 Open Access Article
Open Access ArticleMicroscopic and spectroscopic studies of thermally enhanced electrospun PMMA micro- and nanofibers
Sean
Pelfrey
a,
Travis
Cantu
a,
Michael R.
Papantonakis
b,
Duane L.
Simonson
b,
R. Andrew
McGill
b and
Javier
Macossay
*a
aDepartment of Chemistry, University of Texas Pan American, Edinburg, TX 78539, USA. E-mail: jmacossay@utpa.edu; Fax: (+956) 384-5006; Tel: (+956) 381-3377
bNaval Research Laboratory Code 6365, 4555 Overlook Avenue, SW, Washington, DC 20375, USA
First published on 26th March 2010
Abstract
Carbon nanofibers (CNFs) have been incorporated into poly(methyl methacrylate) (PMMA) through electrospinning. The resulting micro- and nanofibers have been characterized by scanning electron microscopy (SEM), which confirmed fiber formation and demonstrated a core-sheath structure of the PMMA fibers. thermogravimetric analysis (TGA) was used to obtain the thermal properties of the materials, indicating an enhancement in the thermal properties of the composite fibers. In addition, Fourier transform infrared spectroscopy (FTIR) was utilized to investigate the interactions of PMMA micro- and nanofibers with CNFs, demonstrating the preferred sites of intermolecular interactions between the polymer matrix and the filler.
Introduction
Electrospinning is a process used to produce micro- and nanofibers that was originally reported by Formhals in the 1930s and 1940s.1–5 The electrospinning process consists of placing a polymer solution in a syringe, which is directly connected to a high power supply. This high power supply is set to generate a high voltage difference, usually between 5 kV and 30 kV, between the syringe needle and a grounded target. As the polymer solution is ejected from the syringe, the electrical charges on the polymer promote the evaporation of the solvent and the traveling of the polymer through a chaotic pattern that is known as the Taylor cone, thus forming a dry polymer fiber which deposits on the grounded target (Fig. 1). | ||
| Fig. 1 The electrospinning setup. | ||
However, electrospinning was not investigated thoroughly until the advent of nanotechnology, when it was recognized that the process is ideal for obtaining inorganic and polymeric fibers with micrometre and nanometre size diameters.6–8 Moreover, it is recognized that smaller fiber diameters provide high surface areas7,8 and enhanced mechanical properties,9–11 and since electrospinning is currently the only available technique that can provide fibers in the nanometre scale7 in significant amounts, an enormous amount of research using the electrospinning process is being performed. The potential uses of nanofibers produced by electrospinning are quite extensive, such as tissue scaffolds, enzyme and catalyst supports, filtration media, military protective clothing, sensors, electronic and optical devices, textiles and reinforced nanocomposites.7,8,12–20
Carbon nanomaterials include single walled carbon nanotubes (SWNTs), multiwalled carbon nanotubes (MWNTs) (known together as CNTs) and carbon nanofibers (CNFs). These compounds have different structures and morphologies, so while SWNTs are materials made of hollow single rolled up graphene sheets, MWNTs consist of multiple concentric sheets with a hollow core. In contrast, CNFs include hollow or solid graphene sheets with stacked cones, stacked cups or plate morphologies.21 CNTs and CNFs are materials with conjugated C–C sp2 bonds, which result in strong covalent bonds and a remarkable hexagonal network that is capable of distortions for relaxing stress.21,22 As a consequence, these carbon allotropes have a unique combination of high strength, flexibility and stiffness, which makes them quite attractive for reinforcing polymer matrices.23–28 Theoretical and experimental studies have indicated that SWNTs have a Young modulus of approximately 1 TPa and tensile strength from 13–53 GPa, while MWNTs possess a Young modulus at 0.9 TPa and tensile strength up to 150 GPa. In contrast, CNFs present modest mechanical properties with Young modulus varying from 4–6 GPa and tensile strength around 0.2–0.3 GPa.21,22,29,30
Most of the CNTs incorporation into polymers has been to investigate the mechanical properties of the composite materials. Furthermore, CNFs are more readily available than their analogous CNTs, but despite this fact, CNFs are not being thoroughly studied because of the enhanced properties encountered in CNTs. However, it is likely that CNFs will have an important impact in the short term as a polymer reinforcement additive. Therefore, this paper reports on the incorporation of CNFs into PMMA through electrospinning with the objective of obtaining thermally enhanced polymer fibers. Moreover, the properties of the resulting micro- and nanofibers were investigated by SEM, TGA, and FTIR spectroscopy.
Results and discussion
Micro- and nanofiber morphology
Fig. 2 shows an SEM micrograph of a PMMA microfiber, which presents a homogeneous smooth surface and diameter of 1.4 μm. Average diameters of PMMA nanofibers electrospun in our laboratory are 543 ± 140 nm and is reported elsewhere.31Fig. 3(a and b) presents PMMA-CNFs nanofibers which have a non uniform “trunk-like” morphology. Non uniform and rough surface morphologies with variation in fiber diameters have been reported in polymer-CNTs nanofibers produced by electrospinning,32–34 but similar “trunk-like” morphologies had not been observed. Electrospinning is a process that is affected by changes in the electrical conductivity of the polymer solutions, which can cause variations in fiber diameters and morphologies.35 Since CNTs and CNFs do alter the electrical conductivity of solvents and polymers, it is probable that addition of CNFs into the PMMA solution changed the electrical conductivity, resulting in the observed morphologies. | ||
| Fig. 2 PMMA microfiber presenting a 1.4 μm diameter. Scale bar 1 μm; magnification 19K ×. | ||
 | ||
| Fig. 3 PMMA-CNFs nanofibers. (a) Scale bar 200 nm; magnification 35K ×. (b) Scale bar 1 μm; magnification 9K ×. | ||
Fig. 4 shows additional PMMA-CNFs nanofibers, where interesting features are observed. This micrograph demonstrates a sheath (a) and core structure in the electrospun nanofibers, which to the knowledge of the authors, had not been reported for nanofibers. The formation of two distinct sheath and core structures in fibers was first reported in 1970.9 In this paper, the authors proposed that the thin textured sheath was formed because the shearing and tensile forces exerted on the fiber surface during extrusion, and that the texture was “frozen” because of the rapid cooling of the fiber's exposed area. Therefore, the formation of a two component system (sheath and core) is a direct consequence of the different conditions encountered by the inner and outer layers of the fibers. Electrospinning, similar to some extent to extrusion, is a process that exerts shear stresses on the fibers as these are being obtained. Moreover, solvents evaporate readily when high voltages are applied, so formation of sheath and core structures in nanofibers obtained by electrospinning seem to have the same origin as fibers obtained by extrusion.
 | ||
| Fig. 4 PMMA-CNFs nanofibers with their sheath structure (a); evidence of a CNFs bundle (b) and individual CNFs (c) are also observed. Scale bar 1 μm; magnification 12K ×. | ||
Furthermore, research on microfibers demonstrated a strong correlation of the relative ratio of the sheath and core components of the fiber with the tensile strength and Young's modulus, which resulted in a marked increase of these properties with smaller fiber diameters.9,10 In a few words, the sheath structure contained well aligned crystallites along the fiber axis, while the core contained misaligned crystallites due to anisotropic thermal contractions. Consequently, the smaller fiber diameter causes a higher sheath/core ratio that is reflected in higher Young's modulus and tensile strengths. Therefore, observation of these two phases in nanofibers is significant and could spark research efforts to understand and control them. It is important to mention that the PMMA used in these studies is atactic and there is no crystallinity to be determined in these systems, but nevertheless, the sheath and core structures are observed in the nanofibers. Furthermore, Fig. 4 presents a CNF bundle (b) and individual CNFs (c) aligned with the fiber axis, which indicate that the sonication conditions need to be improved in order to achieve complete CNF dispersion and therefore the full benefits of their incorporation into the PMMA matrix.
Thermal properties
Thermogravimetric analysis of PMMA and PMMA-CNFs micro- and nanofibers (Fig. 5) demonstrate that both materials decompose in a single step. However, the onset decomposition temperature for PMMA is 293 °C, while PMMA-CNFs decompose at 348 °C. It is evident from these results that incorporation of CNFs causes a significant increase in thermal stability, suggesting the possibility of preparing electrospun micro- and nano- composite fibers with enhanced thermal properties. Moreover, it is known that poor dispersion is detrimental for thermal and mechanical properties of polymer-carbon nanotubes composites,26 and since SEM in Fig. 4 revealed that some CNFs nanofibers were not completely dispersed, there is a possibility of obtaining even higher thermal stabilities in the materials reported in this paper.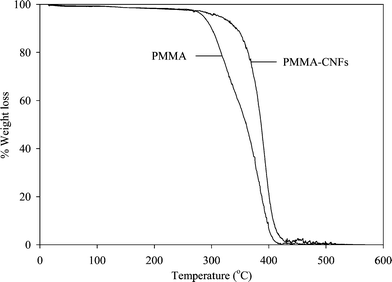 | ||
| Fig. 5 Thermogravimetric analysis of electrospun PMMA and PMMA-CNFs. | ||
Spectroscopic analysis
In order to learn and understand the intermolecular interactions between the polymer and CNFs, FTIR analysis was performed. There were no major differences in the spectra of PMMA and PMMA-CNFs above 1800 cm−1, so this part of the spectra is not included in the following discussion. However, there are interesting changes in the region from 1800 cm−1 to 900 cm−1 (Fig. 6), and the PMMA peaks were assigned based on previously reported work.36,37 The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O ester in PMMA appears at 1710 cm−1 with a small shoulder at 1722 cm−1. However, this peak shifted to 1727 cm−1 for PMMA-CNFs, clearly indicating the existence of chemical interactions between the CO group and CNFs. Furthermore, the CO band for PMMA-CNFs is broader than the corresponding band for electrospun PMMA, which suggests two different chemical environments in the CO.37 It is proposed that one of the chemical environments be attributed to the CO of PMMA molecules in the bulk of the polymer matrix, while the other chemical environment should be the CO of PMMA at the interface, which are the molecules directly interacting with the π bonds of CNFs. A similar increase in peak width was observed in the band at 1429 cm−1 for PMMA, which corresponds to the skeletal CH2 deformation.37 The analogous band for PMMA-CNFs was moved towards higher wavenumbers and broadened to a 1432 cm−1–1444 cm−1 range. The bands at 1233 cm−1 and 1265 cm−1 for PMMA, which correspond to the C–C–O in the ester group,36 did not suffer significant modifications of their width, but were shifted to 1238 cm−1 and 1268 cm−1 for PMMA-CNFs. Finally, the bands that correspond to C–O–C of the methoxy group and skeletal C–C for PMMA appear at 1137 cm−1 and 1186 cm−1. These bands were strongly affected in their shape and shifted towards 1146 cm−1 and 1191 cm−1 for PMMA-CNFs. It is evident from these FTIR data that there are several types of chemical interactions between PMMA and CNFs, which include π–π interactions between the CO and CNFs, σ–π interactions between the skeletal C–C and CNFs and n electrons–π between C–O–C and CNFs. Nevertheless, the most significant shift towards higher wavenumbers was observed in the CO group (Δν + 17), suggesting that the strongest chemical interactions are between the π–π bonds of the CO and the CNFs.
O ester in PMMA appears at 1710 cm−1 with a small shoulder at 1722 cm−1. However, this peak shifted to 1727 cm−1 for PMMA-CNFs, clearly indicating the existence of chemical interactions between the CO group and CNFs. Furthermore, the CO band for PMMA-CNFs is broader than the corresponding band for electrospun PMMA, which suggests two different chemical environments in the CO.37 It is proposed that one of the chemical environments be attributed to the CO of PMMA molecules in the bulk of the polymer matrix, while the other chemical environment should be the CO of PMMA at the interface, which are the molecules directly interacting with the π bonds of CNFs. A similar increase in peak width was observed in the band at 1429 cm−1 for PMMA, which corresponds to the skeletal CH2 deformation.37 The analogous band for PMMA-CNFs was moved towards higher wavenumbers and broadened to a 1432 cm−1–1444 cm−1 range. The bands at 1233 cm−1 and 1265 cm−1 for PMMA, which correspond to the C–C–O in the ester group,36 did not suffer significant modifications of their width, but were shifted to 1238 cm−1 and 1268 cm−1 for PMMA-CNFs. Finally, the bands that correspond to C–O–C of the methoxy group and skeletal C–C for PMMA appear at 1137 cm−1 and 1186 cm−1. These bands were strongly affected in their shape and shifted towards 1146 cm−1 and 1191 cm−1 for PMMA-CNFs. It is evident from these FTIR data that there are several types of chemical interactions between PMMA and CNFs, which include π–π interactions between the CO and CNFs, σ–π interactions between the skeletal C–C and CNFs and n electrons–π between C–O–C and CNFs. Nevertheless, the most significant shift towards higher wavenumbers was observed in the CO group (Δν + 17), suggesting that the strongest chemical interactions are between the π–π bonds of the CO and the CNFs.
 | ||
| Fig. 6 FTIR spectra of electrospun PMMA and PMMA-CNFs. | ||
Experimental
Materials
PMMA Mw = 350 K was purchased from Alfa Aesar. CNFs PR-24-XT LHT LD were a kind donation from Applied Sciences, Inc., Cedarville, OH. Tetrahydrofuran 99+% (THF) and N,N-dimethylformamide 99.8% (DMF) were acquired from Sigma-Aldrich and used without further purification.PMMA and PMMA-CNFs dispersions preparation
Polymer samples were prepared by dissolving 7.5 wt% PMMA in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 THF–DMF solvent mixture. To promote CNFs dispersion within the PMMA solution, samples containing 1 wt% CNFs/7.5 wt% PMMA in a 1:1 THF–DMF solvent system were prepared and sonicated for 15 min in a Hielscher Ultrasound Technology UIP1000hd sonicator.
:1 THF–DMF solvent mixture. To promote CNFs dispersion within the PMMA solution, samples containing 1 wt% CNFs/7.5 wt% PMMA in a 1:1 THF–DMF solvent system were prepared and sonicated for 15 min in a Hielscher Ultrasound Technology UIP1000hd sonicator.
Electrospinning process
The polymer and composite fibers were obtained using a 10 mL glass syringe with a 22 needle gauge (0.7 mm OD × 0.4 mm ID) at a flow rate of 0.05 mL min−1, which was controlled using a KDS 210 pump, KD Scientific Holliston Inc., MA. The high power supply was an ES30P-5W purchased from Gamma High Voltage Research, Ormond Beach, FL. The equipment was attached to the needle tip through an alligator clip and a voltage difference of 15 kV was used; the grounded target was placed at 24.5 cm from the needle tip.Characterization
Scanning electron microscopy (SEM) was used to investigate the surface morphology of the fibers obtained. The samples were sputter-coated with 10 nm of gold and observed using a Carl Zeiss SMT Supra 55 SEM. Thermogravimetric analysis (TGA) was performed using a Perkin Elmer TGA 7 under nitrogen with a flow rate of 20 mL min−1. The samples were ramped from room temperature to 600 °C at a scanning rate of 10 °C min−1 using platinum pans. Fourier transform infrared (FTIR) spectra were obtained after 10 scans from 4000 cm−1 to 400 cm−1 using a Bruker Equinox 55.Conclusion
PMMA micro- and nanofibers were obtained through electrospinning. Incorporation of individual and bundles of CNFs within these PMMA fibers was confirmed by SEM. Furthermore, SEM analysis evidenced the presence of a core-sheath structure in the electrospun nanofibers. Incorporation of CNFs into PMMA resulted in a 55 °C increase on the onset decomposition temperature, demonstrating an enhancement on the thermal properties of composite fibers. Furthermore, FTIR spectra confirmed shifts in the wavenumbers of several PMMA groups, suggesting numerous intermolecular interactions between the polymer matrix and CNFs. However, the most prominent changes indicate that the strongest chemical interactions are between the π–π bonds of the CO and the CNFs.
Acknowledgements
Financial support for this work from the Welch Foundation grant # BG-0017 and NIH-NIGMS-NIA grant # 1SC2AG036825-01 for JM are gratefully acknowledged. The authors are also indebted for the support from the Office of Naval Research.References
- A. Formhals, US Pat., 1,975,504, 1934.
- A. Formhals, US Pat., 2,160,962, 1939.
- A. Formhals, US Pat., 2,187,306, 1940.
- A. Formhals, US Pat., 2,323,025, 1943.
- A. Formhals, US Pat., 2,349,950, 1944.
- D. Li and Y. Xia, Adv. Mater., 2004, 16, 1151–1170 CrossRef CAS.
- A. Greiner and J. H. Wendorff, Angew. Chem., Int. Ed., 2007, 46, 5670–5703 CrossRef CAS.
- Z. M. Huang, Y. Z. Zhang, M. Kotaki and S. Ramakrishna, Compos. Sci. Technol., 2003, 63, 2223–2253 CrossRef CAS.
- K. Kawamura and G. M. Jenkins, J. Mater. Sci., 1970, 5, 262–267 CrossRef CAS.
- B. F. Jones and R. G. Duncan, J. Mater. Sci., 1971, 6, 289–293 CrossRef CAS.
- M. K. Shin, S. I. Kim, S. J. Kim, S. K. Kim, H. Lee and G. M. Spinks, Appl. Phys. Lett., 2006, 89, 231929 CrossRef.
- J. A. Matthews, G. E. Wnek, D. G. Simpson and G. L. Bowlin, Biomacromolecules, 2002, 3, 232–238 CrossRef CAS.
- G. E. Wnek, M. E. Carr, D. G. Simpson and G. L. Bowlin, Nano Lett., 2003, 3, 213–216 CrossRef CAS.
- H. Yoshimoto, Y. M. Shin, H. Terai and J. P. Vacanti, Biomaterials, 2003, 24, 2077–2082 CrossRef CAS.
- E. R. Kenawy, G. L. Bowlin, K. Mansfield, J. Layman, D. G. Simspon, E. H. Sanders and G. E. Wnek, J. Controlled Release, 2002, 81, 57–64 CrossRef CAS.
- A. G. MacDiarmid, W. E. Jones Jr., I. D. Norris, J. Gao, A. T. Johnson Jr., N. J. Pinto, J. Hone, B. Han, F. K. Ko, H. Okuzaki and M. Llaguno, Synth. Met., 2001, 119, 27–30 CrossRef CAS.
- B. Ding, J. Kim, Y. Miyazaki and S. Shiratori, Sens. Actuators, B, 2004, 101, 373–380 CrossRef.
- R. Kessick and G. Tepper, Sens. Actuators, B, 2006, 117, 205–210 CrossRef.
- J. Macossay, J. H. Leal, A. Kuang and R. E. Jones, Polym. Adv. Technol., 2006, 17, 391–394 CrossRef CAS.
- Y. Dror, W. Salalha, R. L. Khalfin, Y. Cohen, A. L. Yarin and E. Zussman, Langmuir, 2003, 19, 7012–7020 CrossRef CAS.
- J. P. Salvetat, J. M. Bonard, N. H. Thomson, A. J. Kulik, L. Forró, W. Benoit and L. Zuppiroli, Appl. Phys. A: Mater. Sci. Process., 1999, 69, 255–260 CrossRef CAS.
- J. P. Salvetat-Delmotte and A. Rubio, Carbon, 2002, 40, 1729–1734 CrossRef CAS.
- D. Qian, E. C. Dickey, R. Andrews and T. Rantell, Appl. Phys. Lett., 2000, 76, 2868–2870 CrossRef CAS.
- P. Calvert, Nature, 1999, 399, 210–211 CrossRef CAS.
- P. M. Ajayan, O. Stephan, C. Colliex and D. Trauth, Science, 1994, 265, 1212–1214 CrossRef CAS.
- X. L. Xie, Y. W. Mai and X. P. Zhou, Mater. Sci. Eng., R, 2005, 49, 89–112 CrossRef.
- L. Jin, C. Bower and O. Zhou, Appl. Phys. Lett., 1998, 73, 1197–1199 CrossRef CAS.
- J. Robertson, Mater. Today, 2004, 7, 46–52 CrossRef CAS.
- B. G. Demczyk, Y. M. Wang, J. Cumings, M. Hetman, W. Han, A. Zettl and R. O. Ritchie, Mater. Sci. Eng., A, 2002, 334, 173–178 CrossRef.
- M. Meo and M. Rossi, Compos. Sci. Technol., 2006, 66, 1597–1605 CrossRef CAS.
- C. Carrizales, S. Pelfrey, R. Rincon, T. M. Eubanks, A. Kuang, M. J. McClure, G. L. Bowlin and J. Macossay, Polym. Adv. Technol., 2008, 19, 124–130 CrossRef CAS.
- J. S. Jeong, J. S. Moon, S. Y. Jeon, J. H. Park, P. S. Alegaonkar and J. B. Yoo, Thin Solid Films, 2007, 515, 5136–5141 CrossRef CAS.
- J. H. Sung, H. S. Kim, J. H. Jin, J. H. Choi and I. J. Chin, Macromolecules, 2004, 37, 9899–9902 CrossRef CAS.
- L. Q. Liu, D. Tasis, M. Prato and H. D. Wagner, Adv. Mater., 2007, 19, 1228–1233 CrossRef CAS.
- K. H. Lee, H. Y. Kim, H. J. Bang, Y. H. Jung and S. G. Lee, Polymer, 2003, 44, 4029–4034 CrossRef CAS.
- O. N. Tretinnikov and K. Ohta, Macromolecules, 2002, 35, 7343–7353 CrossRef CAS.
- A. Matsushita, Y. Ren, K. Matsukawa, H. Inoue, Y. Minami, I. Noda and Y. Ozaki, Vib. Spectrosc., 2000, 24, 171–180 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |