynaphthalene or dialkoxyphenylene moieties. After demonstrating the controlled character of the RAFT polymerization in the presence of these CTAs, well-defined functionalized poly(NIPAM)s with low PDIs and similar molecular weights were selected and subjected to lower critical solution temperature (LCST) measurements using UV-vis spectroscopy. We have investigated the complexation of the polymers with the tetracationic cyclophane cyclobis(paraquat-p-phenylene) (CBPQT4+) and specifically the role the counter anion (Cl−, Br−, I−) of the cyclophane has on the LCST. Moreover, we have shown that the addition of a competing end-functionalized naphthalene poly(NIPAM) guest to a (CBPQT4+)–end-functionalized phenylene poly(NIPAM) complex results in the dethreading of the original architecture.
Introduction
Smart polymers that can respond to an external stimuli (e.g. temperature, pH, ionic strength, light), have attracted considerable attention in recent years.1,2 In particular, thermoresponsive materials that undergo a solubility change as a function of temperature have found applications in various fields such as drug delivery,3–5 paints,6 bioengineering,7 membranes8,9 and nanomaterials.10 It is well established that the lower critical solution temperature (LCST) is strongly affected by many parameters such as concentration,11 structure,7,12 and molecular weight.13,14 In addition, the LCST of a temperature responsive polymer is also influenced by the hydrophilicity–hydrophobicity balance in the polymer. For example, copolymerization of either N-isopropylacrylamide (NIPAM),15N,N-(dimethylamino)ethyl methacrylate (DMAEMA)16,17 or 2-alkyl-2-oxazoline18,19 with hydrophilic or hydrophobic comonomers results in a shift of LCST to higher or lower values, respectively. The most widely studied thermoresponsive polymer is undoubtedly poly(N-isopropylacrylamide) (poly(NIPAM)) as its LCST of 32–34 °C is quite close to the human body temperature20 and that the LCST is largely insensitive to changes in concentration, pH or ionic strength thereby promoting biomedical applications of these polymers. Furthermore, polymers prepared using controlled radical polymerization (CRP) techniques possess sharper LCST transitions than those prepared using standard free radical polymerization methods.21Recently, smart polymers have been developed which combine specific supramolecular interactions (e.g. hydrogen bonds, metal–ligand coordination) and LCST properties in attempts to produce new functional materials. A number of publications describe the incorporation of supramolecular recognition groups in the side-chain of the polymer via the copolymerization of a comonomer bearing, for instance cyclodextrin or adamantyl moieties.22–25 Furthermore, the importance of well-defined polymer architectures has already been demonstrated using different functionalized initiators.14,26,27 Only a limited number of detailed studies have been reported regarding the influence of non-covalent modification on the extremity of well-defined thermosensitive polymers. For example, Chiper et al.28 have recently developed this attractive concept by synthesising a well-defined poly(NIPAM) that was end functionalized with a terpyridine moiety able to participate in metallo-supramolecular interactions. The influence of the metallic centers (FeII, ZnII) associated to different counterions (CH3COO−, PF6−, Cl−) over the LCST was elegantly described.
In this study, we report the design and the synthesis of two novel reversible addition–fragmentation chain transfer (RAFT) agents bearing (dialkoxynaphthalene and dialkoxyphenylene) moieties able to create supramolecular assemblies with the electron-deficient cyclophane cyclobis(paraquat-p-phenylene) (CBPQT4+).29–31 Pseudorotaxanes constructed from CBPQT4+ have attracted interest due to their tuneable complexation properties,32 and formation of coloured guest-specific complexes.33 In a previous communication,34 we reported that a LCST-mediated transition of a naphthalene functionalised poly(NIPAM) derivative could be used as a simple and convenient tool to disrupt complexation with CBPQT4+. In this paper, we have extended this study to include two new types of end-functionalised poly(NIPAM) derivatives (from appropriately functionalised CTAs) to investigate the role these end-groups have on the RAFT polymerisation process, the thermosensitive properties of the polymers and complexation properties with CBPQT4+ in water. The influence the counterion associated with the CBPQT4+ unit has over the LCST has also been investigated.
Results and discussion
CTA synthesis
The good functional group tolerance and the versatility of the RAFT process has led to the synthesis of a wide range of end-chain functionalized polymers.35–37 In this paper, two functional RAFT agents featuring glycol-functionalized naphthalene and phenylene groups (Scheme 1) were designed and successfully synthesized as described in the ESI.† Several parameters can influence the binding ability of the CBPQT4+ unit toward guest molecules. These include the π-electron-donor ability and/or the hydrophobicity of the electron-rich guest and the propensity of ethylene glycol chains to stabilize complexes of this type through hydrogen bonding interactions.32 CTA138 and CTA2 only differ by the number of ethylene glycol units attached to each arm, whereas CTA1 and CTA2 differ from CTA3 in that the complexing agent (see further) is a naphthalene group for CTA1 and CTA2 and a phenylene group in case of CTA3. | ||
| Scheme 1 Structure of the functional chain transfer agents used during the RAFT polymerization of NIPAM. | ||
Reversible addition–fragmentation chain transfer (RAFT) polymerization of NIPAM
RAFT polymerization of NIPAM has been investigated in different reaction conditions using the above mentioned CTAs and resulted in end-chain functionalized poly(NIPAM) derivatives. First, polymerizations of NIPAM were carried out in dimethylformamide (DMF) using AIBN as radical initiator and CTA1 with a constant molar ratio [NIPAM]0 : [CTA1]0 of 100 : 1. The ratio [CTA1]0 : [AIBN]0 has been varied from 1 : 0.1 to 1 : 0.25 either at 70 °C or 80 °C in order to find the optimal polymerization conditions of NIPAM with this functional chain transfer agent. Results are summarized in Table 1.| T (°C) | AIBN (equiv.) | Time (min) | Conversiona (%) | M nth (g mol−1) | M nGPC (g mol−1) | PDIc |
|---|---|---|---|---|---|---|
| a Determined by GC. b M nth = conv. × ([M]/[CTA]) × M(NIPAM) + M(CTA) where M(NIPAM) and M(CTA) correspond to molecular weight of NIPAM and the chain transfer agent CTA, respectively. c Determined by GPC calibrated with polystyrene standards. Solvent: tetrahydrofuran (THF). | ||||||
| 70 | 0.10 | 35 | 21.0 | 3000 | 2900 | 1.04 |
| 190 | 77.5 | 9350 | 9900 | 1.19 | ||
| 420 | 83.9 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 100 |
10800 |
1.21 | ||
| 70 | 0.25 | 35 | 24.6 | 3400 | 5100 | 1.10 |
| 190 | 86.0 | 10300 |
10100 |
1.19 | ||
| 80 | 0.10 | 35 | 62.4 | 7650 | 7250 | 1.12 |
| 190 | 92.0 | 11000 |
10300 |
1.19 | ||
| 420 | 92.7 | 11100 |
10300 |
1.25 | ||
| 80 | 0.25 | 35 | 77.9 | 9400 | 7100 | 1.10 |
| 190 | 94.4 | 11260 |
9000 | 1.20 | ||
| 420 | 95.5 | 11400 |
9600 | 1.16 |
Pseudo-first order rate plots for NIPAM polymerizations mediated by CTA1 are shown in Fig. 1. As expected for a controlled radical polymerization, a linear increase in the semilogarithmic kinetic plot was observed (Fig. 1) for all the four polymerization reactions until high monomer conversion occurred. Higher apparent rate constants were observed when temperature was increased from 70 °C to 80 °C, due to the higher primary radical concentration at early stages and the increase of all rate constants involved during the process. The relative linearity of the kinetic plots is indicative of a constant concentration of radicals during the polymerization process.
 | ||
| Fig. 1 Kinetic plots for the RAFT polymerization of NIPAM in DMF using CTA1 under different conditions. | ||
As expected, at 80 °C, a higher AIBN concentration ([CTA1] : [AIBN] = 1 : 0.25, curve ×, Fig. 1) resulted in a faster polymerization rate, with a loss of control over the polymerization since the first-order kinetic plot was non-linear from the beginning of the polymerization. Nevertheless, when the molar ratio [CTA1] : [AIBN] was lowered to 1 : 0.1 (curve △, Fig. 1), the linearity remained until high monomer conversion was reached (i.e. 86.8%). Over this limit, non-linear first-order kinetic plots could be observed at high monomer conversion (>90%) when the reaction mixture becomes highly viscous. On the other hand, when the polymerization was carried out at 70 °C, despite a consistently lower polymerization rate, the first-order kinetic plot showed a straight line for both concentrations of AIBN, indicating a better control over the polymerization of NIPAM using CTA1. As already observed at 80 °C, the rate of polymerization is higher when the molar ratio [CTA1] : [AIBN] is fixed at 1 : 0.25 compared to 1 : 0.1. It is noteworthy that a short inhibition period of approximately 10–15 minutes could be detected as already observed for acrylamides.39–41 This short retardation period is probably due to the decomposition of AIBN and the time for the whole reaction mixture to be heated at the desired temperature.
The dependence of molecular weight and PDI with monomer conversion was also investigated for CTA1. The results are shown in Fig. 2. At 80 °C, even if the kinetic plots were not strictly linear, the evolution of molecular weights with conversion was not linear with a molar ratio [CTA1] : [AIBN] of 1 : 0.25 (●,○, Fig. 2) while a [CTA1] : [AIBN] ratio of 1 : 0.1 (▲, △, Fig. 2) led to a linear trend and the PDIs remained quite low (PDI ≈ 1.2). The non-linearity for 80 °C ratio 0.25 clearly indicates the occurrence of extensive chain transfer. When the polymerization was carried out at 70 °C with a [CTA1] : [AIBN] molar ratio fixed at 1 : 0.25 (◆, ◇, Fig. 2), the molecular weights did not increase linearly with conversion at the beginning of the polymerization. On the contrary, when the molar ratio is lowered to 1 : 0.1 (■, □, Fig. 2), a linear behaviour is observed even at high monomer conversion rate, as expected for a controlled radical polymerization.
 | ||
| Fig. 2 Dependence of number-average molecular weight (Mn) and polydispersity index (PDI) on conversion for RAFT polymerization of NIPAM using CTA1 under different conditions (shaded symbols refer to molecular weight and non-shaded symbols refer to PDI). | ||
The influence of both temperature and [CTA1] : [AIBN] molar ratio is not clear when polydispersity indices are considered. As shown in Fig. 2, PDIs remained quite low during all four polymerization processes even if a slight increase of PDI values could be noticed at high monomer conversion rate (i.e. over 80%). This normal behaviour at elevated monomer conversion rate is attributed to the higher probability of termination and irreversible transfer reactions. Additionally, the relatively elevated viscosity of the polymerization media leads to an heterogeneous growth of polymer chains.42
The optimal polymerization conditions determined using CTA1 were applied for the RAFT polymerization of NIPAM using CTA2 (see Scheme 1) to obtain a well-defined poly(NIPAM) (P2, MnNMR = 15800 g mol−1, PDI = 1.12) and with CTA3 (see Scheme 1) providing a phenylene-functionalized NIPAM (P3, MnNMR = 15200 g mol−1, PDI = 1.15). Polymers P2 and P3 (see ESI† for the figures representing the NMR spectra of P1, P2 and P3) due to their similar Mn and PDIs were then used in comparison with P1, a well-defined poly(NIPAM) synthesized from CTA1 (MnNMR = 14900 g mol−1, PDI = 1.16).
Study of thermoresponsive polymers in water
We have investigated the thermoresponsive properties of the three end-functionalized poly(NIPAM)s, where only the R group of CTA differs (Scheme 1): naphthalene group bearing 4 ethylene groups in CTA1, naphthalene group bearing 6 ethylene groups in CTA2 and a phenyl group bearing 4 ethylene groups in CTA3. All polymers were selected for their similar molecular weights and their low polydispersity indices, so that the impact of the end group could be efficiently studied near-independently of the molecular weight. The properties of such functionalized poly(NIPAM) in water solution (2 g L−1) were elucidated by measuring the cloud point by UV-vis spectroscopy at 700 nm (determined at 50% transmittance).Furthermore, we have investigated the ability34 of polymers of this type to form a 1 : 1 complex with the electron-deficient cyclophane cyclobis(paraquat-p-phenylene) (CBPQT4+). We have also investigated the influence the four counterions of the CBPQT4+ has in controlling the LCST. All turbidimetry results are summarized in Table 2 and Fig. 3; the latter represents the transmittance versus temperature curves for the polymers functionalized with three different end groups (CTA1, CTA2 and CTA3).
| Referencea | M n poly(NIPAM)b (g mol−1) | X− | LCSTc (°C) | K a, M−1 (ΔH, kcal mol−1)d |
|---|---|---|---|---|
| a P stands for poly(NIPAM). Numbers 1, 2 and 3 correspond to the CTA used during the RAFT process, CTA1, CTA2 and CTA3, respectively. Cl, Br and I correspond to the counterion of CBPQT4+, 4X−. b Determined by 1H NMR in D2O. c LCST determined by UV-vis spectroscopy at 700 nm at 50% transmittance, concentration: 2 g L−1 in water. d Determined by ITC. | ||||
| P1 | 14900 |
— | 27.5 | |
| P1–CBPQT–Cl | 4Cl− | 28.1 | 1.54 × 105 (−12.9) | |
| P1–CBPQT–Br | 4Br− | 27.8 | — | |
| P1–CBPQT–I | 4I− | 27.5 | — | |
| P1–CBPQT–PF6 | 4PF6− | 27.2 | — | |
| P2 | 15800 |
— | 27.2 | |
| P2–CBPQT–Cl | 4Cl− | 27.7 | 2.62 × 105 (−13.6) | |
| P2–CBPQT–Br | 4Br− | 27.7 | — | |
| P2–CBPQT–I | 4I− | 27.4 | — | |
| P3 | 15200 |
— | 28.8 | |
| P3–CBPQT–Cl | 4Cl− | 28.9 | 2.51 × 104 (−0.919) | |
| P3–CBPQT–Br | 4Br− | 28.7 | — | |
| P3–CBPQT–I | 4I− | 28.8 | — |
 | ||
| Fig. 3 Phase transitions of end-functionalized poly(NIPAM) synthesized via the RAFT process using CTA1 (●), CTA2 (■) and CTA3 (◆). Determined using UV-vis spectroscopy at 700 nm, 2 g L−1. Values in brackets correspond to the cloud point. | ||
Sharp transitions could be observed for all functionalized polymers, P1, P2 and P3 obtained from the RAFT process using CTA1, CTA2 and CTA3, respectively. From these measurements, it appears that P1 and P2 have a similar LCST, 27.5 °C (curve ●, Fig. 3 and Table 2) and 27.2 °C (curve ■, Fig. 3 and Table 2) respectively, which is consistent with their similar naphthalene end groups which only differ by the number of ethylene glycol units. It is noteworthy that these LCST values are lower than that of most poly(NIPAM)s described in literature,20 which is most likely due to the hydrophobic nature of the end-groups. On the other hand, when a phenylene group is present at the extremity of the polymer (P3), a slightly higher cloud point was determined at 28.8 °C (curve ◆, Fig. 3 and Table 2). This can be attributed to the more hydrophobic character of the naphthalene unit compared to the phenylene unit.
Complexes of the polymers were prepared by mixing 1 equivalent of a functionalized poly(NIPAM) (P1, P2 and P3) with one equivalent of the electron-deficient cyclophane CBPQT4+, 4X− (X = Cl, Br, I) and their LCSTs were determined as described above34 (see ESI† for the figures representing the phase transitions). When P1 was mixed with CBPQT4+, 4Cl− or CBPQT4+, 4Br−, the corresponding P1–CBPQT–Cl and P1–CBPQT–Br complexes have a cloud point of 28.1 °C and 27.8 °C (Table 2), respectively, which are slightly higher than the cloud point of naphthalene functionalized poly(NIPAM) P1 (i.e. 27.5 °C). This trend suggests that the hydrophilicity–hydrophobicity balance of the material was displaced toward a more hydrophilic structure due to the presence of the cyclophane, which to some extent masks the hydrophobicity of the naphthalene moiety. Interestingly, it appears that the cloud points of P1 and P1–CBPQT–I are identical (Table 2), showing that the “mask effect” of the CBPQT4+ unit seems to be neutralized by the four soft iodide ions. With regard to P2 and its complexed systems (Table 2), the same tendency as for P1 and P1–CBPQT–X (X = Br, Cl, I) can be observed. Indeed, P2–CBPQT–Cl displayed a slightly higher LCST at 27.7 °C than P2 (27.2 °C).
Polymer P3, bearing a phenylene end group as complexing agent (LCST of 28.8 °C, Table 2), showed similar results for LCST whatever the nature of counterions associated with the CBPQT4+ unit (cloud points between 28.7 °C and 28.9 °C). These results clearly show that the hydrophilicity–hydrophobicity balance of the phenylene functionalized polymer is not influenced significantly by the formation of the complex. It is noteworthy that all prepared aqueous solutions comprising functionalized poly(NIPAM) and the complexing agent CBPQT4+, 4X− were coloured (due to the formation of complexes) below their LCST and no colour was observed over the cloud point, thereby suggesting that complexation between the polymeric material and the tetracationic cyclophane no longer occurs.
The cyclophane CBPQT4+, 4PF6− is a non-soluble compound in aqueous media. However, thanks to the presence of the naphthalene moiety attached onto the well-defined hydrophilic poly(NIPAM) polymer backbone, we were able to solubilize the cyclophane by forming its corresponding complex (1 : 1) with P1. Indeed, adding a solution of CBPQT4+, 4PF6− in acetone to a solution of P1 in water produced a purple coloured solution characteristic of complex of this type, without any precipitation. After removal of acetone by nitrogen bubbling, a cloud point of 27.2 °C for P1–CBPQT–PF6 was determined by UV-vis spectroscopy (Table 2), which is slightly lower than the cloud point of uncomplexed P1. This can be attributed to the hydrophobic PF6− ions of the cyclophane, which despite the presence of the hydrophilic tetracationic cyclophane, led a more hydrophobic polymer material compared to P1.
To provide further insight of the nature of complexes, we have recorded 1H-NMR spectra of P3 and P1 (dialkoxyphenylene- and dialkoxynaphthalene-functionalized poly(NIPAM) systems, respectively) with various molecular ratios of CBPQT4+, 4Cl− and compared them to spectra obtained for the uncomplexed CBPQT4+, 4Cl− (Fig. 4). Addition of aliquots of either P1 or P3 to a solution of CBPQT4+, 4Cl− showed significant changes in the chemical shifts relative to those of the non-complexed macrocyclic host (Fig. 4C). For the P1–CBPQT–Cl complex, two sets of signals for cyclophane protons Hα, Hβ and –C6H4– are observed, and significant chemical shift changes for the Hβ (Δδ ≈ −0.8 ppm) and –C6H4– (Δδ ≈ +0.4 ppm) occur relative to the “free” macrocycle (Fig. 4A and B). Spectra for the P3–CBPQT–Cl complex (Fig. 4D and E) displayed significant changes in the chemical shifts of Hα, Hβ and –C6H4– upon the addition of aliquots of the polymer. Thus, NMR spectroscopy is consistent with pseudorotaxane-like complexation between the polymers and the cyclophane.43,44 The association constants determined by isothermal titration calorimetry (ITC) in water for P3–CBPQT–Cl is significantly lower (Ka = 2.51 × 104 M−1, ΔHP3 = −0.919 kcal mol−1) than the naphthalene-functionalized poly(NIPAM)s (Ka = 1.54 × 105 M−1, ΔHP1 = −12.9 kcal mol−1 for P1–CBPQT–Cl and 2.62 × 105 M−1, ΔHP2 = −13.6 kcal mol−1 for P2–CBPQT–Cl).
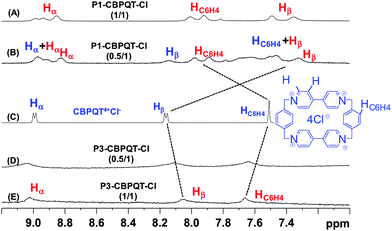 | ||
| Fig. 4 Partial 1H NMR spectra of: (A) P1-CBPQT-Cl (1 : 1 molar ratio); (B) P1-CBPQT-Cl (0.5 : 1); (C) CBPQT4+; (D) P3-CBPQT-Cl (0.5 : 1); (E) P3-CBPQT-Cl (1 : 1). Recorded in D2O at 25 °C. Protons written in blue and red correspond to non-complexed protons and complexed protons of CBPQT4+ respectively. | ||
In the last part of this study, we have taken the above mentioned complexation results into consideration to develop an interesting supramolecular system in which the more strongly binding P1 can displace P3 from the cavity of CBPQT–Cl. We have explored the addition of naphthalene functionalized P1 to an aqueous solution containing the complex P3–CBPQT–Cl. The formation of complex P3–CBPQT–Cl was beforehand confirmed by ITC (Ka = 2.51 × 104 M−1) and UV-vis measurements by the presence of a weak absorption band between 420 nm and 480 nm (black curve, Fig. 5). To a cuvette containing the complex P3–CBPQT–Cl were added aliquots of naphthalene functionalized poly(NIPAM) P1 and the yellow-brownish solution turned immediately purple, presumably resulting from complexation between P1 and CBPQT4+, 4Cl−. UV-vis measurements revealed a red shift in the charge transfer band to 520 nm. Thus UV-vis data are consistent with complex P3–CBPQT–Cl being disrupted upon the addition of the more strongly binding P1 to the mixture.
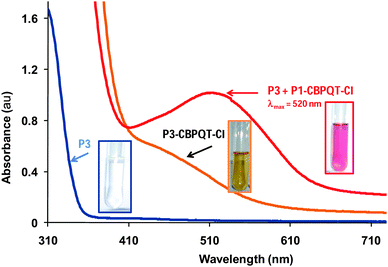 | ||
| Fig. 5 UV-vis spectra of: phenylene based poly(NIPAM) (P3, 5 × 10−4 M, blue curve); P3-CBPQT-Cl (5 × 10−4 M orange curve); and upon the addition of P1 (5 × 10−3 M) (red curve). Photographs were taken of the solutions at 15 °C. | ||
This CBPQT4+ “transfer” can also be monitored using square wave voltammetry (SWV, Fig. 6). To a solution of CBPQT4+, 4Cl− in water (containing 0.1 M NaCl, blue line, Fig. 6), were added 10 equiv. of P3 which resulted in a small negative shift (−5 mV) in the first reduction wave and in a drop of the corresponding current intensity, suggesting a decrease of diffusion coefficient upon complexation with P3 occurs (orange line, Fig. 6).45 When the SWV experiments of P3–CBPQT–Cl were recorded in the presence of P1 (purple line, Fig. 6), a significantly larger shift of −54 mV in the first reduction wave and a more marked drop in peak current was observed, presumably due to the formation of the P1–CBPQT–Cl complex and disassembly of the initial P3–CBPQT–Cl complex.
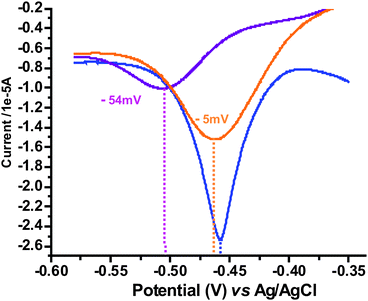 | ||
| Fig. 6 Square wave voltammograms of: CBPQT4+,Cl− (1.5 × 10−3 M) in water (containing 0.1 M NaCl) (blue line); upon the addition of P3 (1.5 × 10−2 M) (orange line); and upon the addition of P1 (purple line). Scan rate = 10 mV s−1. Working electrode: platinum. | ||
Conclusion
In this work, we have synthesized three chain transfer agents bearing electron-rich aromatic recognition motifs such as phenylene and naphthalene groups. After demonstrating the controlled character of the RAFT polymerizations, three functionalized poly(NIPAM) derivatives (P1, P2 and P3) were synthesized using the same optimal conditions to obtain similar molecular weights. We have investigated the ability of these polymers to form thermoresponsive complexes with the tetracationic cyclophane (CBPQT4+, 4X−). In particular, our study revealed that the CTA structure (naphthalene group or a phenylene group) has an influence over the LCST cloud point in water, even for relatively high molecular weights of poly(NIPAM)s (around 15000 g mol−1). Moreover, we have shown that the cyclophane CBPQT4+ has the propensity to be transferred from a well-defined phenylene-based polymer to a well-defined naphthalene-functionalized polymer.
Acknowledgements
Patrice Woisel thanks the CNRS and l'Agence Nationale de la Recherche (ANR JC-0032-01 Electrotunepoly) for funding. Graeme Cooke thanks EPSRC for funding. We thank Aurélie Malfait for GPC analysis.Notes and references
- I. M. Khan, Field Responsive Polymers: Electroresponsive, Photoresponsive, and Responsive Polymers in Chemistry and Biology, ACS Symp. Ser., 1999 Search PubMed.
- S. Minko, Responsive Polymer Material: Design and Applications, Blackwell Publishing, 2006 Search PubMed.
- R. A. Jones, M. H. Poniris and M. R. Wilson, J. Controlled Release, 2004, 96, 379–391 CrossRef CAS.
- D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS.
- E. S. Gil and S. M. Hudson, Prog. Polym. Sci., 2004, 29, 1173–1222 CrossRef CAS.
- M. Camail, H. Essaoudi, A. Margaillan and J. L. Vernet, Eur. Polym. J., 1995, 31, 1119–1125 CrossRef CAS.
- Z. M. O. Rzaev, S. Dinçer and E. Piskin, Prog. Polym. Sci., 2007, 32, 534–595 CrossRef CAS.
- R. Du and J. Zhao, J. Membr. Sci., 2004, 239, 183–188 CrossRef CAS.
- W. Lequieu, N. I. Shtanko and F. E. Du Prez, J. Membr. Sci., 2005, 256, 64–71 CAS.
- P.-W. Zhu, J. Mater. Sci.: Mater. Med., 2004, 15, 567–573 CrossRef CAS.
- Z. Tong, F. Zeng, X. Zheng and T. Sato, Macromolecules, 1999, 32, 4488–4490 CrossRef CAS.
- I. Dimitrov, B. Trzebicka, A. H. E. Muller, A. Dworak and C. B. Tsvetanov, Prog. Polym. Sci., 2007, 32, 1275–1343 CrossRef CAS.
- Y. Xia, X. Yin, N. A. D. Burke and H. D. H. Stover, Macromolecules, 2005, 38, 5937–5943 CrossRef CAS.
- Y. Xia, N. A. D. Burke and H. D. H. Stöver, Macromolecules, 2006, 39, 2275–2283 CrossRef CAS.
- P. Kujawa, C. C. Ester Goh, D. Calvet and F. M. Winnik, Macromolecules, 2001, 34, 6387–6395 CrossRef CAS.
- D. Fournier, R. Hoogenboom, H. M. L. Thijs, R. M. Paulus and U. S. Schubert, Macromolecules, 2007, 40, 915–920 CrossRef CAS.
- S. Creutz, P. Teyssié and R. Jérôme, Macromolecules, 1997, 30, 6–9 CrossRef CAS.
- R. Hoogenboom, H. M. L. Thijs, D. Wouters, S. Hoeppener and U. S. Schubert, Soft Matter, 2008, 4, 103–107 RSC.
- R. Hoogenboom, H. M. L. Thijs, M. J. H. C. Jochems, B. M. van Lankvelt, M. W. M. Fijten and U. S. Schubert, Chem. Commun., 2008, 5758–5760 RSC.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- H. Mori, H. Iwaya, A. Nagai and T. Endo, Chem. Commun., 2005, 4872–4874 RSC.
- Mei Yang, L.-Y. Chu, R. Xie and C. Wang, Macromol. Chem. Phys., 2008, 209, 204–211 CrossRef CAS.
- H. Ohashi, Y. Hiraoka and T. Yamaguchi, Macromolecules, 2006, 39, 2614–2620 CrossRef CAS.
- O. Kretschmann, S. W. Choi, M. Miyauchi, I. Tomatsu, A. Harada and H. Ritter, Angew. Chem., Int. Ed., 2006, 45, 4361–4365 CrossRef.
- S. Amajjahe and H. Ritter, Macromolecules, 2008, 41, 3250–3253 CrossRef CAS.
- P. Kujawa, F. Segui, S. Shaban, C. Diab, Y. Okada, F. Tanaka and F. M. Winnik, Macromolecules, 2006, 39, 341–348 CrossRef CAS.
- S. Jana, S. P. Rannard and A. I. Cooper, Chem. Commun., 2007, 2962–2964 RSC.
- M. Chiper, D. Fournier, R. Hoogenboom and U. S. Schubert, Macromol. Rapid Commun., 2008, 29, 1640–1647 CrossRef CAS.
- M. B. Nielsen, J. O. Jeppesen, J. Lau, C. Lomholt, D. Damgaard, J. P. Jacobsen, J. Becher and J. F. Stoddart, J. Org. Chem., 2001, 66, 3559–3563 CrossRef CAS.
- Y. Liu, A. H. Flood, R. M. Moskowitz and J. F. Stoddart, Chem.–Eur. J., 2005, 11, 369–385 CrossRef.
- M. Bria, J. Bigot, G. Cooke, J. Lyskawa, G. Rabani, V. M. Rotello and P. Woisel, Tetrahedron, 2009, 65, 400–407 CrossRef CAS.
- P. L. Anelli, P. R. Ashton, R. Ballardini, V. Balzani, M. Delgado, M. T. Gandolfi, T. T. Goodnow, A. E. Kaifer and D. Philp, J. Am. Chem. Soc., 1992, 114, 193–218 CrossRef CAS.
- P. R. Ashton, M. Gómez-López, S. Iqbal, J. A. Preece and J. F. Stoddart, Tetrahedron Lett., 1997, 38, 3635–3638 CrossRef CAS.
- J. Bigot, M. Bria, S. T. Caldwell, F. Cazaux, A. Cooper, B. Charleux, G. Cooke, B. Fitzpatrick, D. Fournier, J. Lyskawa, M. Nutley, F. Stoffelbach and P. Woisel, Chem. Commun., 2009, 5266–5268 RSC.
- J. Skey and R. K. O'Reilly, Chem. Commun., 2008, 4183–4185 RSC.
- S. R. Gondi, A. P. Vogt and B. S. Sumerlin, Macromolecules, 2007, 40, 474–481 CrossRef CAS.
- L. Barner and S. Perrier, in Handbook of RAFT Polymerization, ed. C. Barner-Kowollik, Wiley-VCH, 2008, pp. 455–482 Search PubMed.
- X.-P. Qiu, F. Tanaka and F. M. Winnik, Macromolecules, 2007, 40, 7069–7071 CrossRef CAS.
- C. Schilli, M. G. Lanzendorfer and A. H. E. Muller, Macromolecules, 2002, 35, 6819–6827 CrossRef CAS.
- T. Tang, V. Castelletto, P. Parras, I. W. Hamley, S. M. King, D. Roy, S. Perrier, R. Hoogenboom and U. S. Schubert, Macromol. Chem. Phys., 2006, 207, 1718–1726 CrossRef CAS.
- M. Moad and C. Barner-Kowollik, in Handbook of RAFT polymerization, ed. C. Barner-Kowollik, Wiley-VCH, 2008, pp. 51–104 Search PubMed.
- A. Favier, M.-T. Charreyre and C. Pichot, Polymer, 2004, 45, 8661–8674 CrossRef CAS.
- M. Bria, G. Cooke, A. Cooper, J. F. Garety, S. G. Hewage, M. Nutley, G. Rabani and P. Woisel, Tetrahedron Lett., 2007, 48, 301–304 CrossRef CAS.
- M. Asakawa, W. Dehaen, G. L'Abbe, S. Menzer, J. Nouwen, F. M. Raymo, J. F. Stoddart and D. J. Williams, J. Org. Chem., 1996, 61, 9591–9595 CrossRef CAS.
- J. Bigot, B. Charleux, G. Cooke, F. Delattre, D. Fournier, J. Lyskawa, F. Stoffelbach and P. Woisel, Macromolecules, 2010, 43, 82–90 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of the CTAs, general procedure for polymerisation, NMR spectra of polymers, % transmittance versus temperature curves. See DOI: 10.1039/c0py00085j |
| This journal is © The Royal Society of Chemistry 2010 |