Swelling and degradation of hydrogels synthesized with degradable poly(β-amino ester) crosslinkers
Ryan A.
McBath
and
Devon A.
Shipp
*
Department of Chemistry & Biomolecular Science, and Center for Advanced Materials Processing Clarkson University, Potsdam, NY 13669-5810, USA. E-mail: dshipp@clarkson.edu; Fax: +1-315-268-6610; Tel: +1-315-268-2393
First published on 1st April 2010
Abstract
The synthesis of diacrylated poly(β-amino ester) crosslinkers, obtained from the oligomerization of di(ethylene glycol) diacrylate (DEGDA) and piperazine (PIPz), and their use in hydrogels are reported. In particular, the poly(β-amino ester) crosslinkers were used to study swelling and degradation properties of hydrogels made of N-vinylpyrrolidone and 2-hydroxy ethyl methacrylate. Poly(β-amino ester) crosslinkers with two different molecular weights (800 and 3000 g mol−1) and hydrogels with 1 and 3% crosslinker were studied. Swelling and degradation of the gels were monitored in phosphate buffered saline (pH = 7.4) at 37 °C. The swelling and degradation characteristics of the poly(β-amino ester) crosslinked hydrogels were then compared to hydrogels crosslinked with poly(ethylene glycol) diacrylate (PEGDA) of similar molecular weights. It was found that both swelling and degradation were faster in the poly(β-amino ester) crosslinked hydrogels, particularly for the highest molecular weight poly(β-amino ester) crosslinker.
Introduction
Hydrogels have been a focus of attention for many years and are widely used in a variety of bio-related applications since they are typically biocompatible; furthermore, their moisture content can mimic the natural water content of human tissue.1,2 In addition, they are capable of swelling to several times their original size and often experience weight gain of well over several times their original mass. Hydrogels are normally composed of a crosslinked polymer system, where the polymer may be either naturally derived or from synthetic feedstock. Common synthetic monomers/polymers used to produce hydrogels include poly(ethylene glycol) (PEG),3N-substituted acrylamides,3–5 2-hydroxy ethyl methacrylate (HEMA),6N-vinylpyrrolidone (NVP),6,7 and poly(lactic acid) (PLA) or poly(glycolic acid) (PGA).8There are cases in which the slow degradation of the hydrogel can be useful, such as drug delivery and soft tissue replacement applications, among others. For such materials biodegradable crosslinkers can be incorporated into hydrogels to facilitate and control the degradation. Some examples include work by Kelner and Schacht9 who investigated materials containing poly(ethylene oxide) (PEO) that entrap guest drug molecules within the matrix because of pores that are smaller than the size of the drug molecule. This then allowed the release of the drug molecule to be entirely controlled by the degradation of the matrix. The amount of crosslinker used in the hydrogel synthesis can influence the degradation rate; an increase in degradable crosslinker increases the number of degradable moieties in addition to increasing the crosslink density. The rate of degradation is influenced by the bulkiness and hydrophobicity of the polymer side chains.
Thomas et al.10 outlined three methods that lead to the degradation of hydrogels: (1) solubilization, (2) enzyme catalysis, and (3) chemical hydrolysis. The latter method occurs with polymers containing esters, lactones, orthoesters, carbonates, and anhydrides. Thomas et al.10 synthesized biodegradable crosslinkers with lactate esters and glycolate esters. Hydrogels were made of 1.5 mol% crosslinker and 2-hydroxypropyl methacrylate (HPMA). The degradation of these hydrogels was investigated at 37 °C and a pH of 7.4. In agreement with Kelner and Schacht,9 it was found that sterically bulky crosslinkers hydrolyzed slowly. It was also determined that as degradation occurred the gels imbibed more fluid until final degradation.10
Poly(β-amino esters) (PBAEs) have emerged in recent years as potentially excellent biodegradable crosslinker materials. Langer et al.11–17 have explored the synthesis of PBAEs made from the Michael addition of diamines and diacrylates, a step-growth polymerization process that will result in PBAEs with acrylate end groups if the diacrylate is used in slight molar excess. Although not formally hydrogels, Langer et al. have explored using these gel materials as drug and/or DNA delivery vehicles. In these applications, the degradation of the amino ester moiety in the crosslinked PBAE compared with regular ester groups is typically faster, and can be tuned over a wide range of times by selecting particular amine and diacrylate monomers.14,16
While recent focus on PBAEs has been in crosslinked materials for biomedical applications, they have in fact been studied by a variety of research groups. The earliest publication was from 1968 by Yoda and Toda,18 who described the production of a PBAE from piperazine or trans-2,5-dimethylpiperazine and divinyl sulfone. A short time later, Danusso and Ferruti19 examined many options of reacting amines with vinyl groups, as well as acrylic diesters. As stated above, more recently Langer et al.11–17 have published several articles investigating the synthesis and use of PBAEs, in particular examining the application of PBAEs as DNA carrier vehicles.11 The appeal of PBAEs in this type of application lies in the readily degradable linkages, the wide range of monomers that can be used, and the ability to choose amines of appropriate pKa that allows them to degrade at physiologically relevant pH.
In none of the previous work documented above is PBAEs utilized in hydrogel formulations. In an effort to explore these crosslinkers in hydrogels, we report here the synthesis, swelling and degradation behavior of hydrogels made from the PBAE crosslinking of NVP and HEMA. These PBAE-crosslinked hydrogels are compared to hydrogels crosslinked with the commonly used poly(ethylene glycol) diacrylate (PEGDA). The PEGDA crosslinkers may undergo degradation through hydrolysis of the ester bond in aqueous environments and lead to the erosion of the material. However, we expect that the PBAE hydrogels will provide more facile degradation.
Experimental section
Materials
Piperazine (PIPz, 99%), HEMA, NVP, azoisobutyronitrile (AIBN), di(ethylene glycol) diacrylate (DEGDA), and poly(ethylene glycol) diacrylate (PEGDA) (two samples of molecular weights of 575 g mol−1 and 700 g mol−1, PEGDA575 and PEGDA700, respectively) were purchased from Aldrich and used as received except: piperazine was ground into a fine powder and dried under vacuum before use, and AIBN was re-crystallized in methanol. The DEGDA was nominally 75% functionalized with acrylate groups; our characterization with 1H nuclear magnetic resonance (NMR) spectroscopy determined that the actual degree of acrylate functionalization was 83%. Sylgard 184, a PDMS-based mold material, was purchased from Dow Corning and used as directed.Equipment
Gel permeation chromatography (GPC) was performed on a system comprising of a Water 717 auto-sampler, a Waters 515 HPLC pump, two Polymer Labs columns (PLgel Mixed C) and a Viscotek LR40 refractometer. Tetrahydrofuran (THF) was used as a continuous phase at a flow rate of 1.0 mL min−1. The system was calibrated with commercial linear polystyrene standards. 1H and 13C NMR spectra of the polymers were obtained on a Bruker Avance 400 MHz spectrometer in CDCl3.A Haake DC10 water bath was modified to hold 36 samples and was continuously operated at 37 °C. Temperature was controlled by the DC10 module but was checked with a thermometer every 48 hours. Fresh bath water was added as needed in such a manner that no more than a 0.5 °C temperature fluctuation occurred.
Crosslinker synthesis
Crosslinkers were produced by reacting DEGDA with PIPz in a round bottom flask at 25 °C. In order to produce lower molecular weight, ca. 800 g mol−1 (approximately 3 repeat units, thus designated as PBAE3), a mole ratio of ∼1.8 : 1 of DEGDA to PIPz was used. For the crosslinker with molecular weight ca. 3000 (approximately 10 repeat units, PBAE10), the ratio was altered to 11 : 10.PBAE3 was produced by reacting 3 ml of DEGDA dissolved in 3 ml of chloroform with 0.6219 g of PIPz in an additional 3 ml of chloroform. PIPz was added in three equivalent stages at 0, 12, and 24 h. The reaction was then continued at 25 °C for an additional 24 h. The solvent was removed under low pressure in order to concentrate the product. Molecular weight was verified with GPC. The number average weight was also verified by NMR spectroscopy and calculated using integrations of the methylene protons from the ethylene glycol unit as an internal standard relative to acrylate vinyl protons.
PBAE10 was produced by reacting 10 ml of DEGDA dissolved in 5 ml of chloroform with 3.685 g of PIPz in an additional 5 ml of chloroform. Reactants were combined in a 50 ml RB flask and continuously stirred for 6 h at 25 °C. Again the molecular weight was verified using GPC and NMR techniques.
Hydrogel synthesis
Samples were produced at concentration of 1 and 3 wt% crosslinker. HEMA and NVP were used as monomers, and 0.2 wt% AIBN as a thermal initiator. The NVP and HEMA were kept at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mass ratio. Each sample was combined and then bubbled with N2 in order to mix, dissolve the initiator, and remove any dissolved oxygen. After bubbling for 30 minutes the samples were allowed to rest for another 30 minutes prior to casting/curing. 0.2 ml of monomer hydrogel mixture was placed into PDMS molds which were then placed under vacuum at 70 °C and allowed to cure for 2 h. Samples were allowed to cool to room temperature before removal from the molds in order to allow for increased rigidity.
:1 mass ratio. Each sample was combined and then bubbled with N2 in order to mix, dissolve the initiator, and remove any dissolved oxygen. After bubbling for 30 minutes the samples were allowed to rest for another 30 minutes prior to casting/curing. 0.2 ml of monomer hydrogel mixture was placed into PDMS molds which were then placed under vacuum at 70 °C and allowed to cure for 2 h. Samples were allowed to cool to room temperature before removal from the molds in order to allow for increased rigidity.
Swelling and degradation studies
The hydrogel materials were swollen in aqueous phosphate buffered saline (PBS) solution at pH = 7.4. Four different crosslinkers were used in this investigation: PEGDA575 (Mn ≈ 575), PEGDA700 (Mn ≈ 700), PBAE3 (Mn ≈ 800), and PBAE10 (Mn ≈ 3000) each at a concentration of 1 and 3 wt%. Five samples of each hydrogel were cast (10 × 10 × 2 mm), soaked in ethanol for 48 hours, and then allowed to air dry before swelling studies were undertaken. Once dried, materials were placed in 5 ml of phosphate buffered saline (PBS) solution at 37 °C and allowed to swell for 16 hours. Readings were taken every 30 minutes for the first four hours, then every hour for the next four hours, then every two hours for the remaining eight hours. After the initial 16 hours of swelling the samples were monitored for degradation. Weights were taken every 24 hours for the next 30 days. Fresh PBS, previously equilibrated at 37 °C, was replaced every 24 hours at the time of measurement.Results and discussion
Crosslinker synthesis
Two PBAE samples were made by the addition of DEGDA and PIPz (Scheme 1); these two are referred to as PBAE3 and PBAE10, given their respective degrees of polymerization. The samples were characterized by GPC and NMR spectroscopy (vide infra). No side reactions were observed, and both liquid samples were yellow/orange in color when concentrated.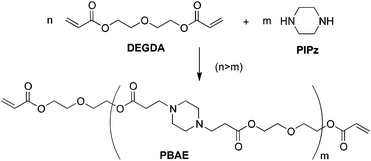 | ||
| Scheme 1 Synthesis of PBAE crosslinkers. | ||
GPC analysis of PBAE3 gave Mn of 830 g mol−1 and a polydispersity (PD) of 1.6. GPC analysis of the PBAE10 gave Mn of 2350 g mol−1 and PD = 2.57. The GPC traces are shown in Fig. 1A and B, respectively. The GPC trace of the PBAE3 clearly shows a series of peaks due to PBAE oligomers. The GPC trace of the PBAE10 shows a peak at lower elution volumes compared with the PBAE3 sample, indicating a higher molecular weight.
 | ||
| Fig. 1 GPC traces of both poly(β-amino ester) crosslinkers: (A) PBAE3 and (B) PBAE10. | ||
The 1H NMR spectra of the same samples are shown in Fig. 2A and B. Integration of the NMR data in Fig. 2 allows for the determination of the molecular weight for each of the crosslinker samples. In particular, this can be achieved using the integrations of the vinyl protons (peaks from ∼5.8 to 6.5 ppm, labeled a′, a and b) and the ethylene protons (peaks from ∼3.5–4.5 ppm, labeled c and d). A comparison of these integrations provides an average molecular weight of 560 g mol−1 for the PBAE3 and 3050 g mol−1 for the PBAE10. These are similar to the Mn values determined by GPC.
 | ||
| Fig. 2 1H NMR spectra of (A) PBAE3 and (B) PBAE10. | ||
Gel fraction
The gel fractions of each hydrogel were measured by determining the weights of the gels before and after soaking in ethanol. Specifically, the initial mass was taken after casting (M0), at 48 hours in ethanol (MEtOH), and once dried (MD). From these data it was determined how much non-covalently bound material (e.g. monomers) was removed due to swelling in ethanol, and thus the gel fraction. Network gel fractions were calculated from eqn (1) and are presented in Table 1. In each case the gel fractions were quite high, with fractions being lower at the higher crosslinker concentration. The gel fraction for the PBAE3 and PBAE10 was slightly higher at the 3% crosslinker when compared to the PEG crosslinkers.| Gel fraction | ||
|---|---|---|
| Crosslinker | 1% | 3% |
| PEGDA575 | 93 | 89 |
| PEGDA700 | 98 | 90 |
| PBAE3 | 97 | 95 |
| PBAE10 | 100 | 98 |
In accounting for the soluble fraction, it should be noted that given the differing reactivities of NVP and HEMA, as observed in their reactivity ratios (rNVP = 0.05 and rHEMA = 3.12),20 one expects HEMA to be consumed well before NVP in a copolymerization. As previously discussed in similar hydrogels based on terpolymerizations of NVP, HEMA and ethylene glycol dimethacrylate by Davis and Huglin,21 this could lead to compositional drift and hence homopolymer (non-crosslinked) poly(NVP) being produced towards the end of the polymerization. This poly(NVP) would be extractable from the gel fraction. In our case, even where the crosslinker is an acrylate, such an effect could be in operation; in fact, the soluble fraction was analyzed by 1H NMR spectroscopy and was found to contain a mixture of NVP and poly(NVP).
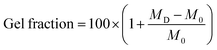 | (1) |
Swelling studies
The hydrogel materials were swollen in phosphate buffered saline (PBS) solution, pH = 7.4, in order to observe the influence of increasing crosslinker chain length on swelling, as well as the significance of the amine in the polymer backbone. PBS solution mimics the physiological conditions of the human body and thus swelling and degradation were both studied under these conditions. Each of the four crosslinkers was used to investigate both swelling and degradation (viz.PEGDA575, PEGDA700, PBAE3, and PBAE10) at concentrations of 1 wt% and 3 wt%. Analysis of hydrogel swelling was undertaken by following methods developed by Peppas et al.2 and Baysal et al.22 through the use of eqn (2). This fits hydrogel swelling percentage (SP) to a Fickian model.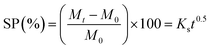 | (2) |
Swelling was also fitted to a non-Fickian (anomalous) model using eqn (3):
 | (3) |
Swelling percent trends for 1 wt% and 3 wt% crosslinker are presented in Fig. 3 and 4, respectively. From these data it is observed that for all samples swelling is achieved in approximately 8 h and then plateau. The PBAE10 crosslinker resulted in a greater degree of swelling over the same period of time compared to PBAE3. Hydrogels crosslinked with 1% PBAE10 absorbed their own weight in water (SP = 100%) in an hour and a half compared to PBAE3 which required two hours. Similarly, at 3% crosslinker the PBAE10 doubled in mass at one and a half hours while PBAE3 took two hours. These results are due to the lower crosslink density found in the PBAE10-crosslinked gels. A comparison of the two PEGDA-crosslinked gels finds a smaller difference than that found with the PBAE-based gels. This may be due to the difference in the molecular weight of the two PEGDA crosslinkers being smaller than the difference in molecular weight of the PBAE crosslinkers. Interestingly, PEGDA575 swells slightly faster and to a greater extent than PEGDA700 at 1% loading while the trend is reversed at 3% crosslinker.
 | ||
| Fig. 3 Hydrogel swelling percent (SP) over initial 16 h with 1% crosslinker. | ||
 | ||
| Fig. 4 Hydrogel swelling percent (SP) over initial 16 h with 3% crosslinker. | ||
Increasing crosslinker from 1 to 3 wt% showed a small decrease in the hydrogels ability to swell. PBAE3 and PBAE10 obtained maximum swelling percentages of 161 and 262% at a concentration of 1% and only 161 and 233% at a concentration of 3%, respectively. These findings reflect that an increased amount of crosslinker leads to a higher crosslinking density and thus a stiffer material which is not capable of imbibing as much solvent.
Swelling profiles were modeled as Fickian by plotting of swelling percent against time to the half power. A straight line was obtained for the first 25% of swelling time (4 h), which represents the Ks for each hydrogel. The model fit was excellent with all r2 values being above 0.9.
Non-Fickian curve fits were also constructed and the values of K and n found that best fit eqn (3). Values for Ks, K, and n are reported below in Table 2. For each of the samples, values of n are in the 0.55 to 0.63 range, indicating a slight deviation from Fickian behavior. For the PEGDA samples, the value of n decreases slightly when the amount of crosslinker is increased from 1 to 3 wt%. The opposite trend is observed with the PBAE samples. This may be due to the greater hydrophilicity of the PBAE chains because of the presence of the H-bonding amino groups; when the PBAE concentration is increased, a more pronounced hydrophilic effect is expected to be observed, hence a move away from Fickian diffusion.
| Crosslinker | Cross-linker (%) | K s/s−1 | K/s−1 | n |
|---|---|---|---|---|
| PEGDA575 | 1 | 1.11 | 3.90 × 10−3 | 0.58 |
| PEGDA575 | 3 | 1.08 | 6.57 × 10−3 | 0.55 |
| PEGDA700 | 1 | 1.00 | 3.93 × 10−3 | 0.58 |
| PEGDA700 | 3 | 1.12 | 5.39 × 10−3 | 0.55 |
| PBAE3 | 1 | 1.17 | 3.99 × 10−3 | 0.57 |
| PBAE3 | 3 | 1.11 | 2.31 × 10−3 | 0.63 |
| PBAE10 | 1 | 1.60 | 2.23 × 10−3 | 0.61 |
| PBAE10 | 3 | 1.52 | 2.05 × 10−3 | 0.63 |
Degradation studies
After the initial 16 hours of swelling the samples were monitored for degradation. The mass for each sample was measured every 24 hours for the next 30 days. The PBS solution was replaced every 24 hours, at the time of measurement. The mass loss was monitored as a percentage of peak weight as shown in eqn (4). | (4) |
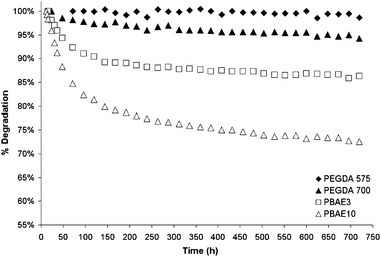
Degradation of hydrogels was monitored from the point of maximum water absorption, which varied for each material. The degradation curves are presented below in Fig. 5 and 6. The final mass loss was determined through two methods: (1) in the swollen state and (2) by drying all samples for several days and measuring a final mass. The results are summarized in Table 3.
| Crosslinker | Cross-linker (%) | Swollen | Dry mass |
|---|---|---|---|
| PEGDA575 | 1 | 7 | 7 |
| PEGDA700 | 1 | 5 | 11 |
| PBAE3 | 1 | 11 | 11 |
| PBAE10 | 1 | 31 | 20 |
| PEGDA575 | 3 | 1 | 3 |
| PEGDA700 | 3 | 6 | 4 |
| PBAE3 | 3 | 14 | 14 |
| PBAE10 | 3 | 27 | 20 |
 | ||
| Fig. 5 Degradation (represented as a percent mass lost from maximum swollen mass) of hydrogels with 1% crosslinker. | ||
 | ||
| Fig. 6 Degradation (represented as a percent mass lost from maximum swollen mass) of hydrogels with 3% crosslinker. | ||
Similar trends are found for degradation as was observed for the swelling. Longer crosslinkers degraded much more rapidly and to a greater extent than crosslinkers with a shorter chain length (i.e. PEGDA). The materials with longer crosslinkers absorbed much more water and thus increase the hydrolysis (and hence degradation) rates. Increasing from 1% to 3% crosslinker reduced the degree of degradation much the same as it did the swelling. This is once again most likely due to the increase rigidity of the material and the lower amount of water present to facilitate degradation.
Conclusions
We have shown that diacrylated poly(β-amino ester)s, produced from DEGDA and PIPz, can be used as crosslinkers in the synthesis of NVP- and HEMA-based hydrogels. Hydrogels were synthesized at 1 and 3 wt% crosslinker and found to undergo faster swelling and reached a greater final swelling percentage than hydrogels made with similar molecular weight diacrylate poly(ethylene glycol)-based crosslinkers. Degradation rates of the PBAE-based hydrogels were also faster than those made by the PEGDA crosslinkers, in part due to their greater water uptake facilitating hydrolysis within the network.Acknowledgements
We thank the Center for Advanced Materials Processing (CAMP) at Clarkson University, a New York State Center for Advanced Technology, and the GK-12 Project-Based Learning Partnership Program at Clarkson University, funded by the NSF (Program # DGE-0338216), for support. We also thank Brodie Rutherglen for technical assistance in some of the polymer syntheses.Notes and references
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1880 CrossRef.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- M. Iza, G. Stoianovici, L. Viora, J. L. Grossiord and G. Couarraze, J. Controlled Release, 1998, 52, 41–51 CrossRef CAS.
- L. M. Geever, D. M. Devine, M. J. D. Nugent, J. E. Kennedy, J. G. Lyons and C. L. Higginbotham, Eur. Polym. J., 2006, 42, 69–80 CAS.
- J. S. Oh, J. M. Kim, K.-J. Lee and Y. C. Bae, Eur. Polym. J., 1999, 35, 621–630 CrossRef CAS.
- L. M. Muratore, K. Steinhoff and T. P. Davis, J. Mater. Chem., 1999, 9, 1687–1691 RSC.
- D. M. Devine and C. L. Higginbotham, Polymer, 2003, 44, 7851–7860 CrossRef CAS.
- K. Guo and C.-C. Chu, Biomaterials, 2007, 28, 3284–3294 CrossRef CAS.
- A. Kelner and E. H. Schacht, J. Controlled Release, 2005, 101, 13–20 CrossRef.
- A. A. Thomas, I. T. Kim and P. F. Kiser, Tetrahedron Lett., 2005, 46, 8921–8925 CrossRef CAS.
- D. M. Lynn and R. Langer, J. Am. Chem. Soc., 2000, 122, 10761–10768 CrossRef CAS.
- D. M. Lynn, D. G. Anderson, D. Putnam and R. Langer, J. Am. Chem. Soc., 2001, 123, 8155–8156 CrossRef CAS.
- A. Akinc, D. G. Anderson, D. M. Lynn and R. Langer, Bioconjugate Chem., 2003, 14, 979–988 CrossRef CAS.
- D. G. Anderson, D. M. Lynn and R. Langer, Angew. Chem., Int. Ed., 2003, 42, 3153–3158 CrossRef CAS.
- S. R. Little, D. M. Lynn, Q. Ge, D. G. Anderson, S. V. Puram, J. Chen, H. N. Eisen and R. Langer, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9534–9539 CrossRef CAS.
- D. G. Anderson, C. A. Tweedie, N. Hossain, S. M. Navarro, D. M. Brey, K. J. Van Vliet, R. Langer and J. A. Burdick, Adv. Mater., 2006, 18, 2614–2618 CrossRef CAS.
- G. T. Zugates, D. G. Anderson, S. R. Little, I. E. B. Lawhorn and R. Langer, J. Am. Chem. Soc., 2006, 128, 12726–12734 CrossRef CAS.
- K. Yoda and T. Toda, Bull. Chem. Soc. Jpn., 1968, 41, 2519–2521.
- F. Danusso and P. Ferruti, Polymer, 1970, 11, 88–113 CrossRef CAS.
- M. A. Al-Issa, T. P. Davis, M. B. Huglin and D. C. F. Yip, Polymer, 1985, 26, 1869–1874 CAS.
- T. P. Davis and M. B. Huglin, Macromolecules, 1989, 22, 2824–2829 CrossRef CAS.
- B. Tasdelen, N. Kayaman-Apohan, O. Guven and B. M. Baysal, J. Appl. Polym. Sci., 2004, 91, 911–915 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |