DOI:
10.1039/C0PY00025F
(Paper)
Polym. Chem., 2010,
1, 854-859
Received
22nd January 2010
, Accepted 15th February 2010
First published on
29th March 2010
Abstract
Well-defined temperature-responsive polymers were covalently conjugated to model proteins by two consecutive Michael addition thiol–ene reactions. Poly(N-isopropylacrylamide) (PNIPAM) prepared by reversible addition–fragmentation chain transfer (RAFT) polymerization was aminolyzed to yield thiol-terminated chains that were subsequently reacted with excess 1,8-bis-maleimidodiethyleneglycol. The resulting maleimide-terminated polymer was reacted with bovine serum albumin and ovalbumin to yield polymer–protein conjugates by a “grafting-to” approach. The thermoresponsive nature of PNIPAM was conferred to the conjugate, as demonstrated by dynamic light scattering analysis that indicated the formation of intermolecular aggregates at elevated temperatures.
Introduction
Modification of biomacromolecules (e.g., proteins, nucleic acids, polysaccharides) with synthetic polymers is a viable means to increase efficacy for in vivo and in vitro applications. In many cases, important characteristics (e.g., solubility, biocompatibility, stability, activity) of the biological component are conserved or enhanced as a result of the nature of the synthetic component.1–11 Typical conjugation methods primarily consist of grafting-from, which describes the polymerization of monomer from a biomolecule capable of initiation, and grafting-to, which involves immobilization of preformed polymer by reactive coupling.9,10 Both methods are accompanied by advantages and disadvantages, and each has proven viable for the efficient preparation of well-defined bioconjugates. Herein, we describe a method for the facile preparation of polymer–protein conjugates by grafting well-defined maleimide-terminated polymers to thiol-containing proteins by highly efficient thiol–ene reactions.
The preparation of well-defined polymers for conjugation to biomolecules by a grafting-to approach requires polymerization conditions that ensure the synthesis of low polydispersity, controlled molecular weight polymer with the functionality necessary to allow subsequent conjugation. Due to a proven ability to yield a variety of tailor-made and well-defined polymers under mild conditions, controlled radical polymerization (CRP) techniques have become invaluable in the field of bioconjuation.7,8,11 The most commonly employed techniques to prepare polymers for subsequent immobilization to biomacromolecules have included atom transfer radical polymerization (ATRP)12,13 and reversible addition–fragmentation chain transfer (RAFT)14 polymerization. The former has proven highly successful for the synthesis of polymer–protein conjugates.15–18 RAFT polymerization has also demonstrated promise in this area,19–28 though few reports29,30 have capitalized on the sulfur chemistry inherent to the polymerization process. The most common RAFT chain transfer agents (CTAs) contain thiocarbonylthio groups that are retained as the ω-terminus in the resulting polymers. These terminal functionalities are easily converted to thiols31–44 or other functional end groups45,46 that can be exploited for a variety of purposes.47
Thiol chemistry is also extensively applied for modification of biological macromolecules, since thiols participate in several particularly efficient chemical transformations, including disulfide coupling/exchange and alkylation by addition to halogenated alkanes, alkenes, or α–β unsaturated carbonyl compounds (i.e., Michael addition). The latter is an excellent example of an efficient thiol–ene reaction suitable for the preparation and functionalization of a wide variety of macromolecules.48 Among various amino acids commonly targeted for modification of proteins, cysteine is particularly attractive for site-specific attachment since there are generally very few reactive cysteine residues in native proteins. Therefore, reactions that target only these thiol groups can facilitate site-specific functionalization and enable regulation of binding events and the rational optimization of protein properties.49 Vinyl sulfone,29,50–53 maleimide,16–19,27,41,54 and activated disulfide17,20,21,26,55–59 end groups are among the most common thiol-reactive groups to have been successfully employed in bioconjugation procedures.
Polymers with thiol-reactive end groups can be prepared by either postpolymerization modification of a preformed polymer16,20,28,29,54 or polymerization with an initiating species that contains an active or latent thiol-reactive moiety.21 Previously we demonstrated that difunctional bismaleimides could be employed to introduce maleimide end groups to RAFT-generated polymers.60 This method enabled the synthesis of various functional telechelics and modular block copolymers and should be extendable to the preparation of polymer conjugates with thiol-containing proteins.61,62 Herein, we demonstrate that polymers prepared by RAFT can be readily coupled to model thiol-containing proteins. End group aminolysis of RAFT-generated poly(N-isopropylacrylamide) (PNIPAM) led to thiol-terminated chains that were subsequently reacted with an excess of a bismaleimide to yield maleimide-terminated PNIPAM (PNIPAM–M). The functional polymer was successfully conjugated to bovine serum albumin (BSA) or ovalbumin (OVA), and the resulting conjugates demonstrated thermoresponsive behavior and retention of bioactivity. Given the facility with which other stimuli-responsive polymers can be prepared by RAFT polymerization,63–66 this method should enable the preparation of a variety of “smart” polymer–protein conjugates.67
Experimental
Materials
2-Dodecylsulfanylthiocarbonylsulfanyl-2-methyl propionic acid (DMP) was synthesized as previously reported.67 2,2′-Azobisisobutyronitrile (AIBN, Sigma, 98%) was recrystallized from ethanol, and N-isopropylacrylamide (NIPAM, TCI America) was recrystallized from hexanes. Sephacryl S200HR chromatography resin and BSA (98%) were purchased from Aldrich and used as received. OVA was purchased from Worthington Biochemical and used as received. All other chemicals were purchased from VWR and used without further purification, unless otherwise noted.
RAFT polymerization of NIPAM was conducted by a method similar to that previously reported ([NIPAM]0 : [CTA]0 : [I]0 = 200 : 1 : 0.2).60 Briefly, NIPAM (3.47 g, 30.7 mmol), DMP (56 mg, 0.15 mmol), 1,3,5-trioxane (35 mg, 0.39 mmol, internal standard), AIBN (5.0 mg, 0.030 mmol), and 1,4-dioxane (15 mL) were sealed in a 20 mL vial. The reaction solution was purged with nitrogen for 30 min, and the vial was placed in a preheated reaction block at 60 °C. After 150 min, the polymerization was stopped by rapid cooling and exposure of the polymerization solution to air (monomer conversion = 83%). The polymerization solution was diluted with tetrahydrofuran (THF), precipitated into hexanes (×3), and dried under vacuum to yield trithiocarbonate terminated polymer (PNIPAM–CTA) (Mn = 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 g mol−1; polydispersity index (PDI) = 1.25).
100 g mol−1; polydispersity index (PDI) = 1.25).
Synthesis of thiol-terminated PNIPAM
PNIPAM–CTA (2.0 g, 0.10 mmol) was dissolved in 1,4-dioxane (10 mL) and stirred for 2 h under a nitrogen atmosphere in the presence of 1-hexylamine and tributylphosphine ([PNIPAM–CTA] : [1-hexylamine] : [tributylphosphine] = 1 : 50 : 10). Thiol-terminated PNIPAM (PNIPAM–SH) was obtained as a white powder after precipitating into ice-cold ether (×3) and drying under vacuum. Yield = 77%; Mn = 19600 g mol−1; PDI = 1.27.
Synthesis of maleimide-terminated PNIPAM
1,8-Bis-maleimidodiethyleneglycol (BM, 0.16 g, 0.51 mmol) was dissolved in 1,4-dioxane (1 mL). PNIPAM–SH (1.0 g, 0.051 mmol) and triethylamine (TEA, 16 mg, 0.16 mmol) were dissolved in 1,4-dioxane (14 mL), and the resulting solution was added drop-wise to the above BM solution while stirring. The solution was stirred at room temperature for 13 h under a nitrogen atmosphere. The unreacted BM was removed by precipitation into ice-cold ether (×6), and the precipitate, maleimide-terminated PNIPAM (PNIPAM–M), was dried under vacuum. Yield = 60%; Mn = 20800 g mol−1; PDI = 1.24.
BSA (66 mg, 1.0 × 10−3 mmol) was mixed with PNIPAM–M (55 mg, 2.7 × 10−3 mmol) in nitrogen-purged phosphate buffer (PB, 10 mL, 0.1 M, pH 7.2) containing 1 mM ethylene diamine tetraacetic acid (EDTA). The reaction solution was stirred for 4 h at room temperature under a nitrogen atmosphere. The reaction solution was dialyzed against DI water (molecular weight cut-off (MWCO) = 10 kDa) for 24 h and lyophilized. The product was dissolved in the eluent NaCl (150 mM) and purified through a gel filtration column. The collected fractions were analyzed by aqueous SEC and SDS-PAGE.
OVA (45 mg, 1.0 × 10−3 mmol) was mixed with tris(2-carboxyethyl)phosphine hydrochloride (TCEP) (28.6 mg, 100 × 10−3 mmol) in nitrogen-purged PB (10 mL, 0.1 M, pH 7.2) containing 1 mM EDTA at room temperature. After 24 h, the solution was centrifuged (×3) using a tube with a membrane of MWCO = 10 kDa to remove excess TCEP. Ellman's assay revealed that the reduced OVA contained an average of three free thiols per OVA molecule. The resulting OVA and PNIPAM–M (170 mg, 8.17 × 10−3 mmol) were then dissolved in nitrogen-purged PB (10 mL) and incubated at room temperature for 24 h under a nitrogen atmosphere. The reaction solution was dialyzed against DI water (MWCO = 10 kDa) for 24 h and lyophilized. The product was purified by gel filtration, and the fractions were analyzed by aqueous SEC and SDS-PAGE.
Determination of free thiol content via Ellman's assay
5,5′-Dithio-bis-(2-nitrobenzoic acid) (Ellman's reagent, 4.0 mg, 0.010 mmol) was dissolved in PB (1 mL, pH 7.2, 0.1 M) to prepare a stock solution. The sample (5.0 mg) was dissolved in PB (2.5 mL) and mixed with the stock solution (50 µL). After 45 min incubation at room temperature, the absorbance at 412 nm was measured by UV-Vis spectroscopy. The thiol concentration was calculated using Beer–Lambert's law (molar extinction coefficient of 2-nitro-5-thiobenzoic acid = 16560 M−1 cm−1 at 412 nm).
Bioactivity assay
The bioactivity of BSA and the BSA conjugates was determined by observing the absorbance associated with the hydrolysis product of 4-nitrophenyl acetate in a manner similar to that previously reported.68 A BSA or BSA conjugate in PB solution (25 µL, pH 8.0, BSA concentration: 0.27 mM) was mixed with a solution of 4-nitrophenyl acetate (10 mM) dissolved in acetonitrile (5 µL) and PB (470 µL, pH 8.0), followed by centrifugation for 5 min at 6000 rpm. After incubating at room temperature for 30 min, the absorbance at 405 nm was measured for each sample to evaluate activity. For the bioactivity assay at 45 °C, the sample solution was centrifuged for 5 min at room temperature and incubated for 25 min at 45 °C. After cooling at room temperature for 5 min, the absorbance at 405 nm was measured.
Cloud point measurements
Turbidity measurements of sample solutions (0.05 w/v% in 150 mM NaCl solution) were monitored by recording the solution absorbance at 600 nm. The temperature was gradually raised from 26 to 50 °C by increments of 2 °C with 5 min equilibration time at each increment. The cloud point was defined as the temperature at 10% of the maximum absorbance.
Particle size measurements
Samples were dissolved in a NaCl solution (150 mM) at a concentration of 0.05 w/v% and filtered through 0.22 µm mixed cellulose ester filters. Prior to each measurement by dynamic light scattering (DLS), the sample solutions were incubated for 30 min at the targeted temperature (25 or 50 °C).
Thermoprecipitation
PNIPAM-containing bioconjugates were dissolved in DI water at a concentration of 2.0 w/v%. The solution was incubated at 40 °C for 15 min, followed by centrifugation for 3 min at 40 °C. The supernatant and the precipitate were collected for analyses.
Analyses
Polymer molecular weights and molecular weight distributions were obtained by organic size exclusion chromatography (SEC) in N,N-dimethylformamide (DMF) with 50 mM LiBr at 55 °C using a flow rate of 1.0 mL min−1 (Viscotek SEC pump; columns: ViscoGel I-series G3000 and G4000 mixed bed columns: molecular weight range 0–60 × 103 and 0–400 × 103 g mol−1, respectively). Detection consisted of a Viscotek refractive index detector operating at λ = 660 nm, and a Viscotek model 270 series platform, consisting of a laser light scattering detector (operating at 3 mW, λ = 670 nm with detection angles of 7° and 90°) and a four-capillary viscometer. Molecular weights were determined by the triple detection method using a dn/dc = 0.077 mL g−1 for PNIPAM. Aqueous (0.5 w/v% NaN3) SEC characterization of proteins and conjugates were conducted at 25 °C with a flow rate of 0.7 mL min−1 (Viscotek SEC pump; column: Biosep-SEC-S 3000). Detection consisted of a Viscotek refractive index detector operating at λ = 660 nm and a Viscotek UV-Vis detector operating at λ = 280 nm. UV-Vis spectroscopic measurements were carried out on a Beckman Coulter DU Series 800 UV-Vis spectrometer equipped with a Peltier temperature controller. 1H NMR spectroscopy was conducted with a Jeol Delta 500 spectrometer operating at 500 MHz. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed with a Bio-Rad Laboratories Mini-Protean 3 Cell System using pre-cast 12% polyacrylamide gels (Pierce). Staining was accomplished with Coomassie blue. Centrifugation and thermoprecipitation were conducted on Sorvall LEGEND RT and Eppendorf 5415R centrifuges, respectively. DLS was conducted with a Malvern Zetasizer Nano-ZS equipped with a 4 mW, 633 nm He–Ne laser, and an Avalanche photodiode detector at 173°.
Results and discussion
Preparation of ω-maleimide-terminated polymer
RAFT polymerization of NIPAM was conducted with a trithiocarbonate CTA and AIBN as the initiator in 1,4-dioxane at 60 °C (Scheme 1). The polymerization was stopped at 83% to yield PNIPAM-CTA with Mn,SEC = 19100 g mol−1, which was in an excellent agreement with the theoretical value of Mn,theo = 18700 g mol−1. The resulting polymer possessed a terminal dodecyl trithiocarbonate (–S–C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S)–S–C12H25) moiety with a characteristic –CH31H NMR resonance at δ = 0.88 ppm. Aminolysis of the terminal trithiocarbonate end groups was accomplished by reacting with 1-hexylamine in the presence of tributylphosphine to limit disulfide formation. After addition of the amine, the yellow color of the polymer solution rapidly disappeared, and UV-Vis spectroscopy verified consumption of the CTA group by reduction of the absorption at 310 nm. NMR analysis of the purified product demonstrated the absence of the CTA-derived terminal methyl, and SEC analysis indicated that disulfide homocoupling of PNIPAM–SH was significantly limited, though not completely inhibited (Fig. 1).
S)–S–C12H25) moiety with a characteristic –CH31H NMR resonance at δ = 0.88 ppm. Aminolysis of the terminal trithiocarbonate end groups was accomplished by reacting with 1-hexylamine in the presence of tributylphosphine to limit disulfide formation. After addition of the amine, the yellow color of the polymer solution rapidly disappeared, and UV-Vis spectroscopy verified consumption of the CTA group by reduction of the absorption at 310 nm. NMR analysis of the purified product demonstrated the absence of the CTA-derived terminal methyl, and SEC analysis indicated that disulfide homocoupling of PNIPAM–SH was significantly limited, though not completely inhibited (Fig. 1).
 |
| | Fig. 1 Organic SEC traces for PNIPAM–CTA, PNIPAM–SH, and PNIPAM–M. | |
Maleimide functional groups were introduced by reacting PNIPAM–SH with a bismaleimide. To avoid coupling of two polymeric thiols, which would result in polymer incapable of further functionalization, a dilute solution of polymer was slowly added to a large excess of the difunctional compound. The final product contained 80% terminal maleimide functionalization, calculated by using the Mn determined by SEC and ratio of peak areas determined by 1H NMR spectroscopy of the vinylogous protons at δ = 6.77 ppm of the maleimide end groups and the –NH–CH(CH3)2 protons at δ = 3.82 ppm in the monomer repeat units.
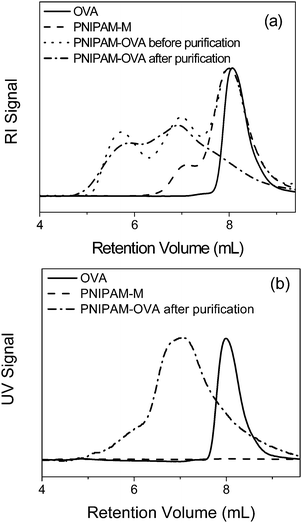
BSA was selected as a model protein for conjugation with the maleimide-terminated polymers because of its ready availability, robust nature, and the presence of a readily accessible free thiol at Cys-34 that is suitable for site-specific conjugation. PNIPAM–M was reacted with BSA in phosphate buffer for 4 h at room temperature. During the conjugation reaction, oxygen-free conditions were employed to prevent BSA aggregation69 or the formation of additional BSA dimer through thiol–disulfide interchange or oxidation reactions. The reaction product was purified by gel filtration, and successful conjugate formation was confirmed by SDS-PAGE (Fig. 2) and SEC (Fig. 3) by the appearance of a conjugate band or peak with higher MW than native BSA. As evident in the SDS-PAGE (Fig. 2, lane c) and SEC (Fig. 3, trace of PNIPAM–BSA before purification) results, incomplete consumption of the protein was observed. This observation is consistent with only about 50% of the Cys-34 residues in commercial BSA batches being available for reaction.17,19,27,69 Indeed, Ellman's analysis of the starting material indicated approximately 45% of the native BSA contained a free thiol. However, the conjugates were readily separated from unreacted PNIPAM–M and BSA by gel filtration, as demonstrated by only one high molecular weight species being observed for the purified product by SDS-PAGE (Fig. 2, lane d) and SEC with RI and UV detection (Fig. 3). The PNIPAM–BSA conjugate was tested for its ability to hydrolyze 4-nitrophenyl acetate, an esterase-like activity known to require the structural integrity of the protein.68 At 25 °C, the conjugate demonstrated quantitative retention of activity, with respect to native BSA. When the assay was conducted at 45 °C, a temperature above the cloud point of the conjugate (vide infra), the activity was observed to be 89% of that for native BSA. It has been previously reported that the activity of proteins conjugated with thermoresponsive polymers can be conveniently tuned by changes in temperature.27,65,66,70,71 In the current study, the modest reduction in bioactivity may be attributable to the rather low molecular weight of the conjugated polymer or the polar ethylene glycol linker.24
 |
| | Fig. 2 SDS-PAGE results of PNIPAM–BSA conjugates: lane a = molecular weight markers (from bottom to top: 50, 60, 70, 85, 100, 120, 150, 200 kDa); lane b = native BSA; lane c = PNIPAM–BSA before purification; lane d = PNIPAM–BSA after purification by gel filtration (PNIPAM Mn = 19100 g mol−1; PDI = 1.25). | |
The dominant egg white protein, OVA, was also conjugated with PNIPAM–M. A mild reduction with TCEP resulted in an average of three free thiols per protein. The reduced OVA was conjugated with maleimide-terminated PNIPAM. As evidenced by SDS-PAGE (Fig. 4) and SEC (Fig. 5), the purified products consisted of conjugates with multiple polymers attached, as expected based on the plurality of available cysteine residues.
 |
| | Fig. 4 SDS-PAGE results of PNIPAM–OVA conjugates: lane a = molecular weight markers (from bottom to top: 30, 40, 50, 60, 70, 85, 100, 120, 150, 200 kDa); lane b = native OVA; lane c = PNIPAM–OVA before purification; lane d = PNIPAM–OVA after purification by gel filtration (PNIPAM Mn = 20800 g mol−1; PDI = 1.24). | |
Temperature responsiveness
The thermoresponsive aqueous solution behavior of the conjugates was also investigated. As determined by turbidimetry measurements at a concentration of 0.05 w/v% in 150 mM NaCl, both PNIPAM–M and the corresponding conjugates with BSA and OVA demonstrated a cloud point of approximately 32–35 °C, indicating the protein had relatively minimal effect on the phase transition temperature of the conjugated PNIPAM (Fig. 6). DLS was used to investigate the size of the conjugates below and above the transition temperature of the immobilized PNIPAM. As expected, at 25 °C the conjugates were larger than the individual polymer or protein components. After heating dilute solutions (0.05 w/v%) above the transition temperature of the conjugate, a dramatic increase in hydrodynamic diameter (Dh) was observed, consistent with self-assembly of the conjugates upon dehydration and aggregation of the tethered PNIPAM chains (Table 1). While we are not certain of the morphology, it is likely the conjugates form polymer–protein aggregates composed of hydrophobic PNIPAM cores stabilized by hydrophilic protein. As compared to the PNIPAM–BSA, the PNIPAM–OVA conjugates formed larger aggregates upon heating, which is to be expected since the lower molecular weight of OVA and multiple polymers attached led to the proportion of PNIPAM chains being greater in this case. A related effect was observed when conjugate solutions of significantly higher concentrations (2.0 w/v%) were heated, with the OVA conjugates being efficiently precipitated, while aggregates of the BSA conjugates remained partially suspended.
|
|
D
h at 25 °C/nm |
D
h at 50 °C/nm |
|
PNIPAM–M: maleimide-terminated poly(N-isopropylacrylamide); BSA: bovine serum albumin; PNIPAM–BSA: poly(N-isopropylacrylamide)–BSA; OVA: ovalbumin; PNIPAM–OVA: poly(N-isopropylacrylamide)–OVA. Concentration = 0.05 w/v%.
|
| PNIPAM–M |
7 |
240 |
| BSA |
3 |
3 |
| PNIPAM–BSA |
17 |
56 |
| OVA |
4 |
4 |
| PNIPAM–OVA |
12 |
180 |
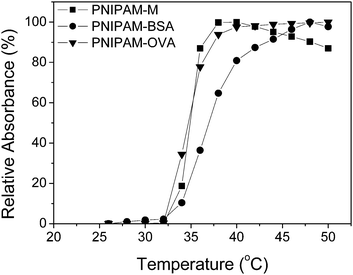 |
| | Fig. 6 Cloud point determination of aqueous solutions of PNIPAM–M, PNIPAM–BSA, and PNIPAM–OVA by turbidimetry. | |
Conclusions
End group aminolysis and subsequent reaction with an excess of a bismaleimide yield ω-maleimide-terminated polymers that can be readily coupled to thiol-containing biomacromolecules. While the proteins considered here were functionalized through thiols present natively in their structure, this general route can be extended to other proteins modified or engineered to contain free cysteine residues. This approach of coupling two thiol-containing macromolecules by consecutive thiol–ene reactions to a bismaleimide leads to adducts connected via reductively stable thioether linkages, a potential benefit for applications in which irreversible conjugation is desired. Preliminary studies indicate the responsive nature of the attached PNIPAM is conferred to the conjugate.
Acknowledgements
Acknowledgement is made to the National Science Foundation (DMR-0846792) and Oak Ridge Associated Universities (Ralph E. Powe Junior Faculty Enhancement Award) for partial support of this research. We thank Dr Sudershan R. Gondi for synthesizing DMP.
Notes and references
- H.-A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 1–17 CrossRef CAS.
- P. Thordarson, B. Droumaguet and K. Velonia, Appl. Microbiol. Biotechnol., 2006, 73, 243–254 CrossRef CAS.
-
J. A. Opsteen and J. C. M. van Hest, in Macromolecular Engineering, ed. K. Matyjaszewski, Y. Gnanou and L. Leibler, Wiley-VCH, Weinheim, 2007, vol. 4, pp. 2645–2687 Search PubMed.
- H. G. Boerner, Macromol. Chem. Phys., 2007, 208, 124–130 CrossRef.
- J.-F. Lutz and H. G. Boerner, Prog. Polym. Sci., 2008, 33, 1–39 CrossRef CAS.
- H. G. Boerner, Prog. Polym. Sci., 2009, 34, 811–851 CrossRef.
- J. Nicolas, G. Mantovani and D. M. Haddleton, Macromol. Rapid Commun., 2007, 28, 1083–1111 CrossRef CAS.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS.
- K. L. Heredia and H. D. Maynard, Org. Biomol. Chem., 2007, 5, 45–53 RSC.
- M. A. Gauthier and H.-A. Klok, Chem. Commun., 2008, 2591–2611 RSC.
- B. L. Droumaguet and J. Nicolas, Polym. Chem., 2010 10.1039/b9py00363k.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689–3745 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- B. S. Lele, H. Murata, K. Matyjaszewski and A. J. Russell, Biomacromolecules, 2005, 6, 3380–3387 CrossRef CAS.
- G. Mantovani, F. Lecolley, L. Tao, D. M. Haddleton, J. Clerx, J. J. L. M. Cornelissen and K. Velonia, J. Am. Chem. Soc., 2005, 127, 2966–2973 CrossRef CAS.
- K. L. Heredia, D. Bontempo, T. Ly, J. T. Byers, S. Halstenberg and H. D. Maynard, J. Am. Chem. Soc., 2005, 127, 16955–16960 CrossRef CAS.
- J. Geng, G. Mantovani, L. Tao, J. Nicolas, G. Chen, R. Wallis, D. A. Mitchell, B. R. G. Johnson, S. D. Evans and D. M. Haddleton, J. Am. Chem. Soc., 2007, 129, 15156–15163 CrossRef CAS.
- M. Li, P. De, S. R. Gondi and B. S. Sumerlin, Macromol. Rapid Commun., 2008, 29, 1172–1176 CrossRef CAS.
- C. Boyer, V. Bulmus and T. P. Davis, Macromol. Rapid Commun., 2009, 30, 493–497 CrossRef.
- L. Tao, J. Liu and T. P. Davis, Biomacromolecules, 2009, 10, 2847–2851 CrossRef CAS.
- S. Kulkarni, C. Schilli, B. Grin, A. H. E. Mueller, A. S. Hoffman and P. S. Stayton, Biomacromolecules, 2006, 7, 2736–2741 CrossRef CAS.
- J. Liu, V. Bulmus, D. L. Herlambang, C. Barner-Kowollik, M. H. Stenzel and T. P. Davis, Angew. Chem., Int. Ed., 2007, 46, 3099–3103 CrossRef CAS.
- S. Kulkarni, C. Schilli, A. H. E. Mueller, A. S. Hoffman and P. S. Stayton, Bioconjugate Chem., 2004, 15, 747–753 CrossRef CAS.
- S. M. Henry, A. J. Convertine, D. S. W. Benoit, A. S. Hoffman and P. S. Stayton, Bioconjugate Chem., 2009, 20, 1122–1128 CrossRef CAS.
- C. Boyer, V. Bulmus, J. Liu, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, J. Am. Chem. Soc., 2007, 129, 7145–7154 CrossRef CAS.
- P. De, M. Li, S. R. Gondi and B. S. Sumerlin, J. Am. Chem. Soc., 2008, 130, 11288–11289 CrossRef CAS.
- L. Tao, C. S. Kaddis, R. R. O. Loo, G. N. Grover, J. A. Loo and H. D. Maynard, Chem. Commun., 2009, 2148–2150 RSC.
- G. N. Grover, S. N. S. Alconcel, N. M. Matsumoto and H. D. Maynard, Macromolecules, 2009, 42, 7657–7663 CrossRef CAS.
- P. J. Roth, F. D. Jochum, R. Zentel and P. Theato, Biomacromolecules, 2010, 11, 238–244 CrossRef CAS.
- A. B. Lowe, B. S. Sumerlin, M. S. Donovan and C. L. McCormick, J. Am. Chem. Soc., 2002, 124, 11562–11563 CrossRef CAS.
- B. S. Sumerlin, A. B. Lowe, P. A. Stroud, P. Zhang, M. W. Urban and C. L. McCormick, Langmuir, 2003, 19, 5559–5562 CrossRef CAS.
- H. Gemici, T. M. Legge, M. Whittaker, M. J. Monteiro and S. Perrier, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2334–2340 CrossRef CAS.
- B. D. Mather, K. Viswanathan, K. M. Miller and T. E. Long, Prog. Polym. Sci., 2006, 31, 487–531 CrossRef CAS.
- X.-P. Qiu and F. M. Winnik, Macromol. Rapid Commun., 2006, 27, 1648–1653 CrossRef CAS.
- R. Narain, M. Gonzales, A. S. Hoffman, P. S. Stayton and K. M. Krishnan, Langmuir, 2007, 23, 6299–6304 CrossRef CAS.
- V. S. Khire, D. S. W. Benoit, K. S. Anseth and C. N. Bowman, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 7027–7039 CrossRef CAS.
- A. P. Vogt, S. R. Gondi and B. S. Sumerlin, Aust. J. Chem., 2007, 60, 396–399 CrossRef CAS.
- C. W. Scales, A. J. Convertine and C. L. McCormick, Biomacromolecules, 2006, 7, 1389–1392 CrossRef CAS.
- N. Kanayama, H. Shibata, A. Kimura, D. Miyamoto, T. Takarada and M. Maeda, Biomacromolecules, 2009, 10, 805–813 CrossRef CAS.
- X. Lou, G. Zhang, I. Herrera, R. Kinach, O. Olga, V. Baranov, M. Nitz and M. A. Winnik, Angew. Chem., Int. Ed., 2007, 46, 6111–6114 CrossRef CAS.
- C. Boyer, A. Granville, T. P. Davis and V. Bulmus, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3773–3794 CrossRef CAS.
- P. J. Roth, D. Kessler, R. Zentel and P. Theato, Macromolecules, 2008, 41, 8316–8319 CrossRef CAS.
- P. J. Roth, D. Kessler, R. Zentel and P. Theato, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3118–3130 CrossRef CAS.
- P. J. Roth, K. T. Wiss, R. Zentel and P. Theato, Macromolecules, 2008, 41, 8513–8519 CrossRef CAS.
- F. D. Jochum, L. zur Borg, P. J. Roth and P. Theato, Macromolecules, 2009, 42, 7854–7862 CrossRef CAS.
- H. Willcock and R. K. O'Reilly, Polym. Chem., 2010, 1, 149 RSC.
- A. B. Lowe, Polym. Chem., 2010, 1, 17 RSC.
- G. G. Kochendoerfer, Curr. Opin. Chem. Biol., 2005, 9, 555–560 CrossRef CAS.
- T. Shimoboji, E. Larenas, T. Fowler, A. S. Hoffman and P. S. Stayton, Bioconjugate Chem., 2003, 14, 517–525 CrossRef CAS.
- T. Shimoboji, Z. L. Ding, P. S. Stayton and A. S. Hoffman, Bioconjugate Chem., 2002, 13, 915–919 CrossRef CAS.
- T. Shimoboji, Z. Ding, P. S. Stayton and A. S. Hoffman, Bioconjugate Chem., 2001, 12, 314–319 CrossRef CAS.
- V. Bulmus, Z. Ding, C. J. Long, P. S. Stayton and A. S. Hoffman, Bioconjugate Chem., 2000, 11, 78–83 CrossRef CAS.
- K. Velonia, A. E. Rowan and R. J. M. Nolte, J. Am. Chem. Soc., 2002, 124, 4224–4225 CrossRef CAS.
- V. Vazquez-Dorbatt, Z. P. Tolstyka, C.-W. Chang and H. D. Maynard, Biomacromolecules, 2009, 10, 2207–2212 CrossRef CAS.
- K. L. Heredia, T. H. Nguyen, C.-W. Chang, V. Bulmus, T. P. Davis and H. D. Maynard, Chem. Commun., 2008, 3245–3247 RSC.
- D. Bontempo, K. L. Heredia, B. A. Fish and H. D. Maynard, J. Am. Chem. Soc., 2004, 126, 15372–15373 CrossRef CAS.
- Z. Jia, J. Liu, C. Boyer, T. P. Davis and V. Bulmus, Biomacromolecules, 2009, 10, 3253–3258 CrossRef CAS.
- L. Tao, J. Liu, J. Xu and T. P. Davis, Chem. Commun., 2009, 6560–6562 RSC.
- M. Li, P. De, S. R. Gondi and B. S. Sumerlin, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5093–5100 CrossRef CAS.
- C. L. McCormick, B. S. Sumerlin, B. S. Lokitz and J. E. Stempka, Soft Matter, 2008, 4, 1760–1773 RSC.
-
C. M. Schilli, A. H. E. Mueller, E. Rizzardo, S. H. Thang and Y. K. Chong, Advances in Controlled/Living Radical Polymerization, American Chemical Society, Washington, DC, 2003 Search PubMed.
- C. d. l. H. Alarcon, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- P. S. Stayton, A. S. Hoffman, M. El-Sayed, S. Kulkarni, T. Shimoboji, N. Murthy, V. Bulmus and C. Lackey, Proc. IEEE, 2005, 93, 726–736 CrossRef CAS.
- A. S. Hoffman and P. S. Stayton, Macromol. Symp., 2004, 207, 139–151 CrossRef CAS.
- A. S. Hoffman and P. S. Stayton, Prog. Polym. Sci., 2007, 32, 922–932 CrossRef CAS.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS.
- J. T. Tildon and J. W. Ogilvie, J. Biol. Chem., 1972, 247, 1265–1271.
- D. C. Carter and J. X. Ho, Adv. Protein Chem., 1994, 45, 153–203 CAS.
- A. S. Hoffman, P. S. Stayton, T. Shimoboji, G. Chen, Z. Ding, A. Chilkoti, C. Long, M. Miura, J. Chen, T. Park, N. Monji, C. A. Cole, J. M. Harris and K. Nakamae, Macromol. Symp., 1997, 118, 553–563 CAS.
- A. S. Hoffman, P. S. Stayton, V. Bulmus, G. Chen, J. Chen, C. Cheung, A. Chilkoti, Z. Ding, L. Dong, R. Fong, C. A. Lackey, C. J. Long, M. Miura, J. E. Morris, N. Murthy, Y. Nabeshima, T. G. Park, O. W. Press, T. Shimoboji, S. Shoemaker, H. J. Yang, N. Monji, R. C. Nowinski, C. A. Cole, J. H. Priest, J. Milton Harris, K. Nakamae, T. Nishino and T. Miyata, J. Biomed. Mater. Res., Part A, 2000, 52, 577–586 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.