DOI:
10.1039/C0PY00065E
(Paper)
Polym. Chem., 2010,
1, 1013-1023
Multicolor electrochromic poly(amide-imide)s with N,N-diphenyl-N′,N′-di-4-tert-butylphenyl-1,4-phenylenediamine moieties†
Received
25th February 2010
, Accepted 6th April 2010
First published on
10th May 2010
Abstract
A new imide ring-preformed dicarboxylic acid monomer bearing the N,N,N′,N′-tetraphenyl-1,4-phenylenediamine (TPPA) unit, i.e., N,N-bis(4-tert-butylphenyl)-N′,N′-bis(4-carboxyphthalimido)-1,4-phenylenediamine (2), was synthesized from the condensation of N,N-bis(4-aminophenyl)-N′,N′-bis(4-tert-butylphenyl)-1,4-phenylenediamine (1) and two equivalent amount of trimellitic anhydride (TMA). A new series of aromatic poly(amide-imide)s (PAIs) 4a–4g containing the electroactive TPPA moiety were prepared from the diimide-diacid 2 with various aromatic diamines by the phosphorylation polyamidation reaction. All the polymers were readily soluble in many organic solvents and could be solution-cast into tough and flexible polymer films. They displayed moderate to high glass-transition temperatures (280–320 °C) and good thermal stability. For the PAIs derived from common aromatic diamines, cyclic voltammograms of their cast films on the indium–tin oxide (ITO)-coated glass substrate exhibited two reversible oxidation redox couples around 0.68 and 1.10 V vs. Ag/AgCl in acetonitrile solution, and they revealed high redox and electrochromic stability with a color change from colorless neutral form to yellow and blue oxidized forms at applied potentials scanning from 0.0 to 1.3 V. Cyclic voltammograms of PAIs 4f and 4g obtained from diimide-diacid 2 and triphenylamine-based diamines exhibited three and four reversible oxidation redox couples, respectively. Because there are more than two redox states are electrochemically accessible from PAIs 4f and 4g, they could form tristable and even tetrastable color species and thus could display several colors upon oxidation at varying applied voltages. The electrochromic performance of a representative PAI 4c was investigated. This anodically coloring polymer film showed good electrochromic properties with high coloration efficiency (CE = 268 cm2 C−1 for the yellow coloration) and high optical contrast ratio of transmittance change (ΔT%) up to 33% at 411 nm and 32% at 922 nm for the yellow coloring, and 72% at 698 nm for the blue coloring. After over 100 cyclic switches, the polymer films still retained excellent redox and electrochromic activity.
Introduction
Electrochromic materials exhibit a change in optical absorption or transmittance upon redox switching. There are many chemical systems that are intrinsically electrochromic, such as transition metal oxides, inorganic coordination complexes, organic molecules, and conjugated polymers.1 Traditionally, interest in electrochromic materials has been directed towards optical changes in the visible region, leading to many technological applications such as variable reflectance mirrors, tunable windows, and electrochromic displays.2 Of the available electrochromic materials, conjugated conducting polymers such as poly(3,4-alkylenedioxythiophene) and poly(3,4-alkylenedioxypyrrole) based polymers and other similar systems have become widely researched electrochromic materials because of their fast switching speeds, improved processability, and color tunability through structural modification.3–5 In recent years, we have carried out extensive studies on the design and synthesis of triarylamine-based high-performance polymers such as aromatic polyamides and polyimides for electrochromic applications.6 The electrochromic function of these polymers came from the triarylamine core which can be easily oxidized and the resulting radical cation is stable enough and can undergo long-standing redox cycles.
Aromatic poly(amide-imide)s (PAIs) possess balanced characteristics between polyamides and polyimides such as high thermal stability and good mechanical properties together with ease of processability.7 Since we successfully applied the Yamazaki-Higashi phosphorylation reaction8 to the direct synthesis of high-molecular-weight PAIs from the TMA-derived imide ring-bearing dicarboxylic acids and aromatic diamines using triphenyl phosphite (TPP) and pyridine as condensing agents,9 this efficient synthetic route has proved to exhibit significant advantages in preparing operations as compared with conventional acid chloride or isocyanate methods.10 Thus, many novel PAIs have been readily prepared by this convenient technique in our and other laboratories.11 Furthermore, this synthetic procedure can offer us the option of the incorporation of specific functionalities between amide or imide groups in the PAI backbone. The incorporation of such functional groups may provide a method of controlling certain physical properties or special functions of the resulting PAIs.
The redox properties, ion-transfer process, and electrochromic behavior of N,N,N′,N′-tetraphenyl-1,4-phenylenediamine (TPPA) derivatives are important for technological application.12 The radical cations of TPPA derivatives such as N,N,N′,N′-tetra-4-methoxyphenyl-1,4-phenylenediamine showed rather strong intervalence charge-transfer (IV-CT) bands in near infrared (NIR) region as measured by UV/Vis/NIR spectroelectrochemistry.13 Recently, it has been demonstrated that aromatic polyamides containing TPPA moieties reveal interesting electrochromic characteristics, such as polyelectrochromism, high coloration efficiency, fast response time, high optical contrast, and excellent redox stability.14 Herein, we report the synthesis and characterization of a new family of TPPA-containing aromatic PAIs based on the diimide-diacid 2 which was prepared from the condensation of N,N-bis(4-aminophenyl)-N′N′bis(4-tert-butylphenyl)-1,4-phenylenediamine (1) and TMA. With such a configuration, the electrochemically active sites of the pendent phenyl groups of the TPPA unit are blocked, giving the polymers extra electrochemical stability. As a result, these PAIs were found to possess an enhanced redox and electrochromic stability. In addition, the PAIs obtained from the polymerization reactions of diimide-diacid 2 with triphenylamine-based diamine and TPPA-containing diamine exhibited a multi-colored electrochromic behavior due to the presence of three or four redox-active amino centers in the repeat unit.
Experimental part
Materials
The TPPA-containing diamine monomer 1 was synthesized by a four-step reaction sequence starting from bis(tert-butylphenyl)amine and p-fluoronitrobenzene.15 The commercially available diamine monomers including p-phenylenediamine (3a), m-phenylenediamine (3b), 4,4′-diaminodophenyl ether (3c), and 9,9-bis(4-aminophenyl)fluorene (3e) were used as received from Tokyo Chemical Industry. According to a procedure reported previously,16 2-triflouromethyl-4,4′-diaminodiphenyl ether (3d) (mp: 112–113 °C) was synthesized by the chloro-displacement reaction of 2-chloro-5-nitrobenzotrifluoride with p-nitrophenol in the presence of potassium carbonate, followed by the hydrazine palladium-catalyzed reduction of the intermediate dinitro compound. 4,4′-Diamino-4′′-tert-butyltriphenylamine (3f) (mp: 113–115 °C)6c was synthesized by the caesium fluoride-mediated condensation of 4-tert-butylaniline with p-fluoronitrobenzene, followed by a palladium-catalyzed hydrazine reduction. N-Methyl-2-pyrrolidone (NMP) was dried over calcium hydride for 24 h, distilled under reduced pressure, and stored over 4 Å molecular sieves in a sealed bottle. Commercially obtained calcium chloride (CaCl2) was dried under vacuum at 180 °C for 3 h prior to use. Tetrabutylammonium perchlorate, Bu4NClO4, was recrystallized from ethyl acetate under nitrogen atmosphere and then dried in vacuo prior to use. All other reagents and solvents were used as received from commercial sources.
Monomer synthesis
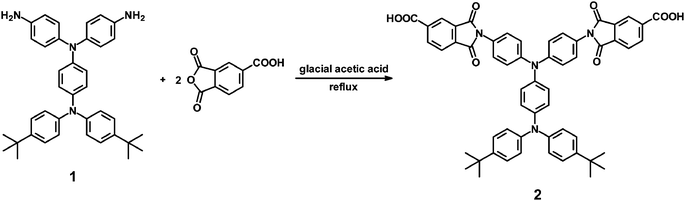
N,N-Bis(4-tert-butylphenyl)-N′,N′-bis(4-carboxyphthalimido)-1,4-phenylenediamine (2).
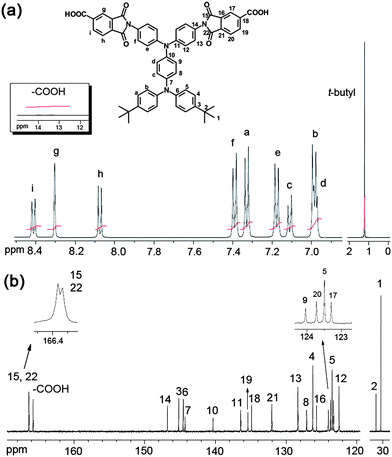
A flask was charged with 5.4 g (10 mmol) of diamine 1, 4.0 g (20 mmol) of trimellitic anhydride, and 50 mL of glacial acetic acid. The heterogeneous mixture was refluxed for 12 h. After cooling, the mixture was poured into 300 mL of methanol, and the precipitate was collected by filtration and washed thoroughly with methanol. The crude product obtained was purified by recrystallization from N,N-dimethylformamide (DMF) and then dried in vacuum to afford 7.2 g (80% yield) of diimide-dicarboxylic acid monomer 2 as pale brown solid, mp = 321–323 °C. IR (KBr): 2700–3400 (O–H str.), 2964 (t-butyl C–H str.), 1778, 1724 cm−1 (imide C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O str.). 1H NMR (500 MHz, DMSO-d6, δ, ppm) (for the peak assignments, see Fig. 1a): 1.27 (s, 18H, t-butyl), 6.98 (d, J = 8.9 Hz 2H, Hd), 6.99 (d, J = 8.6 Hz, 4H, Hb), 7.11 (d, J = 8.8 Hz, 2H, Hc), 7.18 (d, J = 8.8 Hz, 4H, He), 7.33 (d, J = 8.6 Hz, 4H, Ha), 7.39 (d, J = 8.8 Hz, 4H, Hf), 8.07 (d, J = 7.8 Hz, 2H, Hh), 8.30 (s, 2H, Hg), 8.41 (d, J = 7.8 Hz, 2H, Hi). 13C NMR (125 Hz, DMSO-d6, δ, ppm) (for the peak assignments, see Fig. 1b): 31.2 (C1), 34.0 (C2), 122.5 (C12), 123.3 (C17), 123.5 (C5), 123.7 (C20), 124.1 (C9), 125.7 (C16), 126.2 (C4), 127.1 (C8), 128.3 (C13), 132.0 (C21), 134.9 (C18), 135.4 (C19), 136.4 (C11), 140.4 (C10), 144.3 (C7), 144.6 (C6), 145.2 (C3), 146.8 (C14), 165.8 (–COOH), 166.37, 166.38 (imide carbonyl carbons). Anal. Calcd for C56H46N4O8 (903.00): C, 74.49%; H, 5.13%; N, 6.20%. Found: C, 74.15%; H, 5.06%; N, 6.15%.
O str.). 1H NMR (500 MHz, DMSO-d6, δ, ppm) (for the peak assignments, see Fig. 1a): 1.27 (s, 18H, t-butyl), 6.98 (d, J = 8.9 Hz 2H, Hd), 6.99 (d, J = 8.6 Hz, 4H, Hb), 7.11 (d, J = 8.8 Hz, 2H, Hc), 7.18 (d, J = 8.8 Hz, 4H, He), 7.33 (d, J = 8.6 Hz, 4H, Ha), 7.39 (d, J = 8.8 Hz, 4H, Hf), 8.07 (d, J = 7.8 Hz, 2H, Hh), 8.30 (s, 2H, Hg), 8.41 (d, J = 7.8 Hz, 2H, Hi). 13C NMR (125 Hz, DMSO-d6, δ, ppm) (for the peak assignments, see Fig. 1b): 31.2 (C1), 34.0 (C2), 122.5 (C12), 123.3 (C17), 123.5 (C5), 123.7 (C20), 124.1 (C9), 125.7 (C16), 126.2 (C4), 127.1 (C8), 128.3 (C13), 132.0 (C21), 134.9 (C18), 135.4 (C19), 136.4 (C11), 140.4 (C10), 144.3 (C7), 144.6 (C6), 145.2 (C3), 146.8 (C14), 165.8 (–COOH), 166.37, 166.38 (imide carbonyl carbons). Anal. Calcd for C56H46N4O8 (903.00): C, 74.49%; H, 5.13%; N, 6.20%. Found: C, 74.15%; H, 5.06%; N, 6.15%.
 |
| | Fig. 1 (a) 1H-NMR and (b) 13C-NMR spectra of diimide-diacid 2 in DMSO-d6. | |
The synthesis of PAI 4a is used as an example to illustrate the general synthetic route. A mixture of 0.6321 g (0.7 mmol) of the diimide-dicarboxylic acid 2, 0.0757 g (0.7 mmol) of p-phenylenediamine (3a), 0.15 g of calcium chloride, 1.0 mL of triphenyl phosphite, 0.3 mL of pyridine, and 1.7 mL of NMP was heated with stirring at 120 °C for 3 h. The resulting viscous solution was poured slowly with stirring into 150 mL of methanol, giving rise to a tough, fibrous precipitate. The precipitated product was collected by filtration, washed repeatedly with methanol and hot water, and dried to give a quantitative yield of PAI 4a. The inherent viscosity of the polymer was 0.61 dL/g, measured in DMAc (containing 5 wt% LiCl) at a concentration of 0.5 g/dL at 30 °C. The IR spectrum of 4a (film) exhibited characteristic amide absorption bands at 3200–3400 cm−1 (amide N–H str.) and 1675 cm−1 (amide carbonyl str.), together with the imide absorption bands at 1778 cm−1 (asymmetrical C O str.), 1724 cm−1 (symmetrical C O str.), and 725 cm−1 (imide ring deformation).
Preparation of the PAI films
A solution of polymer was made by dissolving about 0.6 g of the PAI sample in 10 mL of DMAc. The homogeneous solution was poured into a 9 cm glass Petri dish, which was placed in a 90 °C oven overnight to remove most of the solvent. The cast film was then released from the glass substrate and was further dried in vacuo at 160 °C for 8 h. The obtained films were about 50–60 μm in thickness and were used for X-ray diffraction measurements, solubility tests and thermal analyses.
Measurements
Infrared (IR) spectra were recorded on a Horiba FT-720 FT-IR spectrometer. Elemental analyses were run in a Heraeus VarioEL III CHNS elemental analyzer. 1H- and 13C-NMR spectra were measured on a Bruker AVANCE 500 FT-NMR system with tetramethylsilane as an internal standard. The inherent viscosities were determined with a Cannon-Fenske viscometer at 30 °C. Wide-angle X-ray diffraction (WAXD) measurements were performed at room temperature (ca. 25 °C) on a Shimadzu XRD-6000 X-ray diffractometer with a graphite monochromator (operating at 40 kV and 30 mA), using nickel-filtered Cu-Kα radiation (λ = 1.5418 Å). The scanning rate was 2° min−1 over a range of 2θ = 10–40°. Thermogravimetric analysis (TGA) was performed with a Perkin-Elmer Pyris 1 TGA. Experiments were carried out on approximately 4–6 mg of samples heated in flowing nitrogen or air (flow rate = 40 cm3 min−1) at a heating rate of 20 °C min−1. DSC analyses were performed on a Perkin-Elmer Pyris 1 DSC at a scan rate of 20 °C min−1 in flowing nitrogen. Thermomechanical analysis (TMA) was determined with a Perkin-Elmer TMA 7 instrument. The TMA experiments were carried out from 50 to 400 °C at a scan rate of 10 °C min−1 with a penetration probe 1.0 mm in diameter under an applied constant load of 10 mN. Softening temperatures (Ts) were taken as the onset temperatures of probe displacement on the TMA traces. Ultraviolet-visible (UV-Vis) spectra of the polymer films were recorded on an Agilent 8453 UV-Visible spectrometer. Electrochemistry was performed with a CHI 611C electrochemical analyzer. Voltammograms are presented with the positive potential pointing to the left and with increasing anodic currents pointing downwards. Cyclic voltammetry was conducted with the use of a three-electrode cell in which ITO (polymer films area about 0.8 cm × 1.25 cm) was used as a working electrode. A platinum wire was used as an auxiliary electrode. All cell potentials were taken with the use of a home-made Ag/AgCl, KCl (sat.) reference electrode. Ferrocene was used as an external reference for calibration (+0.48 V vs. Ag/AgCl). Spectroelectrochemistry analyses were carried out with an electrolytic cell, which was composed of a 1 cm cuvette, ITO as a working electrode, a platinum wire as an auxiliary electrode, and a Ag/AgCl reference electrode. Absorption spectra in the spectroelectrochemical experiments were measured with an Agilent 8453 UV-Visible spectrophotometer. Coloration efficiency is derived from the equation: η = ΔOD/Q,17 ΔOD is optical density change at specific absorption wavelength and Q is ejected charge determined from the in situ experiments. Photoluminescence (PL) spectra were measured with a Varian Cary Eclipse fluorescence spectrophotometer. Fluorescence quantum yields (ΦF) of the samples in NMP were measured by using quinine sulfate in 1 N H2SO4 as a reference standard (ΦF = 54.6%).18 All corrected fluorescence excitation spectra were found to be equivalent to their respective absorption spectra.
Results and discussion
Monomer synthesis
The TPPA-containing diimide-dicarboxylic acid monomer, N,N-bis(4-tert-butylphenyl)-N′,N′-bis(4-carboxyphthalimido)-1,4-phenylenediamine (2), was obtained by reacting diamine 1 with 2 mol equiv of trimellitic anhydride (TMA) in refluxing glacial acetic acid (Scheme 1). IR, 1H-NMR, and 13C-NMR spectroscopic techniques were used to identify the structure of the targeted diimide-dicarboxylic acid 2. The IR spectrum of 2 (Fig. S1, ESI†) showed absorption bands around 2700–3400 (–OH, carboxylic acid), 1778 (imide CO asymmetrical stretching), and 1724 cm−1 (imide CO symmetrical stretching and acid CO stretching), confirming the presence of imide ring and carboxylic acid groups in the structure. The 1H- and 13C-NMR spectra of 2 are illustrated in Fig. 1 with the assignments of all peaks. The assignments of all the resonance signals were assisted by two-dimensional COSY and HMQC NMR spectra shown in Fig. S2, ESI.† The resonance signals appearing at downfield regions (8.41–8.07 ppm) in the 1H-NMR spectrum are ascribed to the trimellitimido protons. The resonance peaks in the region of 6.9–7.5 ppm are assigned to the phenylene protons of the TPPA segment. The signals appeared at 1.27 ppm in the 1H-NMR spectrum and 34.0 ppm (a quaternary carbon) and 31.2 ppm (a methyl carbon) in the 13C-NMR spectrum are peculiar to the tert-butyl groups. These results suggest the successful preparation of the imide ring-preformed dicarboxylic acid monomer 2.
 |
| | Scheme 1 Synthesis of diimide-dicarboxylic acid 2. | |
Synthesis and basic characterization of PAIs
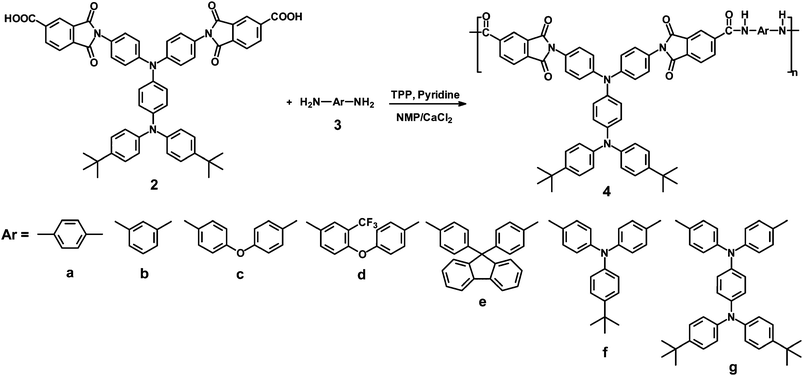
A series of seven PAIs 4a–4g containing tert-butyl-blocked TPPA units were prepared from the diimide-dicarboxylic acid 2 and various aromatic diamines (3a–3g) by the direct polycondensation reaction using triphenyl phosphite (TPP) and pyridine as condensing agents (Scheme 2).8 The polymerization proceeded homogeneously throughout the reaction and afforded highly viscous polymer solutions. The products precipitated in a tough fiber-like form when pouring slowly the resultant polymer solutions into stirred methanol. The obtained PAIs had inherent viscosities in the range of 0.33–0.78 dL g−1. The molecular weights of all the PAIs are sufficiently high to permit the formation of flexible and tough films by casting from their DMAc solutions. Structural features of these PAIs were confirmed by IR and 1H-NMR spectroscopy. A typical IR spectrum for the representative PAI 4a is also included in Fig. S1.† The characteristic absorption bands for imide ring appear around 1778 cm−1, 1724 cm−1 (imide C O), and 725 cm−1 (imide ring deformation), while bands of amide groups appear around 3300 and 1675 cm−1. A typical 1H-NMR spectrum of PAI 4c is shown in ESI Fig. S3.† Assignments of each proton are also given in the figure, and the spectrum is in good agreement with the proposed polymer structure.
 |
| | Scheme 2 Synthesis of PAIs 4a–4g. | |
As indicated by the wide-angle X-ray diffraction (WAXD) patterns shown in Fig. S4 (ESI),† all the polymers were essentially amorphous. Qualitative solubility was determined at 1% w/v concentration, stirring for 24 h at room temperature and heating up to the solvent boiling point for those samples which remained insoluble at room temperature. As shown in Table 1, all the polymers were highly soluble in polar solvents such as NMP, DMAc and N,N-dimethylformamide (DMF). As compared to the corresponding 4′ series PAIs, the present series PAIs exhibited an enhanced solubility because of the increased flexibility or free volume caused by the introduction of the three-dimensional TPPA moiety and bulky pendent tert-butyl substituents into the repeat unit. Thus, the excellent solubility makes these polymers potential candidates for practical application by spin-coating or inkjet-printing processes.
Table 1 Inherent viscosity and solubility of PAIs
| Polymer code |
η
inh
/dL g−1 |
Solubility in various solventsb |
| NMP |
DMAc |
DMF |
DMSO |
m-Cresol |
THF |
|
Measured at a polymer concentration of 0.5 g dL−1 in DMAc–5 wt% LiCl at 30 °C.
The qualitative solubility was tested with 10 mg of a sample in 1 mL of stirred solvent. ++, soluble at room temperature; +, soluble on heating; ±, partially soluble; —, insoluble even on heating.
Values in parentheses are data of analogous polyamides 4′ having the corresponding diacid residue as in the 4 series.
|
|
4a
|
0.61 |
++ (±)c |
++ (—) |
+ (—) |
+ (—) |
+ (—) |
+ (—) |
|
4b
|
0.47 |
++ (++) |
++ (++) |
++ (++) |
± (+) |
++ (+) |
++ (—) |
|
4c
|
0.39 |
++ (++) |
++ (++) |
++ (++) |
++ (+) |
++ (+) |
± (±) |
|
4d
|
0.70 |
++ (++) |
++ (++) |
++ (++) |
± (++) |
++ (+) |
± (±) |
|
4e
|
0.34 |
++ (++) |
++ (++) |
++ (++) |
++ (++) |
++ (+) |
++ (—) |
|
4f
|
0.78 |
++ (++) |
++ (++) |
++ (++) |
± (±) |
++ (+) |
++ (—) |
|
4g
|
0.33 |
++ |
++ |
++ |
++ |
++ |
± |

|
Thermal properties
The thermal properties of the PAIs were investigated by TGA, DSC and TMA, and the results are summarized in Table 2. Typical TGA curves of a representative PAI 4a in both air and nitrogen atmospheres are illustrated in the inset of Fig. 2. TGA measurements reveal that they possess high thermal stability with decomposition temperature above 400 °C. Their 10% weight-loss temperatures in nitrogen and air were recorded in the range of 470–535 and 493–553 °C, respectively. The amount of carbonized residue (char yield) of these polymers in nitrogen atmosphere is more than 62% at 800 °C. The high char yields of these polymers can be ascribed to their high aromatic content. The glass-transition temperatures (Tgs) of all the polymers were observed in the range of 280–320 °C by DSC. All the polymers indicated no clear melting endotherms up to the decomposition temperatures on the DSC thermograms. This result also supports the amorphous nature of these PAIs. Comparing the thermal properties data, one will find that PAI 4e showed the highest Tg due to the attachment of bulky lateral fluorene groups thus restricting the segmental mobility. The softening temperatures (Ts) of the polymer film samples were determined by the TMA method with a loaded penetration probe. They are obtained from the onset temperature of the probe displacement on the TMA trace. A representative TMA thermogram for PAI 4a is illustrated in Fig. 2. In all cases, the Ts values obtained by TMA are lower (by 15–34 °C) than the Tg values measured by the DSC experiments (Table 2). This may indicate that these PAIs exhibited a higher degree of plasticity near Tg because of the increased free volume caused by the bulky TPPA and tert-butyl groups.
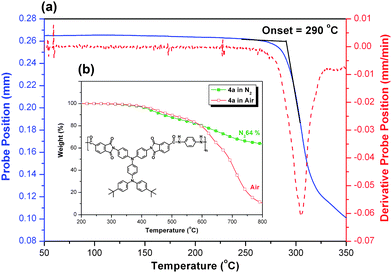 |
| | Fig. 2 (a) TMA and (b) TGA curves of PAI 4a with a heating rate of 10 and 20 °C min−1, respectively. | |
Table 2 Thermal properties of PAIs
| Polymer code |
T
g/°Ca |
T
s/°Cb |
T
d at 5% weight loss/°Cc |
T
d at 10% weight loss/°Cc |
Char yield (wt %)d |
| In N2 |
In air |
In N2 |
In air |
|
Midpoint temperature of the baseline shift on the second DSC heating trace (rate = 20 °C min−1) of the sample after quenching from 400 to 50 °C (rate = −200 °C min−1) in nitrogen.
Softening temperature measured by TMA with a constant applied load of 10 mN at a heating rate of 10 °C min−1.
Decomposition temperature at which a 5% or 10% weight loss was recorded by TGA at a heating rate of 20 °C min−1 and a gas flow rate of 20 cm3 min−1.
Residual weight percentage at 800 °C in nitrogen.
|
|
4a
|
314 |
290 |
423 |
434 |
470 |
493 |
64 |
|
4b
|
299 |
272 |
453 |
455 |
512 |
515 |
65 |
|
4c
|
280 |
260 |
447 |
451 |
495 |
515 |
66 |
|
4d
|
284 |
250 |
449 |
451 |
523 |
498 |
64 |
|
4e
|
320 |
289 |
486 |
481 |
535 |
540 |
68 |
|
4f
|
293 |
278 |
474 |
463 |
533 |
521 |
69 |
|
4g
|
282 |
255 |
474 |
516 |
532 |
553 |
70 |
Optical and electrochemical properties
The optical properties of the PAIs are investigated by UV-vis absorption and photoluminescence (PL) spectroscopy. The results are summarized in Table 3. These polymers exhibit strong UV-vis absorption bands at 306–313 nm in NMP solution, which can be attributed to the combinations of n-π* and π–π* transition originating from the conjugated TPPA moieties. In solid state, these PAIs showed UV-vis absorption characteristics (λmax around 306–317 nm) similar to those in the solution. These PAIs exhibited fluorescence emission maxima around 370–385 nm in NMP solution with extremely low fluorescence quantum yield ranging from 0.23% for 4d to 0.47% for 4e. The low fluorescence efficiency can be attributed to intramolecular charge transfer quenching effect between the TPPA donor and the imide acceptor.
Table 3 Optical and electrochemical properties of PAIs
| Polymer code |
In solutiona |
As film |
Oxidation potentialc/V |
E
g
/eV |
HOMOe/eV |
LUMOf/eV |
|
λ
max
abs/nm |
λ
max
PL/nm |
ΦFb(%) |
λ
max
abs/nm |
λ
onset
abs/nm |
E
onset
|
E
1/2
ox1
|
E
1/2
ox2
|
E
1/2
ox3
|
E
1/2
ox4
|
E
onset
|
E
1/2
ox1
|
E
onset
|
E
1/2
ox1
|
|
Measured in dilute solutions in NMP at a concentration of about 1 × 10−5 mol L−1.
Fluorescence quantum yield calculated in an integrating sphere with quinine sulfate as the standard (ΦF = 54.6%).
Oxidation potentials from cyclic votammograms (vs. Ag/AgCl in CH3CN).
Energy gap = 1240/Abs λonset of the polymer film.
The HOMO energy levels were calculated from E1/2ox1 or Eonset, referenced to ferrocene (4.8 eV).
LUMO = HOMO − Eg.
|
|
4a
|
312 |
369 |
0.53 |
306 |
417 |
0.44 |
0.68 |
1.07 |
— |
— |
2.97 |
4.80 |
5.04 |
1.83 |
2.07 |
|
4b
|
313 |
373 |
0.47 |
307 |
405 |
0.46 |
0.69 |
1.06 |
— |
— |
3.06 |
4.82 |
5.05 |
1.76 |
1.99 |
|
4c
|
306 |
376 |
0.45 |
306 |
401 |
0.42 |
0.69 |
1.04 |
— |
— |
3.09 |
4.78 |
5.05 |
1.69 |
1.96 |
|
4d
|
311 |
385 |
0.23 |
307 |
401 |
0.47 |
0.68 |
1.07 |
— |
— |
3.09 |
4.83 |
5.04 |
1.74 |
1.95 |
|
4e
|
311 |
371 |
0.47 |
311 |
400 |
0.45 |
0.69 |
1.10 |
— |
— |
3.10 |
4.81 |
5.05 |
1.71 |
1.95 |
|
4f
|
310 |
370 |
0.37 |
317 |
428 |
0.47 |
0.69 |
0.89 |
1.05 |
— |
2.89 |
4.83 |
5.05 |
1.94 |
2.16 |
|
4g
|
313 |
376 |
0.26 |
316 |
460 |
0.42 |
0.63 |
0.69 |
0.95 |
1.04 |
2.70 |
4.78 |
4.99 |
2.08 |
2.29 |
|
4′c
|
304 |
368 |
0.22 |
301 |
400 |
0.82 |
1.00 |
— |
— |
— |
2.97 |
4.80 |
5.04 |
2.08 |
2.26 |
|
4″c
|
310 |
435 |
0.97 |
306 |
417 |
0.44 |
0.68 |
1.07 |
— |
— |
3.06 |
4.82 |
5.05 |
1.87 |
2.00 |
|
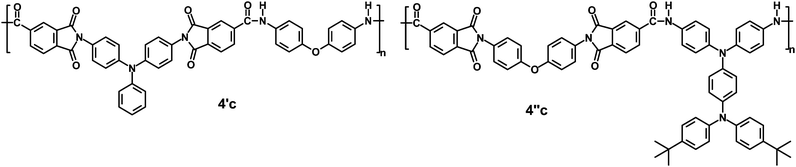
The electrochemical behavior of the PAIs was investigated by cyclic voltammetry (CV) conducted for the cast film on an ITO-coated glass substrate as working electrode in dry acetonitrile (CH3CN) containing 0.1 M of Bu4NClO4 as an electrolyte under nitrogen atmosphere. The derived oxidation potentials are summarized in Table 3. Fig. 3 shows the CV curves for PAIs 4c and 4′c. There are two reversible oxidation redox couples with half-wave potentials (E1/2) of 0.69 V and 1.04 V for PAI 4c and one reversible oxidation redox couple with E1/2 of 1.00 V for PAI 4′c in the oxidative scan. The two oxidation waves of 4c are separated by about 350 mV, emphasizing the strong electronic coupling between the amino groups in the TPPA moiety through the p-phenylene linker. Because of the stability of the films and good adhesion between the polymer and ITO substrate, the 4c exhibited excellent redox stability. PAI 4c preserved excellent elecroactivity after 50 cycles between 0.0 and 1.3 V. Upon oxidation, the polymer film of 4c exhibited a multi-electrochromic behavior; a color change from colorless to yellow at 0.8 V and then to blue at 1.2 V. For PAI 4c, the two oxidations observed correspond to successive one electron removal from the TPPA functionality of 4c to yield, first, a stable, delocalized TPPA radical cation and, second, a quinonediimine-type dication. The first oxidation at E1/2 = 0.69 V for PAI 4c could be assigned to electron loss from the nitrogen atom in the pendent 4,4′-di-tert-butyldiphenylamine unit, which is more electron-rich than the nitrogen atom on the main-chain amino group. The highly stable redox stability of this polymer can be attributed to the presence of tert-butyl group on the active sites of the TPPA moiety. It is well known that triarylamines with substituents in their para-position generally show reversible one-electron oxidation behavior.19
 |
| | Fig. 3 Cyclic voltammograms of (a) ferrocene and the cast films of (b) PAI 4c and (c) PAI 4′c on the ITO-coated glass substrate in 0.1 M Bu4NClO4/CH3CN at a scan rate of 100 mV s−1. | |
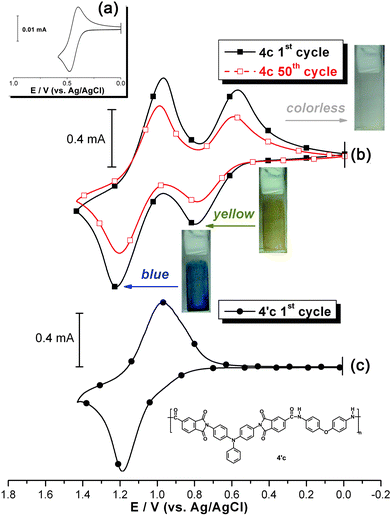
The CV curve of PAI 4f is illustrated in Fig. 4, where those of PAIs 4c and 4′c are also included for comparison. PAI 4f exhibits three redox amino centers in each repeat unit and shows three corresponding quasireversible anodic redox couples. The second oxidation at E1/2 = 0.89 V appears to involve one electron loss from the triphenylamine unit between the amide linkages. The first (E1/2 = 0.69 V) and third (E1/2 = 1.05 V) oxidation waves are related to the electron losses from the electroactive TPPA unit of 4f. These potentials are similar to those associated with monocation and dication formations from the TPPA unit in PAI 4c. Therefore, a possible anodic oxidation pathway of PAI 4f is proposed as shown in Scheme S1 (ESI).† The electrochemical stability of PAI 4f could also be ascribed to the fact that all the electrochemically active sites of the arylamino unit are blocked with tert-butyl groups.
 |
| | Fig. 4 Cyclic voltammograms of (a) ferrocene and (b) the cast films of PAIs 4′c, 4c and 4f on the ITO-coated glass substrate in 0.1 M Bu4NClO4/CH3CN at a scan rate of 50 mV s−1. | |
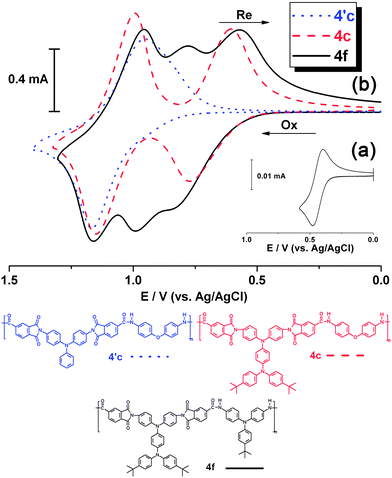
Fig. 5 compares the cyclic voltammograms and differential pulse voltammograms of PAI 4g and structurally related PAIs 4c and 4′′c. PAIs 4c and 4′′c, which have isomeric repeating units with the TPPA unit in different segments, each revealed two well-resolved, reversible redox couples in their CV diagrams, corresponding to two sequential electron removals from the electroactive TPPA unit in their polymer backbones. The onset potentials of the oxidative processes of PAI 4′′c are lower than those of PAI 4c. This can be rationalized because the TPPA segment is incorporated in the amide portion in the former case. Therefore, the amino units became more easily oxidized. PAI 4g also shows two apparent redox couples. However, the peak is broader than that of PAIs 4c and 4′′c due to close overlapping of the redox waves. From the differential pulse voltammograms of 4g, we can find three separate oxidation peaks at 0.65, 1.0, and 1.2 V, together with a shoulder around 0.75 V. Therefore, we propose a possible four-step reaction sequence for the electrochemical oxidation of PAI 4g as shown in Scheme S2 (ESI).† The E1/2 values of these four oxidations are estimated to be 0.63, 0.69, 0.95, and 1,04 V, respectively. However, the first two oxidations occurred almost simultaneously by a two-electron loss event. Therefore, the two oxidation waves merged and became indistinguishable in the CV curve. Similar two-electron oxidation event also occurred in the latter two oxidation processes.
 |
| | Fig. 5 (a) Cyclic voltammograms and (b) differential pulse voltammograms of the cast films of PAIs 4c, 4′′c and 4g on the ITO-coated glass substrate in 0.1 M Bu4NClO4/CH3CN solution. Scan rate, 50 mV s−1; pulse amplitude, 50 mV; pulse width, 50 ms; pulse period, 0.2 s. | |
The HOMO (highest occupied molecular orbital) energy levels of the investigated PAIs were calculated from the oxidation onset potentials (Eonset) or half-wave potentials of the first oxidation wave (E1/2ox1) and by comparison with ferrocene (4.8 eV).20 These data together with absorption spectra were then used to obtain the LUMO (lowest unoccupied molecular orbital) energy levels (Table 3). According to the HOMO and LUMO energy levels obtained, the PAIs in this study appear to be appropriate as hole injection and transport materials.
Spectroelectrochemical and electrochromic properties
The electro-optical properties of the polymer films were investigated using the changes in electronic absorption spectra under a voltage pulse. The electrode preparations and solution conditions were identical to those used in the CV experiments. In the following study, we first examined the spectral change of PAI 4c film (Fig. 6) during electrochemical oxidation. In the neutral state, PAI 4c exhibited strong absorption at wavelength around 313 nm, characteristic for triarylamine π–π* transitions, but it was almost transparent in the visible and near infrared (NIR) regions. When the applied potential was gradually raised from 0.8 to 1.0 V, the absorption intensity at 313 nm slightly dropped and a new shoulder band appeared at 411 nm accompanied with a broad band having its maximum around 922 nm in the NIR region. We attribute this spectral change to the formation of a stable monocation radical from the TPPA moiety. The absorption band in the NIR region is assigned to an intervalence charge-transfer (IV-CT) between states in which the positive charge is centered at different amino centers.13,21 The intensity of the IV-CT band gradually decreased when the TPPA radical cation was further oxidized to the dication (at electrode potential of 1.3 V), with a formation of a new strong absorption band centered at about 698 nm. The observed absorption changes of the film of PAI 4c are fully reversible and are associated with strong color changes. From the photos shown in Fig. 6, it can be seen that the film switches from a transmissive neutral state (nearly colorless) to a highly absorbing semioxidized state (yellow) and a fully oxidized state (blue).
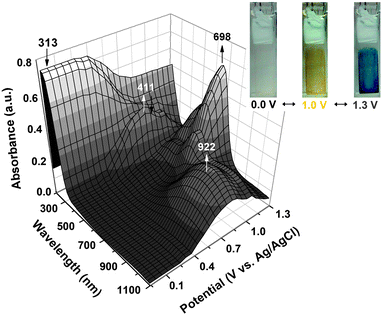 |
| | Fig. 6 The 3-D spectroelectrochemical behavior of a PAI 4c thin film on the ITO-coated glass substrate (in CH3CN containing 0.1 M Bu4NClO4 as the supporting electrolyte) between 0 and 1.3 V. The photo shows the color change of the film on an ITO electrode at indicated potentials. | |
Next, PAIs 4f and 4g were examined. Their spectral changes upon oxidation are illustrated in Fig. 7 and 8, respectively. In the neutral state, these two PAIs revealed an absorption tail in the region of 400–550 nm in addition to the strong π–π* transition bands at 313 nm. Therefore, in the neutral state their films exhibited a pale yellow color, not completely colorless. The absorption tail may be accounted for a stronger interchain charge-transfer interaction between the arylamino donor (between the amide groups) and the trimellitimido acceptor. Similar to that observed for PAI 4c, as the applied potential was raised to 0.8 V PAI 4f showed a decrease in absorption at 313 nm with a concomitant appearance of a peak at 411 nm and a broad IV-CT band centered around 922 nm. This spectral change corresponds to the first oxidation process of 4f as shown in Scheme S1.† When the electrode potential was adjusted to 1.0 V, another absorption band appeared at 792 nm and the strong IV-CT band at the NIR region still could be observed. This clearly provides evidence that the second oxidation originates from the triphenylamine core between the amide groups in the 4f main chain. We believe that the polymer has been oxidized to one with the bis(radical cation) repeating unit as the third structure shown in Scheme S1.† When the applied potential was set at 1.3 V, a strong absorption band in the 450–900 nm region centering at around 685 nm was observed, whereas the intensity of the absorption peak at 411 nm and the IV-CT band gradually decreased. The almost disappearance of the IV-CT absorption indicates the full oxidation of all the three amino centers in the repeat unit of 4f. As can be seen from the inset in Fig. 7, the film of PAI 4f displayed multi-electrochromic behavior with coloration change from pale yellow to yellowish green, green, and blue along with increasing of the applied potential.
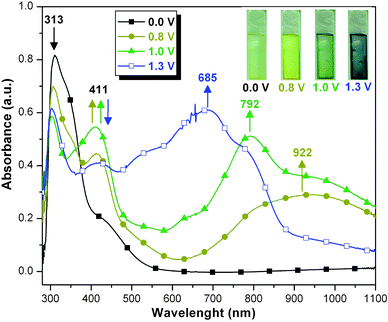 |
| | Fig. 7 The spectral change and electrochromic behavior of PAI 4f thin film on the ITO-coated glass slide in 0.1 M Bu4NClO4/CH3CN at various applied potentials (vs. Ag/AgCl). | |
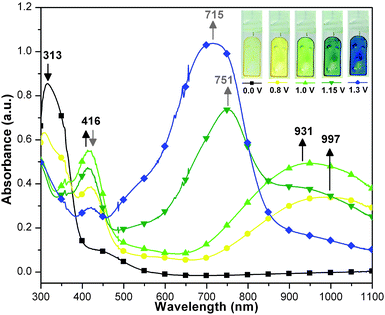 |
| | Fig. 8 The spectral change and electrochromic behavior of PAI 4g thin film on the ITO-coated glass slide (in CH3CN containing 0.1 M Bu4NClO4 as the supporting electrolyte) at various electrode potentials (vs. Ag/AgCl). | |
A similar spectral change was observed for PAI 4g at early stage of oxidation (Fig. 8). The absorption peak at 416 nm and the IV-CT band at 997 nm started to appear when the applied potential was set at 0.8 V and reached the maximum when the applied potential was raised to 1.0 V. This result illustrated that the first two oxidations of the TPPA units occurred almost simultaneously. Although the oxidation waves could not be well-resolved in the CV analysis for the third and fourth oxidations, the stepwise oxidation processes were clearly evidenced by the spectroelectrochemistry experiments. Further increase of the applied potential over 1.0 V led to the increase of the absorption intensity between 600 and 850 nm. This absorption change indicated the occurrence of the third oxidation. Upon further oxidation, the absorption band at 416 nm reached the minimum intensity and the band at 500–800 nm further intensified. It was also found that the IV-CT band at the NIR region diminished gradually, implying the occurrence of the fourth oxidation. When the polymer film of 4g was oxidized, its color changed from pale yellow to deeper yellow, yellowish green, greenish blue, and blue, as can be seen from the inset of Fig. 8. The results supported the proposed mechanisms in the oxidative processes of PAI 4g (Scheme S2, ESI†).
For electrochromic switching studies, the polymer film of 4c was cast onto an ITO-coated glass slide in the same manner as described earlier, and the film was potential stepped between its neutral (0 V) and oxidized states (+ 1.0 or 1.3 V). While the potential was switched, the absorbance at 411, 681 and 922 nm was monitored as a function of time with UV-vis-NIR spectroscopy. Fig. 9 and 10 depict the stability and the switching behavior of the polymer for the oxidative process. The switching time was calculated at 90% of the full switch because it is difficult to perceive any further color change with naked eye beyond this point. As illustrated in ESI Fig. S5,† thin film from PAI 4c revealed switching times of 5.1 s for λmax = 922 nm and 2.5 s for bleaching, reflecting the different reaction rates between the neutral and oxidized forms of the film of 4c. When the potential was switched between 0 and 1.3 V, thin film of PAI 4c would require almost 3.7 s for coloration at 681 nm and 1.6 s for bleaching. After over 100 cyclic scans between 0.0 and 1.0 V, the polymer films still exhibited good electrochemical and electrochromic stability. Coloration efficiency (CE; η) was measured by monitoring the amount of ejected charge (Q) as a function of the change in optical density (ΔOD) of the polymer film.17 The electrochromic coloration efficiencies (η = ΔOD/Q) of the polymer films of 4c after various switching cycles are summarized in Table 4. The CE of PAI 4c is high, ranging from 268 cm2 C−1 for the first cycle to 255 cm2 C−1 for the 100th cycle. After switching 100 times between 0 V and 1.0 V, the film of PAI 4c only showed 4.9% decay on CE. Therefore, all these results revealed that the present PAIs exhibit high redox stability and good electrochromic performance.
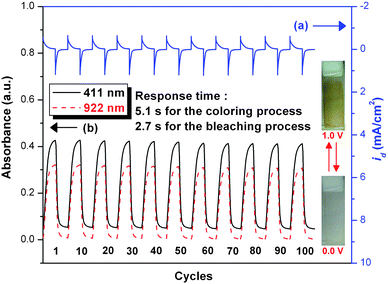 |
| | Fig. 9 (a) Current consumption and (b) electrochromic switching between 0 and 1.0 V and optical absorbance change monitored at 411 and 922 nm for the cast film of PAI 4c on ITO-glass slide (active area ∼1 cm2) in 0.1 M Bu4NClO4/CH3CN with a cycle time of 20 s. | |
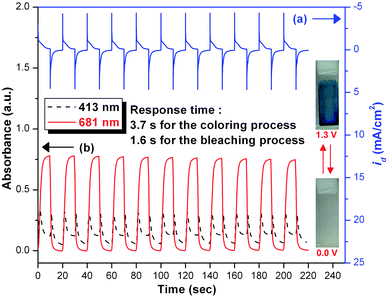 |
| | Fig. 10 (a) Current consumption and (b) electrochromic switching between 0 and 1.3 V and optical absorbance change monitored at 413 and 681 nm for the cast film of PAI 4c on ITO-glass slide (active area ∼1 cm2) in 0.1 M Bu4NClO4/CH3CN with a cycle time of 20 s. | |
Table 4 Coloration efficiency with optical and electrochemical data of PAI 4c
| Cyclesa |
ΔOD922b |
Q
/mC cm−2 |
η
/cm2 C−1 |
Decay (%)e |
|
Times of cyclic scan by applying potential steps between 0 V and 1.0 V (vs. Ag/AgCl).
Optical density change at 922 nm.
Ejected charge, determined from the in situ experiments.
Coloration efficiency is derived from the equation: η = ΔOD922/Q.
Decay of coloration efficiency after various cyclic scans.
|
| 1 |
0.322 |
1.20 |
268 |
0 |
| 10 |
0.321 |
1.25 |
257 |
4.1 |
| 20 |
0.319 |
1.24 |
257 |
4.1 |
| 30 |
0.317 |
1.24 |
256 |
4.5 |
| 40 |
0.315 |
1.23 |
256 |
4.5 |
| 50 |
0.312 |
1.22 |
256 |
4.5 |
| 60 |
0.310 |
1.21 |
256 |
4.5 |
| 70 |
0.310 |
1.21 |
256 |
4.5 |
| 80 |
0.309 |
1.21 |
255 |
4.9 |
| 90 |
0.307 |
1.20 |
255 |
4.9 |
| 100 |
0.305 |
1.19 |
255 |
4.9 |
Conclusions
The diimide-dicarboxylic acid 2 was synthesized as a new PAI building block. A series of novel PAIs with N,N,N′,N′-tetraphenyl-1,4-phenylenediamine (TPPA) units were readily prepared from 2 with various aromatic diamines via the phosphorylation polyamidation reaction. Because of the presence of the bulky tert-butyl and three-dimensional TPPA unit, all the polymers were amorphous, had good solubility in many polar aprotic solvents, and exhibited excellent film-forming ability. In addition to high Tg values and good thermal stability, all the obtained PAIs also revealed good electrochemical and electrochromic stability and multicolor electrochromic behavior. After long-term cyclic switching, the polymer films still preserved high redox and electrochromic activity. The new materials described here represent a novel entry to the polymeric electrochromics that may find potential application in the fabrication of electrochromic devices.
Acknowledgements
We thank the National Science Council of the Republic of China for the financial support of this work.
References
-
(a)
P. M. S. Monk, R. J. Mortimer and D. R. Rosseinsky, Electrochromism: Fundamentals and Applications, VCH, Weinheim, Germany, 1995 Search PubMed;
(b) R. J. Mortimer, Chem. Soc. Rev., 1997, 26, 147 RSC;
(c) D. R. Rosseinsky and R. J. Montimer, Adv. Mater., 2001, 13, 783 CrossRef CAS;
(d)
P. M. S. Monk, R. J. Mortimer and D. R. Rosseinsky, Electrochromism and Electrochromic Devices, Cambridge University Press, Cambridge, UK, 2007 Search PubMed.
-
(a) J. P. Cronin, T. J. Gudgel, S. R. Kennedy, A. Agrawal and D. R. Uhlmann, Mater. Res., 1999, 2, 1 CAS;
(b) D. Cummins, G. Boschloo, M. Ryan, D. Corr, S. Rao and D. Fitzmaurice, J. Phys. Chem. B, 2000, 104, 11449 CrossRef CAS;
(c) H. W. Heuer, R. Wehrmann and S. Kirchmeyer, Adv. Funct. Mater., 2002, 12, 89 CrossRef CAS;
(d) G. Sonmez, C. K. F. Shen, Y. Rubin and F. Wudl, Angew. Chem., Int. Ed., 2004, 43, 1498 CrossRef CAS;
(e) R. J. Mortimer, A. L. Dyer and J. R. Reynolds, Displays, 2006, 27, 2 CrossRef CAS.
-
(a) L. Groenendaal, F. Jonas, D. Freitag, H. Pielartzik and J. R. Reynolds, Adv. Mater., 2000, 12, 481 CrossRef CAS;
(b) L. Groenendaal, G. Zotti, P. H. Aubert, S. M. Maybright and J. R. Reynolds, Adv. Mater., 2003, 15, 855 CrossRef CAS;
(c) R. M. Walczak and J. R. Reynolds, Adv. Mater., 2006, 18, 1121 CrossRef CAS.
-
(a) G. Sonmez, H. Meng and F. Wudl, Chem. Mater., 2004, 16, 574 CrossRef CAS;
(b) G. Sonmez, H. B. Sonmez, C. K. F. Shen and F. Wudl, Adv. Mater., 2004, 16, 1905 CrossRef;
(c) G. Sonmez and F. Wudl, J. Mater. Chem., 2005, 15, 20 RSC;
(d) G. Sonmez, H. B. Sonmez, C. K. F. Shen, R. W. Jost, Y. Rubin and F. Wudl, Macromolecules, 2005, 38, 669 CrossRef CAS.
-
(a) C. G. Wu, M. I. Lu, S. J. Chang and C. S. Wei, Adv. Funct. Mater., 2007, 17, 1063 CrossRef CAS;
(b) A. Durmus, G. E. Gunbas and L. Toppare, Chem. Mater., 2007, 19, 6247 CrossRef CAS;
(c) G. E. Gunbas, A. Durmus and L. Toppare, Adv. Mater., 2008, 20, 691 CrossRef CAS;
(d) A. Cihaner and F. Algi, Adv. Funct. Mater., 2008, 18, 3583 CrossRef CAS;
(e) F. Algi and A. Cihaner, Org. Electron., 2009, 10, 704 CrossRef CAS;
(f) M. Pamuk, S. Tirkes, A. Cihaner and F. Algi, Polymer, 2010, 51, 62 CrossRef CAS.
-
(a) S. H. Cheng, S. H. Hsiao, T. H. Su and G. S. Liou, Macromolecules, 2005, 38, 307 CrossRef CAS;
(b) G. S. Liou, S. H. Hsiao and H. W. Chen, J. Mater. Chem., 2006, 16, 1831 RSC;
(c) S. H. Hsiao, Y. M. Chang, H. W. Chen and G. S. Liou, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4579 CrossRef CAS;
(d) C. W. Chang, S. H. Hsiao and G. S. Liou, J. Mater. Chem., 2007, 17, 1007 RSC;
(e) S. H. Hsiao, G. S. Liou, Y. C. Kung and H. J. Yen, Macromolecules, 2008, 41, 2800 CrossRef CAS;
(f) Y. C. Kung, G. S. Liou and S. H. Hsiao, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1740 CrossRef CAS.
-
Y. Imai, in Synthesis of Polyamideimides in Polyimide: Fundamentals and Applications, ed. M. K. Ghosh and K. L. Mittal, Marcel Dekker, New York, 1996, pp. 49–70 Search PubMed.
-
(a) N. Yamazaki, M. Matsumoto and F. Higashi, J. Polym. Sci., Polym. Chem. Ed., 1975, 13, 1373 CrossRef CAS;
(b) F. Higashi, S. Ogata and Y. Aoki, J. Polym. Sci., Polym. Chem. Ed., 1982, 20, 2081 CrossRef CAS;
(c) W. R. Krigbaum, R. Kotek, Y. Mihara and J. Preston, J. Polym. Sci., Polym. Chem. Ed., 1985, 23, 1907 CrossRef.
-
(a) C. P. Yang and S. H. Hsiao, Makromol. Chem., 1989, 190, 2119 CrossRef CAS;
(b) S. H. Hsiao and C. P. Yang, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 1149 CrossRef CAS;
(c) S. H. Hsiao and C. P. Yang, Makromol. Chem., 1990, 191, 155 CrossRef CAS.
-
(a) W. Wrasidlo and J. M. Augl, J. Polym. Sci., Part A-1: Polym. Chem., 1969, 7, 321 CrossRef CAS;
(b) W. Wrasidlo and J. M. Augl, J. Polym. Sci., Part A-1: Polym. Chem., 1969, 7, 1589 CrossRef CAS;
(c) J. L. Nieto, J. G. de la Campa and J. de Abajo, Makromol. Chem., 1982, 183, 557 CrossRef CAS;
(d) J. G. de la Campa, J. de Abajo and J. L. Nieto, Makromol. Chem., 1982, 183, 571 CrossRef CAS.
-
(a) S. H. Hsiao, C. P. Yang, C. W. Chen and G. S. Liou, Eur. Polym. J., 2005, 41, 511 CrossRef CAS;
(b) C. P. Yang, Y. P. Chen and E. M. Woo, J. Appl. Polym. Sci., 2006, 101, 2854 CrossRef CAS;
(c) Y. T. Chern, C. M. Huang and S. C. Huang, Polymer, 1998, 39, 2929 CrossRef CAS;
(d) D. J. Liaw, P. N. Hsu, W. H. Chen and B. Y. Liaw, Macromol. Chem. Phys., 2001, 202, 1483 CrossRef CAS;
(e) H. Behniafar and A. Banihashemi, Eur. Polym. J., 2004, 40, 1409 CrossRef CAS;
(f) C. Lee, N. P. Iyer, K. Min, H. Pak and H. Han, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 137 CrossRef CAS.
- K. Y. Chiu, T. H. Su, C. W. Huang, G. S. Liou and S. H. Cheng, J. Electroanal. Chem., 2005, 578, 283 CrossRef CAS.
- C. Lambert and G. Noll, J. Am. Chem. Soc., 1999, 121, 8434 CrossRef CAS.
-
(a) G. S. Liou and C. W. Chang, Macromolecules, 2008, 41, 1667 CrossRef CAS;
(b) C. W. Chang and G. S. Liou, J. Mater. Chem., 2008, 18, 5638 RSC;
(c) C. W. Chang, C. H. Chung and G. S. Liou, Macromolecules, 2008, 41, 8441 CrossRef CAS;
(d) H. J. Yen and G. S. Liou, Chem. Mater., 2009, 21, 4062 CrossRef CAS.
- S. H. Hsiao, G. S. Liou and H. M. Wang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2330 CrossRef CAS.
- C. P. Yang, R. S. Chen and K. H. Chen, Colloid Polym. Sci., 2003, 281, 505 CrossRef CAS.
-
(a) C. L. Gaupp, D. M. Welsh, R. D. Rauh and J. R. Reynolds, Chem. Mater., 2002, 14, 3964 CrossRef CAS;
(b) R. J. Mortimer and J. R. Reynolds, J. Mater. Chem., 2005, 15, 2226 RSC.
- J. N. Demas and G. A. Crosby, J. Phys. Chem., 1971, 75, 991 CrossRef.
-
(a) E. T. Seo, R. F. Nelson, J. M. Fritsch, L. S. Marcoux, D. W. Leedy and R. N. Adams, J. Am. Chem. Soc., 1966, 88, 3498 CrossRef CAS;
(b) R. F. Nelson and R. N. Adams, J. Am. Chem. Soc., 1968, 90, 3925 CrossRef CAS;
(c) H. Zhao, C. Tanjutco and S. Thayumanavan, Tetrahedron Lett., 2001, 42, 4421 CrossRef CAS;
(d) S. Amthor, B. Noller and C. Lambert, Chem. Phys., 2005, 316, 141 CrossRef CAS.
- J. Y. Shen, C. Y. Lee, T. H. Huang, J. T. Lin, Y. T. Tao, C. H. Chien and C. Tsai, J. Mater. Chem., 2005, 15, 2455 RSC.
-
(a) C. Lambert and G. Noll, Angew. Chem., Int. Ed., 1998, 37, 2107 CrossRef CAS;
(b) C. Lambert and G. Noll, Synth. Met., 2003, 139, 57 CrossRef CAS;
(c) C. C. Chiang, H. C. Chen, C. S. Lee, M. K. Leung, K. R. Lin and K. H. Hsieh, Chem. Mater., 2008, 20, 540 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: o. See DOI: 10.1039/c0py00065e |
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.