Highly efficient, stoichiometric radical exchange reactions using isoindoline profluorescent nitroxides†
James P.
Blinco‡
*ab,
Kathryn E.
Fairfull-Smith
b,
Aaron S.
Micallef
a and
Steven E.
Bottle*
b
aCentre for Magnetic Resonance and Australian Institute for Bioengineering and Nanotechnology, University of Queensland, Brisbane, Queensland 4702, Australia. Fax: +61 (0)7 3346 3973; Tel: +61 (0)7 3346 3864
bARC Centre of Excellence for Free Radical Chemistry and Biotechnology, Queensland University of Technology, Brisbane, Queensland 4001, Australia. E-mail: s.bottle@qut.edu.au; Fax: +61 (0)7 3138 1804; Tel: +61 (0)7 3138 1356
First published on 8th May 2010
Abstract
Exchange reactions between the isoindoline profluorescent nitroxide 1,1,3,3-tetramethyldibenzo[e,g]isoindolin-2-yloxyl (TMDBIO) and a TEMPO capped polystyrene were carried out. High conversions to the desired products were achieved using only stoichiometric ratios of nitroxide relative to polymer. The scope of this study was expanded by exploiting a di-nitroxide 9,10-bis(5-[1,1,3,3-tetramethylisoindolin-2-yloxy])anthracene (BTMIOA) as a connector between two polymer chains forming PS–nitroxide–PS systems.
The combination of efficient conjugation chemistry with controlled free radical polymerization has increasingly proven to be an important tool in the synthesis of novel polymeric materials. Polymerisation techniques such as Nitroxide Mediated Polymerisation (NMP), Atom Transfer Radical Polymerisation (ATRP) and Reversible Addition-Fragmentation Chain Transfer (RAFT) polymerisation have successfully been combined with conjugation reactions such as the “click” chemistry of copper(I) azide-alkyne cycloaddition (CuAAC) to achieve a wide variety of structures ranging from complex, multi-arm architectures through to the attachment of polymers to fluorescent markers or biologically important molecules with a high degree of selectivity and efficiency.1–6
The nitroxide exchange reaction is a similar conjugation technique involving the trapping of free radicals generated from a small molecule or macromolecular precursor. In this process, a polymer chain with an alkoxyamine cap (usually prepared via NMP) is heated in solution in the presence of a conjugation partner which features a nitroxide moiety (see Scheme 1). As with NMP, the carbon–oxygen bond of the alkoxyamine is thermally cleaved, regenerating what was once the propagating radical and a free nitroxide. This propagating radical is then trapped by the nitroxide arising from the conjugate polymer. The first example of this kind of conjugation was reported by Turro et al.7 and involved the end-functionalization of polystyrene chains with photo- and bio-active molecules. This approach was furthered by Higaki et al. who reported that by heating two homopolymers together, one bearing cap-nitroxide-chain and the other nitroxide-cap-chain, it was possible to obtain a block copolymer with an Mn equal to the sum of the two initial homopolymers.8–10 This group was also able to use similar reactions for the formation of gel networks.11
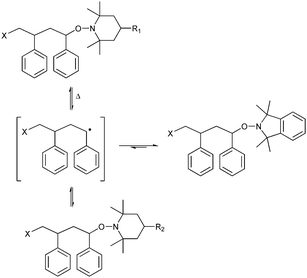 | ||
| Scheme 1 Conjugate exchange of nitroxides in polymer systems where piperidine nitroxides may be statistically exchanged (R1vs. R2) or swapped with a more strongly binding isoindoline nitroxide. | ||
The alkoxyamine cap and the conjugate exchange nitroxide have commonly been piperidine based nitroxides. Consequently the energy required for bond homolysis for the desired conjugate is similar to that of the initial alkoxyamine cap. The product is therefore just as likely to undergo the reverse reaction, giving a statistical mixture of starting material and product. This is clearly shown through the broadened polydispersity of the block copolymers synthesized by Higaki et al.12 It is possible to increase the yield of desired product by using a large excess of the target conjugate. However, even with >15-fold excess of conjugate, it was shown that the exchange is still not quantitative.7
Herein we report a novel and simple means to overcome this limitation by exploiting the differences in C–O bond strengths between different classes of nitroxide alkoxyamines. Recently, we reported examples of the application of profluorescent nitroxides as probes of the thermal degradation of polymers.13–16 Notably in all cases where an isoindoline based profluorescent nitroxide is used, the trapping of the generated radicals is essentially irreversible, even at temperatures reaching 150 °C.16
Investigations by others have shown that the dissociation constant (kd) of the carbon–oxygen bond homolysis energy for the isoindoline alkoxyamine is more than an order of magnitude less than that of the piperidine equivalent.17 This is likely to be due to the reduced steric demand for isoindoline systems compared with the piperidine-based adducts and also accounts for the poor performance of isoindoline alkoxyamines as NMP agents.18–20 This added stability can be advantageous in nitroxide exchange reactions where the high stability of the isoindoline conjugation product relative to the piperidine adduct, avoids the need for a large excess of nitroxide. There are also examples in the literature of molecules containing 2, 3, or 4 isoindoline nitroxides,14,21–23 and so it should be possible to use the nitroxide not only as an end group but also as a linker between multiple-chains to form more complex architectures. Herein, to demonstrate the viability of this approach, we describe the stoichiometric reaction of TEMPO-capped polystyrene with an isoindoline profluorescent nitroxide as well as initial studies into the use of isoindoline di-nitroxides to form polymer–nitroxide–polymer systems.
In this study polystyrene (PS) was chosen as the model polymer system. PS was synthesized via NMP using AIBN as the initiator and 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) as the control agent and is described as PS–TEMPO. The polymerization was stopped at low conversion to maximize end group fidelity. The polymer was shown to have an Mn of 35 kDa with a low polydispersity (1.1). The single and di-isoindoline nitroxides used were 1,1,3,3-tetramethyldibenzo[e,g]isoindolin-2-yloxyl (TMDBIO) and 9,10-bis(5-[1,1,3,3-tetramethylisoindolin-2-yloxy])anthracene (BTMIOA) respectively. These probes have been used in several of our previous studies and their stability and photo-physical capabilities are well understood. BTMIOA was synthesized using literature procedures14,21 and TMDBIO was synthesized in 4 steps from phenanthro[9,10-c]-1,3-anhydride in an overall yield of 24%. Full synthetic details and characterization of this probe can be found in the ESI†.
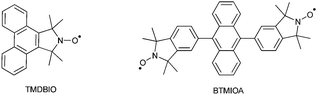
For the exchange reaction, PS–TEMPO and TMDBIO (1.1 equiv.) were dissolved in toluene and degassed via argon bubbling. The vessel was sealed and the mixture heated to 80 °C with samples taken periodically. The conversion of the profluorescent nitroxide (minimal emission) into the fluorescent alkoxyamine end group (high emission, Fig. 1) was monitored by fluorescence spectroscopy. The majority of the exchange occurred within the first 15 minutes with a maximum being reached after ca. 240 minutes. The increase in fluorescence from 60 to 240 minutes is minimal and very slow compared with the rapid initial reaction. This is likely to be due to an increased competition for radical trapping between the shrinking concentration of TMDBIO and the growing concentration of liberated TEMPO radical.
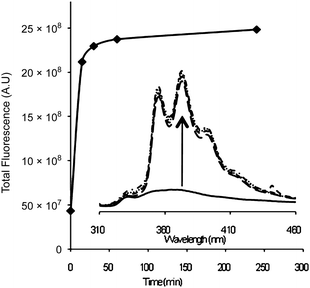 | ||
| Fig. 1 Fluorescence increase with time and the appearance of the phenanthrene fluorescence (inset). | ||
Evidence for the exchange is seen in the 1H NMR (Fig. 2) of the re-precipitated product which, as well as showing the typical PS spectra, clearly shows the appearance of peaks in the aromatic region between 7.2 and 8.8 ppm attributable to the aromatic signals from the phenanthrene moiety. This is also supported by the UV-Vis absorption spectrum (see ESI†) where by the phenanthrene vibronic at 300 nm can be seen well resolved from any PS absorptions. SEC confirms that, while there is little change in the molecular weight or polydispersity of the polymer, there is a large increase in the UV absorbance of the main polymer chain due to the conjugation of the aromatic chromophore. Using the absorbance at 300 nm and the known extinction coefficient of these alkoxyamines16 we are able to calculate that after 60 minutes reaction time there is an 85% exchange efficiency to the TMDBIO capped PS. Calculating the efficiency using NMR is in close agreement (∼80%) with this determination.
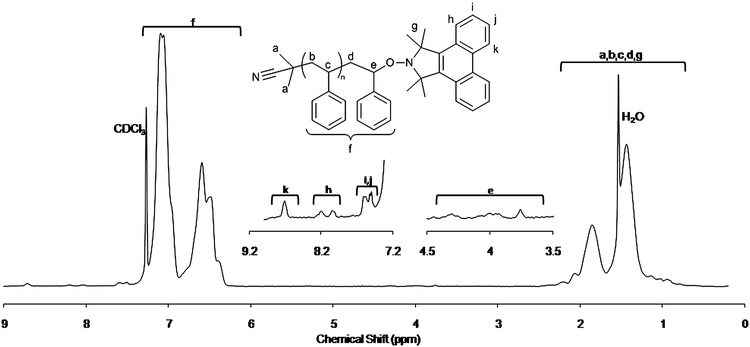 | ||
| Fig. 2 1H NMR spectrum of PS–TMDBIO with expansions. Inset showing signals arising from the TMDBIO end group. | ||
To test the stability of the isoindoline end group to the reverse reaction, PS–TMDBIO was heated in the presence of 1.1 equiv. of TEMPO. After 12 h of heating, the fluorescent signal had dropped <5% overall indicating the majority of the TMDBIO was still attached to the PS. It should be noted that when higher temperatures and a large excess of TEMPO were used, it was possible to force the formation of PS–TEMPO.
Exchange experiments were also carried out using the di-nitroxide BTMIOA which showed similar results. Thus employing BTMIOA in place of TMDBIO (stoichiometry 1 : 0.5, PS–TEMPO : BTMIOA) showed it was possible to use isoindoline nitroxides not only for end-group exchange, but as linkages between two separate chains. As with TMDBIO, a large increase in the fluorescence from the solution and the reprecipitated polymer is observed (see Fig. 3). In the case of BTMIOA, however, for fluorescence to be seen, both radical moieties must undergo trapping.14 SEC also supports the trapping of two chains by BTMIOA, observed as a significant shift in the main peak to approximately double the original molecular weight (Fig. 3). Notably, while the majority of the polymer appears to comprise PS–BTMIOA–PS as expected, a small shoulder evident on the low molecular weight side of the distribution may indicate the presence of an amount of PS–TEMPO or PS–BTMIOA. This may reflect the increased competition at high conversion where the concentration of TEMPO is increased.
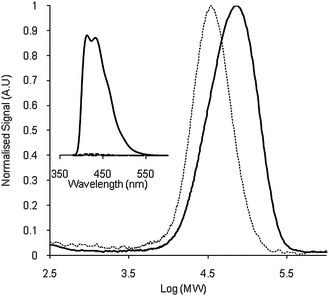 | ||
Fig. 3 Comparison of the SEC chromatograms and fluorescence intensity (inset) of PS–TEMPO (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) Mn 35 ) Mn 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600, Mp 36700, PDI 1.28 and PS–BTMIOA–PS ( 600, Mp 36700, PDI 1.28 and PS–BTMIOA–PS (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ) Mn 63700, Mp 70200, PDI 1.35. ) Mn 63700, Mp 70200, PDI 1.35. | ||
The use of an isoindoline profluorescent nitroxide in exchange reactions has been demonstrated in this preliminary study. Analysis of the end group exchanged polymer by NMR, GPC and UV spectroscopy shows that even in stoichiometric ratios, the exchange occurs with a high level of efficiency. Initial studies using a di-nitroxide show that it can act not only as an end group but also as a link between two polymer chains. There is now potential for isoindoline nitroxides to be used as cores for the timely and efficient formation of complex architectures without the need for further polymer functionalization or catalysts. Further studies into the kinetics of the exchange as well as investigations with different polymer blocks are continuing.
Acknowledgements
We gratefully acknowledge financial support for this work from the Australian Research Council (ARC Centre of Excellence CE0561607 and Linkage International Projects LE0775684 and LE0668517) and thank Lauren Butler and the Queensland node of the Australian National Fabrication Facility for the use of GPC equipment.Notes and references
- B. S. Sumerlin, N. V. Tsarevsky, H. Gao, P. Golas, G. Louche, R. Y. Lee and K. Matyjaszewski, ACS Symp. Ser., 2006, 944, 140–152 CAS.
- G. D. Fu, L. Q. Xu and F. Yao, ACS Appl. Mater. Interfaces, 2009, 1, 239–243 Search PubMed.
- J. Geng, J. Lindqvist, G. Mantovani and D. Haddleton, Angew. Chem., Int. Ed., 2008, 47, 4180–4183 CrossRef CAS.
- X. Zhang, X. Lian, L. Liu, J. Zhang and H. Zhao, Macromolecules, 2008, 41, 7863–7869 CrossRef CAS.
- J. Geng, G. Mantovani, L. Tao, J. Nicolas, G. Chen, R. Wallis, D. A. Mitchell, B. R. G. Johnson, S. D. Evans and D. M. Haddleton, J. Am. Chem. Soc., 2007, 129, 15156–15163 CrossRef CAS.
- H. Gao and K. Matyjaszewski, Macromolecules, 2006, 39, 4960–4965 CrossRef CAS.
- N. J. Turro, G. Lem and I. S. Zavarine, Macromolecules, 2000, 33, 9782–9785 CrossRef CAS.
- Y. Higaki, H. Otsuka and A. Takahara, Macromolecules, 2006, 39, 2121–2125 CrossRef CAS.
- H. Otsuka, K. Aotani, Y. Higaki and A. Takahara, Chem. Commun., 2002, 2838–2839 RSC.
- H. Otsuka, K. Aotani, Y. Higaki and A. Takahara, J. Am. Chem. Soc., 2003, 125, 4064–4065 CrossRef CAS.
- Y. Amamoto, Y. Higaki, Y. Matsuda, H. Otsuka and A. Takahara, J. Am. Chem. Soc., 2007, 129, 13298–13304 CrossRef CAS.
- Y. Higaki, H. Otsuka and A. Takahara, Polymer, 2006, 47, 3784–3791 CrossRef CAS.
- J. P. Blinco, D. J. Keddie, T. Wade, P. J. Barker, G. A. George and S. E. Bottle, Polym. Degrad. Stab., 2008, 93, 1613–1618 CrossRef CAS.
- K. E. Fairfull-Smith, J. P. Blinco, D. J. Keddie, G. A. George and S. E. Bottle, Macromolecules, 2008, 41, 1577–1580 CrossRef CAS.
- A. Micallef, S. Bottle, J. Blinco and G. George, ACS Symp. Ser., 2007, 978, 59–69.
- A. S. Micallef, J. P. Blinco, G. A. George, D. A. Reid, E. Rizzardo, S. H. Thang and S. E. Bottle, Polym. Degrad. Stab., 2005, 89, 427–435 CrossRef CAS.
- S. Marque, J. Org. Chem., 2003, 68, 7582–7590 CrossRef CAS.
- S. P. Cresidio, F. Aldabbagh, W. K. Busfield, I. D. Jenkins, S. H. Thang, C. Zayas-Holdsworth and P. B. Zetterlund, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 1232–1241 CrossRef CAS.
- G. Moad and E. Rizzardo, Macromolecules, 1995, 28, 8722–8728 CrossRef CAS.
- Y. K. Chong, F. Ercole, G. Moad, E. Rizzardo, A. G. Anderson and S. H. Thang, Macromolecules, 1999, 32, 6895–6903 CrossRef CAS.
- K. E. Fairfull-Smith and S. E. Bottle, Eur. J. Org. Chem., 2008, 5391–5400 CrossRef CAS.
- A. G. M. Barrett, G. R. Hanson, A. J. P. White, D. J. Williams and A. S. Micallef, Tetrahedron, 2007, 63, 5244–5250 CrossRef CAS.
- C. D. Smith, R. C. Bott, S. E. Bottle, A. S. Micallef and G. Smith, J. Chem. Soc., Perkin Trans. 2, 2002, 533–537 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental details for TMDBIO synthesis and nitroxide exchange reactions; UV-Vis and fluorescence comparisons of PS–TEMPO with PS–TMDBIO. See DOI: 10.1039/c0py00015a |
| ‡ Current address: Preparative Macromolecular Chemistry, Institut für Technische Chemie und Polymerchemie, Universität Karlsruhe (TH)/Karlsruhe Institute of Technology (KIT), Engesserstr. 18, 76128 Karlsruhe, Germany. Fax: +49 721 608 5740; Tel: +49 721 608 6869; E-mail: E-mail: james.blinco@kit.edu |
| This journal is © The Royal Society of Chemistry 2010 |