DOI:
10.1039/C0PY00028K
(Paper)
Polym. Chem., 2010,
1, 1030-1038
Synthesis and chiral recognition of optically active hydrogels containing helical polymer chains†
Received
25th January 2010
, Accepted 12th April 2010
First published on
11th May 2010
Abstract
Copolymerizations of two novel N-propargylamide monomers [HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCH2NHCOR, in M1, R: CH
CCH2NHCOR, in M1, R: CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2; in M2, R: CH2CH2CH2CHCH2] with the other two N-propargylamide monomers (M3: achiral monomer; M4: chiral monomer), which were synthesized previously, were carried out in varied monomer feed ratios in the presence of (nbd)Rh+B−(C6H5)4, with the aim to prepare optically active polymers simultaneously possessing helical polymer backbones and pendent polymerizable vinyl groups. Among the four homopolymers derived from M1–M4, polymer 1 and polymer 2 cannot dissolve completely in any of the usual solvents, while polymer 3 and polymer 4 are known to form stable helical conformations. The expected copolymers were obtained in high yields (≥95%) and exhibited good solubility in several organic solvents. The obtained copolymers were found to adopt stable helical structures under the investigated conditions. The as-prepared helical copolymers bearing vinyl groups were further employed as macromonomers to copolymerize with N-isopropylacrylamide (NIPAM) in chloroform at 55 °C using 2,2′-azobis(isobutyronitrile) as an initiator and N,N′-methylenebisacrylamide as a cross-linking agent. The copolymerizations took place smoothly, quantitatively providing hydrogels containing helical polymer chains preformed in the first copolymerization step. The helical structures remained in the hydrogels according to circular dichroism spectroscopy measurements. The hydrogels preferentially adsorbed D-tryptophan and (R)-(+)-1-phenylethylamine in the corresponding two enantiomer pairs, demonstrating the potential applications of the novel optically active hydrogels in the areas of chiral recognition and chiral resolution.
CH2; in M2, R: CH2CH2CH2CHCH2] with the other two N-propargylamide monomers (M3: achiral monomer; M4: chiral monomer), which were synthesized previously, were carried out in varied monomer feed ratios in the presence of (nbd)Rh+B−(C6H5)4, with the aim to prepare optically active polymers simultaneously possessing helical polymer backbones and pendent polymerizable vinyl groups. Among the four homopolymers derived from M1–M4, polymer 1 and polymer 2 cannot dissolve completely in any of the usual solvents, while polymer 3 and polymer 4 are known to form stable helical conformations. The expected copolymers were obtained in high yields (≥95%) and exhibited good solubility in several organic solvents. The obtained copolymers were found to adopt stable helical structures under the investigated conditions. The as-prepared helical copolymers bearing vinyl groups were further employed as macromonomers to copolymerize with N-isopropylacrylamide (NIPAM) in chloroform at 55 °C using 2,2′-azobis(isobutyronitrile) as an initiator and N,N′-methylenebisacrylamide as a cross-linking agent. The copolymerizations took place smoothly, quantitatively providing hydrogels containing helical polymer chains preformed in the first copolymerization step. The helical structures remained in the hydrogels according to circular dichroism spectroscopy measurements. The hydrogels preferentially adsorbed D-tryptophan and (R)-(+)-1-phenylethylamine in the corresponding two enantiomer pairs, demonstrating the potential applications of the novel optically active hydrogels in the areas of chiral recognition and chiral resolution.
1. Introduction
Hydrogels are water-swollen, cross-linked polymeric structures produced by the simple reaction of one or more monomers, and have found an extremely wide range of applications in the fields of, for instance, pharmacy, biotechnology, agriculture, and controlled release of drugs.1,2 One category of the most commonly studied hydrogels is made from poly(N-isopropylacrylamide) (PNIPAM), which undergoes a phase transition from a hydrophilic, water-swollen state to a hydrophobic, globular state when heated to above its lower critical solution temperature (LCST) of approx. 32 °C in water. Incorporation of other co-monomers in PNIPAM can produce a wide variety of stimuli responsivities such as pH, ionic strength, and light.3,4 More recently, supramolecular hydrogels are drawing increasing attention due to their scientific interest and technological applications. Deng et al.5 prepared chemical-responsive supramolecular hydrogels following the concepts of self-assembly and hydrophobic interactions by using cyclodextrins. Also from cyclodextrins and together with azobenzene groups, Zhao and Stoddart6 prepared light-responsive hydrogel systems, taking advantage of the light-induced trans-cis isomerization of azobenzene units. Besides the rapid responsivity to external stimuli, hydrogels have also been extensively investigated as separating materials. Zhuang and coworkers7 examined the separation efficiency of bovine serum albumin using the hydrogels they particularly designed and prepared. Hydrogels with the ability to absorb polysaccharide and protein were reported by Dong et al.8 Some other hydrogels showed separation and concentration ability toward peptides,9 cell lysate propeins,10 and lignin.11 Using hydrogels to extract and remove heavy metals12 and carboxylic acids13 from aqueous solutions were also reported. However, up to date only a few reports14–16 have been found to focus on chiral recognition and/or enantioselective adsorption in literature. The present article reports novel optically active hydrogels containing helical polymer chains, which act as chiral selectors for enantioselective adsorption. Such hydrogels demonstrated considerable chiral recognition ability and therefore possess potential applications in chiral resolution.
The synthesis of artificial helical polymers has always been an attractive research topic,17–22 not only due to their visually beautiful structures but also due to their intriguing properties. Thanks to the advances in organic chemistry and polymer chemistry, a great many helical polymers have been successfully synthesized so far, in addition to the relatively well-studied helical polymers.23–26 The Percec group22 reported on polyisocyanides with fan-structured pendent groups, in which the crowded bulky side chains facilitated the polymer backbones to adopt helical conformations. Yashima and coworkers27 prepared helical polymers from substituted acetylenes with branched structures, and also created the elegant double-helix polymers.28 Also of great interest are the helical polymers reported by Novak and coworkers.29 Helical polymers are full of promise for a variety of practical applications in chiral catalysis, chiral recognition/resolution, optical/electrical materials and so forth.17,30–32 For example, Yashima and coworkers33 and Masuda and coworkers34 prepared interesting polymer gels with chiral-responsivity and chiral recognition abilities. Some other studies of similar topics are also worth mentioning. Okamoto and coworkers35 reported polymer gels with chiral helical cavity by using a molecular imprinting technique from methyl methacrylate derivatives; Liu and Dordick14 reported chiral molecularly imprintable hydrogels based on sugar acrylates. Aoki et al.36 prepared optically active hydrogels with thermosensitivity using methacrylamide derivatives.
In previous studies, we have prepared a series of substituted polyacetylenes, including poly(N-propargylamide)s,37–41 poly(N-propargylsulfamide)s,42 and poly(N-propargylurea)s.43 Most of the obtained polymers can form helical structures under suitable conditions. We also prepared stable emulsions from substituted acetylene monomers44 and grafted the helical polymer chains on vinyl polymer particles.45 The as-prepared functional particles exhibited the expected chiral recognition ability. Based on the investigations above and referring to the leading investigations from Okamoto,46 who reported helical polymers possessing chiral resolution abilities, in this study we designed and prepared functional hydrogels via copolymerizations of NIPAM and a specific macromonomer which is able to form helical structures and simultaneously contain polymerizable vinyl groups in side chains. In the as-prepared novel hydrogels, the helical macromonomers endow the hydrogels with chiral recognition and enantioselective adsorption performances. Accordingly, such optically active hydrogels are considered as being of high promise for applications in chiral resolution.
2. Experimental section
Measurements
1H and 13C-NMR spectra were recorded on a Bruker AV600 spectrometer with CDCl3 as solvent. FT-IR spectra were recorded with a Nicolet NEXUS 670 spectrophotometer (KBr tablet). Molecular weights (Mn) and molecular weight distributions (Mw/Mn) of the (co)polymers were determined by GPC (Waters 515–2410 system), calibrated by using polystyrenes as standards and THF as an eluent. Circular dichroism (CD) and UV-vis absorption spectra were recorded on a JASCO J-810 spectropolarimeter equipped with a JULABO heating immersion circulator. The CD spectra of swollen hydrogels were measured referring to the methods reported by Yashima and coworkers33 and Masuda and coworkers47 A little amount of the swollen hydrogel (approx. 0.02 g) was placed and tightly clamped between two quartz plates. Since the thickness of the hydrogels cannot be kept unified for all the measurements, the CD spectra so-obtained provide qualitative data. Specific rotations were measured on a JASCO P-1020 digital polarimeter with a sodium lamp as the light source at room temperature.
Materials
Solvents were distilled under reduced pressure before use. Propargylamine (Aldrich), isobutyl chloroformate (Aldrich), 4-methylmorpholine (Aldrich), acrylic acid (Beijing Chemistry Reagent Company), 5-hexenoic acid (Alfa Aesar), N-isopropylacrylamide (NIPAM, TCI), N,N′-methylenebisacrylamide (BIS, Beijing Chemistry Reagent Company), (R)-(+)-1-phenylethylamine, (S)-(−)-1-phenylethylamine (Alfa Aesar), D-tryptophan, and L-tryptophan (Alfa Aesar) were used without further purification. (nbd)Rh+B−(C6H5)4 was prepared as reported.48 2,2′-Azobis(isobutyronitrile) (AIBN, TCI) was purified by recrystallization from methanol.
Monomer synthesis
Monomer 3 (M3) and monomer 4 (M4) were synthesized according to the method reported in the previous articles.49,50 Monomer 1 (M1) and monomer 2 (M2) as new compounds were synthesized for the first time by the method introduced earlier.49 Taking the synthesis of M1 as an example, the main synthetic procedure is described as follows. Acrylic acid (2.4 mL, 35 mmol), isobutyl chloroformate (4.6 mL, 35 mmol), and 4-methylmorpholine (4.4 mL, 35 mmol) were added to THF (200 mL) sequentially. The solution was stirred at room temperature for 10 min, and then propargylamine (2.4 mL, 35 mmol) was added to the solution dropwise. The solution was stirred at room temperature for another 4 h. After that, the white precipitate formed during the reaction was filtered off, and the filtrate was collected, to which ethyl acetate (ca. 50 ml) was added to extract the desired product. The combined solution was washed with 2 mol L−1 HCl aqueous solution three times and then washed with saturated aqueous NaHCO3 to neutralize the solution. Then, the solution was dried over anhydrous MgSO4, filtered, and concentrated to give the target monomer. The crude monomer was further purified twice by flash column chromatography on silica gel (hexane–ethyl acetate = 2/1, in ml ml−1). M2 was synthesized in a similar way using the corresponding carboxylic acid. The crude M2 was purified further by recrystallization twice from THF–hexane solvent mixture. The analytical and spectroscopic data of M1 and M2 are as follows:
Monomer 1.
Yield 68%, yellowish solid. IR (KBr): 3292 (H–N), 2118 (H–C), 1659 (CO), 1536, 1406, 1118, 2957 cm−1 (H–CC). 1H-NMR (CDCl3, 600 MHz, 25 °C), δ: 5.63–6.17 (CH2 CH), 6.25–6.28 (CH2 CH), 2.20 (CHC), 3.80–3.81 (CHCCH2), 7.24 ppm (NH). 13C NMR (600 MHz, CDCl3), δ: 29.16, 71.46, 79.42, 127.08, 130.29, 165.72 ppm. Anal. Calcd for C6H7NO: C, 66.04; H, 6.47; N, 12.84. Found: C, 66.25; H, 6.41; N, 12.63.
Monomer 2.
Yield 83%, colorless crystal, IR (KBr): 3295 (N–H), 2115 (H–CC), 1639 (CO), 1533, 1249, 1118, 2955 cm−1 (H–CC). 1H-NMR (CDCl3, 600 MHz, 25 °C), δ: 2.16–2.23 (CHC, H2CCHCH2CH2CH2), 4.00–4.02 (HCCCH2), 1.69–1.74 (H2CCHCH2CH2), 2.04–2.07 (H2CCHCH2), 4.94–5.01 (H2CCH), 5.71–5.78 (H2CCH), 7.25 ppm (NH). 13C NMR (600 MHz, CDCl3), δ: 24.51, 29.09, 33.05, 35.49, 71.47, 79.62, 115.44, 137.77, 172.55 ppm. Anal. Calcd for C9H13NO: C, 71.49; H, 8.64; N, 9.26. Found: C, 71.24; H, 8.57; N, 9.18.
(Co)polymerization
Referring to the earlier studies,49 the (co)polymerizations were carried out with (nbd)Rh+B−(C6H5)4 as the catalyst in dry chloroform under nitrogen for 2 h, with the total monomer concentration being kept at 0.5 M, and the concentration of the catalyst = 5 mM. After (co)polymerization, the reaction mixture was poured into a large amount of hexane to precipitate the formed (co)polymer. Then, the (co)polymer was filtered off, washed with hexane, and dried under reduced pressure.
Preparation of hydrogels
All the polymerizations were carried out in a glass tube equipped with a three-way stopcock under nitrogen. In detail, the helical copolymer synthesized above (0.05 g), N-isopropylacrylamide (NIPAM, 0.95 g), and N,N′-methylenebisacrylamide (BIS, 0.001 g) were dissolved in chloroform. The monomer solution was bubbled with dried nitrogen for 30 min prior to polymerization. Then 2,2′-azobis(isobutyronitrile) (AIBN, 0.001 g) was added to the monomer solution under nitrogen. The polymerization was carried out at 55 °C for 4 h. After that, the resulting hydrogel was taken out and immersed in chloroform for one week to remove the unreacted monomers and initiator. The chloroform was changed everyday. Herein, it should be pointed out that the hydrogel was considered to be pure enough only when its mass change was less than 0.005 g over 24 h. For this purpose, the hydrogels were immersed in solvent for a long period. After purification, the hydrogel was dried under reduced pressure. The yields of the copolymers were found to be nearly 100%.
Swelling ratio of the hydrogels
The swelling ratios of the obtained hydrogels were measured as follows. After immersion in deionized water at a predetermined temperature, hydrogels were taken out from water and the excess water on the surface of the hydrogels was carefully wiped off with a filter paper. The hydrogels were repeatedly weighed and immersed in water at the given temperature until the weight of the hydrogel reached a constant value. The swelling ratio (SR) of the hydrogels can be determined by equation, SR = (Ws−Wd)/Wd, in which Ws and Wd are the weight of the hydrogel after absorbing water and dry weight of the hydrogel, respectively.
Chiral recognition experiments
All the chiral recognition experiments were conducted by immersing the hydrogel in adsorbate solutions at room temperature. In the case of tryptophan, deionized water was used as the solvent, while in the case of phenylethylamine, CHCl3 was used as solvent. With the latter as an example, the main procedure is stated below. A CHCl3 solution (40 mL) of (R)-(+)- or (S)-(−)-1-phenylethylamine (c = 1 mg mL −1) was prepared and its optical activity was measured. Subsequently, a predetermined amount of hydrogel (1 g) was encased in a filter paper and then immerged in the solution. After every certain period of time, the outer solution was subjected to optical rotation measurement with a Jasco digital polarimeter (λ = 589 nm) using a cylindrical quartz cell (10 mL) at room temperature. Then the chiral compound concentration can be determined according to optical rotation data. Meanwhile, hydrogels with adsorbed chiral compounds were subjected to CD spectroscopy measurements.
3. Results and discussion
Strategy for the preparation of optically active hydrogels
The strategy for the preparation of optically active hydrogels containing helical polymer chains is schematically presented in Scheme 1. Helical copolymers are synthesized in the first step by copolymerization with an Rh catalyst from two monomers (cf.Scheme 2): one offers the helical polymer structures while the other provides vinyl groups intended for the subsequent free radical copolymerization. In a second step, the preformed helical copolymers containing vinyl groups undergo free radical copolymerizations with NIPAM and a cross-linker, BIS. From the copolymerizations, the expected hydrogels can be prepared, which contain helical polymer chains of a predominant handedness and thus exhibit optical activity. Due to the optically active helical polymers, the as-prepared hydrogels demonstrate chiral recognition and chiral resolution abilities.
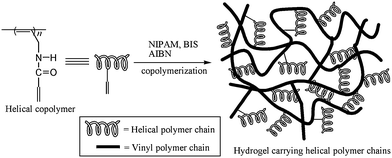 |
| | Scheme 1 A schematic presentation of the PNIPAM hydrogels containing helical polymer chains. | |
Synthesis and (co)polymerization of monomers
Scheme 2 shows the structures of M1 and M2. The two monomers contain the particularly designed vinyl groups in their substituted groups. The two monomers were synthesized smoothly by the method reported elsewhere.49 The synthesis and purification processes and the analytical and spectroscopic data of the two novel monomers are presented in the experimental section above. Both M1 and M2 were subjected to homopolymerization in solvents in the presence of (nbd)Rh+B−(C6H5)4, a well-known catalyst for performing polymerization of mono-substituted acetylenes.37–45 Unfortunately, even though the two corresponding homopolymers (polymer 1 and polymer 2, derived from M1 and M2, respectively) were obtained in a 100% yield (Table 1), they could not dissolve entirely in any of the usual solvents (THF, CHCl3, toluene, DMF, DMSO, acetone, methanol, etc.), as presented in Table 2. Therefore, M1 and M2 are not desirable for the following investigations.
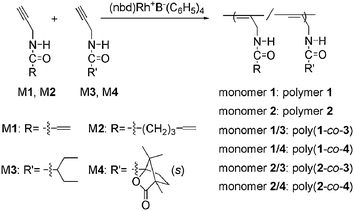 |
| | Scheme 2 The substituted acetylene monomers (M1–M3: achiral monomer; M1 and M2: containing vinyl group; M4: chiral monomer) and (co)polymers. | |
| Copolymer |
Monomer ratio/mol mol−1 |
Yieldb(wt%) |
M
n
|
M
w/Mnc |
[α]Dd/° |
|
With (nbd)Rh+B−(C6H5)4 as the catalyst in CHCl3 at 30 °C for 2 h; [M]total = 1.0 mol L−1; [M]total/[Rh] = 100.
Hexane-insoluble part.
Measured by GPC (polystyrenes as standards; THF as eluent).
Insoluble or only partially soluble in usual solvents.
|
| Polymer 1e |
— |
100 |
— |
— |
— |
| Polymer 2e |
— |
100 |
— |
— |
— |
| Poly(10.1-co-30.9) |
M1/M3 = 1/9 |
95 |
10000 |
1.87 |
−11 |
| Poly(10.1-co-40.9) |
M1/M4 = 1/9 |
98 |
12000 |
2.64 |
−690 |
| Poly(20.1-co-30.9) |
M2/M3 = 1/9 |
96 |
10000 |
1.94 |
−1 |
| Poly(20.1-co-40.9) |
M2/M4 = 1/9 |
99 |
17000 |
2.58 |
−596 |
Table 2 Solubility of the obtained (co)polymersa
Accordingly, M1 and M2 were subjected to copolymerization with an achiral monomer (M3) and a chiral monomer (M4), respectively. The structures of M3 and M4 are illustrated in Scheme 2. In the anticipated copolymers, M1 and M2 units offer vinyl groups, while M3 and M4 units offer helical polymer chain segments. In addition, the content of the co-monomers bearing vinyl groups, i.e., M1 and M2, should be not too high, to decrease the effects of these co-monomers on the helical structures formed by M3 and M4 monomer units. Accordingly, the content of M1 and M2 was fixed at 10 mol%. The copolymerization results are presented in Table 1. The copolymers could be yielded with moderate molecular weights (Mn ≥ 10000) and in high yields (≥96%), and their polydispersity indices (Mw/Mn) were in the range 1.1–2.5. The copolymerizations exhibited similar characteristics to those observed in the copolymerization systems already reported.40,51,52 Taking into account the nearly quantitative yields, the copolymers are considered to have the same compositions as the corresponding monomer feed ratios. This observation was also reported many times in the investigations of similar research topics.37–40,42,43,51,52 Besides, the solubility of the copolymers (as presented in Table 2) was satisfactory, which makes it possible for us to continue our investigations. Particularly importantly, both poly(10.1-co-40.9) and poly(20.1-co-40.9) displayed high specific rotations, due to the helical polymer main chains in the copolymers, as discussed later on.
Secondary structure of the copolymers
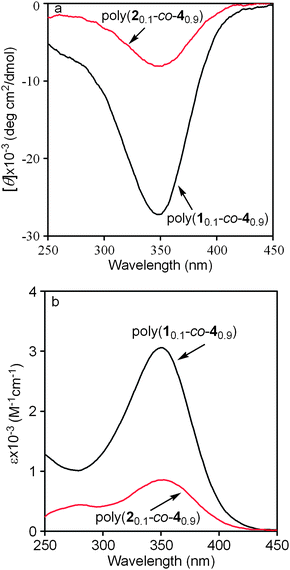
As previously reported, both the two homopolymers from M349 and M450 could form stable helical structures at a wide temperature range (i.e., from 10 to 50 °C) in chloroform. Moreover, the polymer from chiral M4 has strong optical activity, which is assumed to be due to the “chiral amplification” effects frequently observed in helical polymers.53 In addition, CD and UV-vis spectroscopies have been proven to be effective to examine the secondary structures of polymers.23 This is also true in previously investigated helical polymers. When the earlier polymers, poly(N-propargylamide)s, poly(N-propargylsulfamide)s, and poly(N-propargylurea)s with similar structures to the present copolymers, adopt helical conformations in their backbones, intense CD signals and UV-vis absorptions can be observed in a wavelength range from 345–410 nm. But for helical polymers with the equal amount of left- and right-handed helices, only UV absorptions can be observed because of racemization effect.37–43
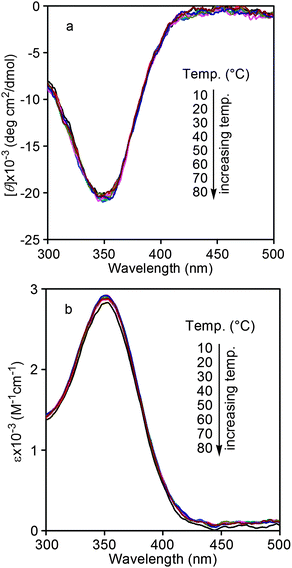
CD and UV-vis spectra of the current copolymers were thus measured. As shown in Fig. 1, strong UV-vis adsorption peaks could be observed around 390 nm in the UV-vis spectra of both poly(10.1-co-30.9) and poly(20.1-co-30.9), and there were no CD effects in their CD spectra. In other words, both poly(10.1-co-30.9) and poly(20.1-co-30.9) could take helical conformations under the examined conditions, but they were not optically active due to the equal amount of left- and right-handed helices. This observation is in accordance with the results in optical rotations (Table 1). Moreover, stronger UV-vis adsorption peaks appeared in poly(20.1-co-30.9) than in poly(10.1-co-30.9). This demonstrates a higher helical content occurred in the former than in the latter.38 In order to investigate the thermal stability of the helical conformations formed in poly(20.1-co-30.9), the copolymer was subjected to UV-vis spectroscopy measurement at elevated temperatures. The relevant results are depicted in Fig. 2. We can see that poly(20.1-co-30.9) formed stable helical structures in a temperature range from 10 to 80 °C in toluene, since the UV-vis absorption changed little with increasing temperature. When compared with our earlier helical polymers, the helical conformations in poly(20.1-co-30.9) are much higher in thermal stability, most likely due to the bulky pendent groups in this copolymer.39,49
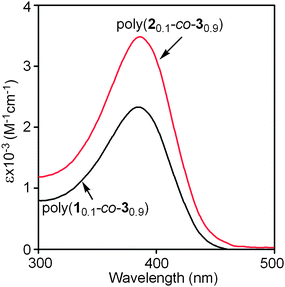 |
| | Fig. 1 UV-vis spectra of poly(10.1-co-30.9) and poly(20.1-co-30.9) in CHCl3, measured at 25 °C, c = 0.1 mM. | |
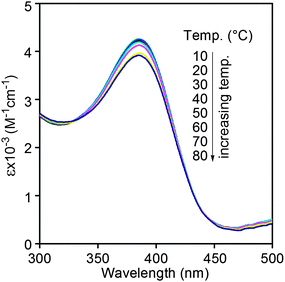 |
| | Fig. 2 UV-vis spectra of poly(20.1-co-30.9) in toluene at varied temperatures (from 10 to 80 °C) (c = 0.1 mM). | |
The above investigations manifest that the copolymers of poly(20.1-co-30.9) can form relatively stable helices. However, the copolymers formed left- and right-handed helices in the equal amount, resulting in polymers without optical activity. This cannot satisfy our need to prepare optically active hydrogels in the next step. Accordingly, we utilized M4 instead of M3 for preparing the optically active helical copolymers, since the former contains chiral groups. In Table 1, both M1/M4 and M2/M4 systems could undergo copolymerization smoothly, providing the two corresponding chiral copolymers, i.e., poly(10.1-co-40.9) and poly(20.1-co-40.9). The relevant data are shown in Table 1. The solubility of the two copolymers is illustrated in Table 2. The secondary structures of the two chiral copolymers were examined by CD and UV-vis spectroscopy analysis. As shown in Fig. 3(a) and 3(b), poly(10.1-co-40.9) and poly(20.1-co-40.9) showed intense CD signals around 350 nm, nearly completely corresponding with the UV-vis absorption peaks. This observation is the same as that observed in the helical homopolymer of M4.49 It is thus indicated that poly(10.1-co- 40.9) and poly(20.1-co-40.9) also formed helices under the examined conditions, and both of them exhibited optical activity due to the predominant handed helices in the copolymers. But poly(10.1-co-40.9), when compared with poly(20.1-co-40.9), exhibits stronger UV-vis absorption and more intense CD effects under the same investigation conditions. Furthermore, Fig. 4(a) and 4(b) show that poly(10.1-co-40.9) formed helical structures with a high thermal stability in the temperature range from 10 to 80 °C in toluene. A combination of the above investigations demonstrated that poly(10.1-co-40.9) is more favorable for use in the preparation of optically active hydrogels, which will be stated below.
Synthesis of hydrogels
Taking advantage of the vinyl groups in the helical copolymer prepared above, poly(10.1-co-40.9), hydrogels were prepared further. The hydrogels were obtained through the free radical copolymerization of the chiral helical copolymer and N-isopropylacrylamide (NIPAM) in chloroform at 55 °C using 2,2′-azobis(isobutyronitrile) (AIBN) as an initiator and N,N′-methylenebisacrylamide (BIS) as a cross-linker. Fig. 5 presents a typical photograph of the resulting hydrogels containing optically active helical polymer of varied content. The three hydrogels exhibit a slight difference in their appearance, namely, with an increase in the content of helical polymer, the hydrogels became darker in color. The yellowish color in the hydrogels is also observed in the corresponding helical polymer and copolymer in the current study; the yellowish color is also observed in the helical polymers of the same type and even in the emulsions consisting of helical substituted polymer acetylenes.44 It seems that when the helical (co)polymers from substituted acetylene monomers adopt helical structures, they frequently show a color of yellow or a color close to yellow.42,49
 |
| | Fig. 5 Photographs of the prepared hydrogels; 1, 2, and, 3 samples contain 5 wt%, 10 wt%, and 15 wt% poly(10.1-co-40.9), respectively. | |
Fig. S1 (in the ESI†) depicts the FT-IR spectra of poly(10.1-co-40.9) (Fig. S1a) and the hydrogel from poly(10.1-co-40.9) (Fig. S1b). In the two spectra, characteristic bands could be observed at 3432 (–NH–), 1788 (–C(=O)O–), 1652 (–C(=O)NH–), 1543 cm−1 (–C(=O)NH–) indicating the functional groups in both poly(10.1-co-40.9) and the hydrogel. Nevertheless, because of the high similarity between poly(10.1-co-40.9) and NIPAM units in the molecular structure, especially as both of them contain –CONH– groups, FT-IR spectra could not offer more information. However, the appearance and the quantitative yield of the product and its insolubility in all solvents confirmed the formation of hydrogels.
Optical activity of the hydrogels
The obtained hydrogels were further studied by measuring their CD and UV-vis spectra to elucidate the existence of helical polymer chains in the hydrogels and their optical activities. As shown in Fig. 6(a), the hydrogels with different helical copolymer content showed intense CD signals at a wavelength between 350 and 410 nm. In more detail, with an increase in the content of poly(10.1-co-40.9), the CD signal showed a blue shift and simultaneously an increase in CD signal strength. It is proposed that the surrounding PNIPAM chain segments prevented the poly(10.1-co-40.9) chains from forming the helical structures as they should in the absence of PNIPAM chains. Resulting from the surrounding hindrance, poly(10.1-co-40.9) chains are assumed to form helices with much larger pitch and larger conjugation length,37,54 when compared with the helical structures in the absence of PNIPAM. When the content of poly(10.1-co-40.9) increased, the hindrance caused by the PNIPAM chains decreased, allowing the poly(10.1-co-40.9) chains to form helical structures as they like. The detailed mechanism involved in this phenomenon needs more investigation. However, it is clear that the hydrogels contain helical poly(10.1-co-40.9) and thus are optically active.
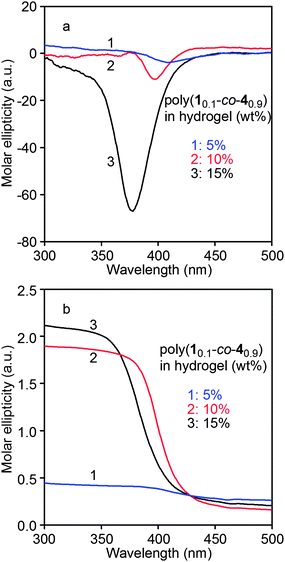 |
| | Fig. 6 (a) CD and (b) UV-vis absorption spectra of the hydrogels containing 5, 10, and 15 wt% of poly(10.1-co-40.9) swelled in deionized water at 25 °C for 24 h. | |
In the UV-vis spectra in Fig. 6b, when the content of poly(10.1-co-40.9) increased to 10 wt% and above, the UV absorption became quite large at the wavelength <410 nm. In spite of the intense CD signal and high optical activity in the hydrogel containing 10 wt% and 15 wt% of poly(10.1-co-40.9), the swelling capacity of these two hydrogels was not very well. Therefore we choose the hydrogel polymerized with 5 wt% poly(10.1-co-40.9) to conduct the chiral recognition and enantioselective adsorption experiments. These investigations will be presented in detail below.
The swelling ratio of the hydrogels was determined, as illustrated in Fig. 7. For the hydrogels with 5 wt% poly(10.1-co-40.9), their swelling ratios dramatically decreased with increasing the cross-linking agent, BIS. This can be simply explained by the increased cross-linking density caused by increasing BIS. More importantly, the swelling ratios demonstrated a sharp variation around the LCST of PNIPAM hydrogel, i.e., 32 °C.
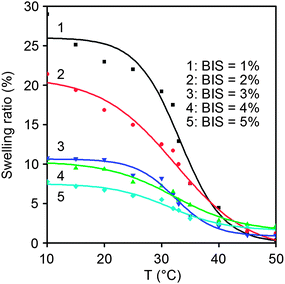 |
| | Fig. 7 Swelling ratios of the optical hydrogels with varied content of BIS in water as a function of temperature. | |
Chiral recognition of the hydrogels
In this study, chiral tryptophan and phenylethylamine were used as model compounds to be recognized. To this end, the hydrogels with 5 wt% poly(10.1-co-40.9) were immersed in chiral tryptophan/water solution and chiral phenylethylamine/CHCl3 solution, respectively. The original concentration of D- and L-tryptophan and (R)-(+)- and (S)-(−)-phenylethylamine was 0.1 mg mL−1. The absorption of the chiral compounds by the poly(10.1-co-40.9) hydrogel lasted for 10 h. The hydrogels separately having absorbed D-tryptophan and (R)-(+)-phenylethylamine were subjected to CD spectroscopy measurement, as shown in Fig. 8. When compared with D-tryptophan and the pure hydrogel (Fig. 8a), the hydrogel after absorbing D-tryptophan showed both the CD signals of the absorbent and adsorbate.
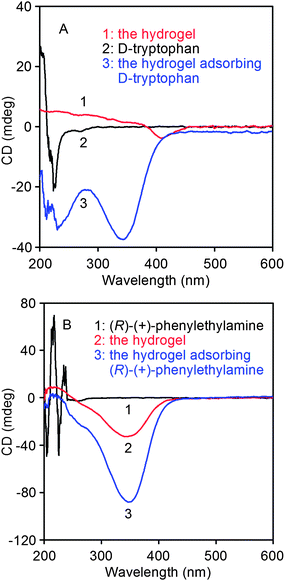 |
| | Fig. 8 CD spectra of the hydrogels having absorbed (A) D-tryptophan (in H2O) and (B) (R)-(+)-phenylethylamine (in CHCl3). | |
This observation is more pronounced in the case of (R)-(+)-phenylethylamine, as evidenced by Fig. 8b. Thus it can be concluded that the hydrogel absorbed D-tryptophan and (R)-(+)-phenylethylamine to some degree. To support this observation, quantitative data will be presented next. In addition, from Fig. 8a and 8b, the pure hydrogel showed different CD spectra in water and in CHCl3. This phenomenon will be investigated in detail in our future studies.
Next, we first investigated the enantioselective adsorption of the hydrogels toward D- and L-tryptophan. A predetermined amount of the hydrogel encapsulated in filter paper was immerged in a mixture consisting of D- and L-tryptophan in water. The hydrogel demonstrated different adsorption to D- and L-tryptophan, as illustrated in Fig. 9a. It is demonstrated that the hydrogel preferentially adsorbed D-tryptophan. Nevertheless, the maximum adsorption of D-tryptophan was only ca. 25%, even though the adsorption equilibrium could be reached within 2 h. Thus it seems that the enantioselective adsorption of the hydrogel toward chiral tryptophan is not satisfactory. Furthermore, we took (R)-(+)- and (S)-(–)-phenylethylamine as model racemates to elucidate the chiral separation ability of the hydrogels.
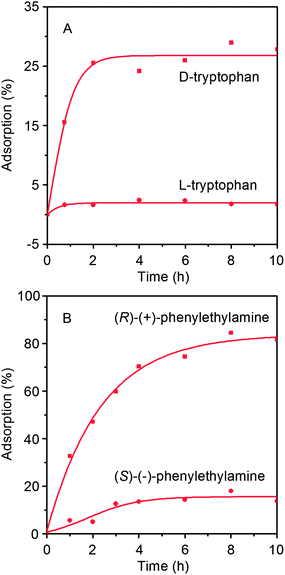 |
| | Fig. 9 Adsorption profiles of (A) tryptophan (in H2O) and (B) phenylethylamine (in CHCl3) enantiomers by the poly(10.1-co-40.9)-based hydrogels at room temperature. | |
In this case, the adsorption was conducted in CHCl3 instead of water since phenylethylamine does not dissolve in water. The hydrogels demonstrated a predominant absorption of (R)-(+)-phenylethylamine rather than the other enantiomer (Fig. 9b). In other words, the hydrogel adsorbed (R)-(+)-phenylethylamine with much more ease relative to (S)-(−)-phenylethylamine. More interestingly, the maximum adsorption was far higher, up to 80%, when compared to the former case of D-tryptophan. This maximum adsorption was achieved after adsorbing for 6 h. It further reflected that the hydrogel is more suited to the resolution of chiral phenylethylamine. Other racemates will be examined in our work. We are convinced that by optimizing the structure and composition of the hydrogels, optically active hydrogels with higher chiral resolution properties will be realized. Such investigations are currently under way.
4. Conclusions
In this article, two novel N-propargylamide monomers were first obtained, from which and the other two monomers already studied, helical copolymers bearing vinyl groups were synthesized. Based on the as-prepared helical copolymers and NIPAM, optically active hydrogels containing helical polymer chains were further obtained. The helical polymer segments can function as chiral selectors, so such optically active hydrogels showed chiral recognition and chiral resolution properties, especially in the chiral resolution of phenylethylamine enantiomers. The hydrogels adsorbed (R)-(+)-phenylethylamine much better than its corresponding enantiomer. These findings provide important information for us to further design and develop new materials for applications in chiral recognition and chiral resolution.
Acknowledgements
The project was supported by “Program for New Century Excellent Talents in University” (NCET-06-0096), “the National Science Foundation of China” (20574004 and 20974007), “Specialized Research Fund for the Doctoral Program of Higher Education” (20060010001), and the “Program for Changjiang Scholars and Innovative Research Team in University” (PCSIRT, IRT0706).
Notes and references
- Y. Tanaka, J. P. Gong and Y. Osada, Prog. Polym. Sci., 2005, 30, 1 CrossRef CAS.
- J. Kopeček, Biomaterials, 2007, 28, 5185 CrossRef CAS.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163 CrossRef CAS.
- Y. Osada, J. P. Gong and Y. Tanaka, J. Macromol. Sci., Polym. Rev., 2004, 44, 87.
- W. Deng, H. Yamaguchi, Y. Takashima and A. Harada, Angew. Chem., Int. Ed., 2007, 46, 5144 CrossRef CAS.
- Y. Zhao and J. F. Stoddart, Langmuir, 2009, 25, 8442 CrossRef CAS.
- Y. Zhuang, L. Chen, Z. Zhu and H. Yang, Polym. Adv. Technol., 2000, 11, 192 CrossRef CAS.
- J. Dong, L. Chen, Y. Ding and W. Han, Macromol. Chem. Phys., 2005, 206, 1973 CrossRef CAS.
- J. Kobayashi, A. Kikuchi, K. Sakai and T. Okano, Anal. Chem., 2003, 75, 3244 CrossRef CAS.
- C. S. Putka, S. H. Gehrke, M. Willis, D. Stafford and J. Bryant, Biotechnol. Bioeng., 2002, 80, 139 CrossRef CAS.
- W. Cai, E. C. Anderson and R. B. Gupta, Ind. Eng. Chem. Res., 2001, 40, 2283 CrossRef CAS.
- H. Tokuyama and T. Iwama, Langmuir, 2007, 23, 13104 CrossRef CAS.
- Y. S. Aşçı and M. J. İHasdemir, J. Chem. Eng. Data, 2008, 53, 2351 CrossRef.
- X. C. Liu and J. S. Dordick, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 1665 CrossRef CAS.
- K. Sugiyama, S. Rikimaru, Y. Okada and K. Shiraishi, J. Appl. Polym. Sci., 2001, 82, 228 CrossRef CAS.
- B. V. Shankar and A. Patnaik, J. Phys. Chem. B, 2007, 111, 9294 CrossRef CAS.
- T. Nakano and Y. Okamoto, Chem. Rev., 2001, 101, 4013 CrossRef CAS.
- E. Yashima, K. Maeda and Y. Furusho, Acc. Chem. Res., 2008, 41, 1166 CrossRef CAS.
- T. Masuda, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 165 CrossRef CAS.
- J. E. Y. Lam and B. Z. Tang, Acc. Chem. Res., 2005, 38, 745 CrossRef CAS.
- M. Fujiki, Top. Curr. Chem., 2007, 284, 119.
- J. G. Rudick and V. Percec, Acc. Chem. Res., 2008, 41, 1641 CrossRef CAS.
- J. H. K. K. Hirschberg, L. Brunsveld, A. Ramzi, J. A. J. M. Vekemans, R. P. Sijbesma and E. W. Meijer, Nature, 2000, 407, 167 CrossRef CAS.
- T. Aoki, T. Kaneko, N. Maruyama, A. Sumi, M. Takahashi, T. Sato and M. Teraguchi, J. Am. Chem. Soc., 2003, 125, 6346 CrossRef CAS.
- K. Terada, T. Masua and F. Sanda, Macromolecules, 2009, 42, 913 CrossRef CAS.
- J. Cui, A. Liu, J. Zhi, Z. Zhu, Y. Guan, X. H. Wan and Q. F. Zhou, Macromolecules, 2008, 41, 5245 CrossRef CAS.
- T. Kajitani, H. Lin, K. Nagai, K. Okoshi, H. Onouchi and E. Yashima, Macromolecules, 2009, 42, 560 CrossRef CAS.
- T. Maeda, Y. Furusho, S. Sakurai, J. Kumaki, K. Okoshi and E. Yashima, J. Am. Chem. Soc., 2008, 130, 7938 CrossRef CAS.
- H. Z. Tang, E. R. Garland, B. M. Novak, J. He, P. L. Polavarapu, F. C. Sun and S. S. Sheiko, Macromolecules, 2007, 40, 3575 CrossRef CAS.
- A. Zhang, F. RodrÍguez-Ropero, D. Zanuy, C. Alemán, E. W. Meijer and A. D. Schlüter, Chem.–Eur. J., 2008, 14, 6924 CrossRef CAS.
- L. Angiolini, C. Carlini and M. Tramontini, Makromol. Chem., 1990, 191, 2083 CrossRef CAS.
- K. Akagi, Polym. Int., 2007, 56, 1192 CrossRef CAS.
- H. Goto, H. Q. Zhang and E. Yashima, J. Am. Chem. Soc., 2003, 125, 2516 CrossRef CAS.
- R. Liu, F. Sanda and T. Masuda, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4175 CrossRef CAS.
- S. Habaue, T. Satonaka, T. Nakano and Y. Okamoto, Polymer, 2004, 45, 5095 CrossRef CAS.
- T. Aoki, M. Muramatsu, A. Nishina, K. Sanui and N. Ogata, Macromol. Biosci., 2004, 4, 943 CrossRef CAS.
- J. P. Deng, J. Tabei, M. Shiotsuki, F. Sanda and T. Masuda, Macromolecules, 2004, 37, 9715 CrossRef CAS.
- J. P. Deng, J. Tabei, M. Shiotsuki, F. Sanda and T. Masuda, Macromolecules, 2004, 37, 1891 CrossRef CAS.
- J. P. Deng, J. Tabei, M. Shiotsuki, F. Sanda and T. Masuda, Macromolecules, 2004, 37, 7156 CrossRef CAS.
- J. P. Deng, W. G. Zhao, J. M. Wang, Z. G. Zhang and W. T. Yang, Macromol. Chem. Phys., 2007, 208, 218 CrossRef CAS.
- L. Ding, X. F. Jiao, J. P. Deng, W. G. Zhao and W. T. Yang, Macromol. Rapid Commun., 2009, 30, 120 CrossRef CAS.
- Z. G. Zhang, J. P. Deng, W. G. Zhao, J. M. Wang and W. T. Yang, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 500 CrossRef CAS.
- J. P. Deng, X. F. Luo, W. G. Zhao and W. T. Yang, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4112 CrossRef CAS.
- J. P. Deng, B. Chen, X. F. Luo and W. T. Yang, Macromolecules, 2009, 42, 933 CrossRef CAS.
- K. Zhou, L. Y. Tong, J. P. Deng and W. T. Yang, J. Mater. Chem., 2010, 20, 781 RSC.
- Y. Okamoto, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1731 CrossRef CAS.
- R. Nomura, K. Yamada, J. Tabei, Y. Takakura, T. Takigawa and T. Masuda, Macromolecules, 2003, 36, 6939 CrossRef.
- R. R. Schrock and J. A. Osborn, Inorg. Chem., 1970, 9, 2339 CrossRef CAS.
- J. P. Deng, J. Tabei, M. Shiotsuki, F. Sanda and T. Masuda, Macromolecules, 2004, 37, 5149 CrossRef CAS.
- J. Tabei, R. Nomura and T. Masuda, Macromolecules, 2002, 35, 5405 CrossRef CAS.
- J. P. Deng, J. Tabei, M. Shiotsuki, F. Sanda and T. Masuda, Macromol. Chem. Phys., 2004, 205, 1103 CrossRef CAS.
- J. P. Deng, B. Chen, Z. G. Zhang and W. T. Yang, Polym. Int., 2007, 56, 1247 CrossRef CAS.
- M. M. Green, J. W. Park, T. Sato, A. Teramoto, S. Lifson, R. L. B. Selinger and J. V. Selinger, Angew. Chem., Int. Ed., 1999, 38, 3138 CrossRef.
- V. Percec, M. Obata, J. Rudick, B. De Binod, M. Glodde, T. K. Bear, S. N. Magonov, V. S. K. Balagurusamy and P. A. Heiney, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3509 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.