Thermoresponsive polyacrylates obtained via a cascade of enzymatic transacylation and FRP or NMP
Dragos
Popescu
,
Richard
Hoogenboom
*,
Helmut
Keul
* and
Martin
Moeller
*
Institute of Technical and Macromolecular Chemistry, RWTH Aachen and DWI an der RWTH Aachen e.V., Pauwelsstr. 8, D-52056, Aachen, Germany. E-mail: moeller@dwi.rwth-aachen.de; keul@dwi.rwth-aachen.de; r.hoogenboom@tue.nl
First published on 7th May 2010
Abstract
Following a cascade reaction comprising enzymatic monomer synthesis and radical polymerization, different functional poly(acrylate)s were obtained. Transacylation of methyl acrylate (MA) with diethylene glycol monoethyl ether, 1,2-propanediol or 1,2,6-hexanetriol in the presence of Novozyme 435 led to functional acrylate monomers. In a subsequent step, after filtration of the enzyme, removal of MA by distillation and eventual addition of a hydrophilic co-monomer, (2-hydroxyethyl acrylate, HEA), the mixture was copolymerized via free radical and nitroxide mediated polymerization resulting in poly(acrylate)s with predefined functionalities. Furthermore the thermoresponsive properties of the resulting copolymers are discussed with respect to the polymer composition and the molecular weight distribution.
1. Introduction
Polymers that undergo phase transitions in response to external stimuli have been widely investigated in various fields.1–3 For the development of robust “smart” materials the response must be reversible, when the stimulus is stopped or another one is applied. Several stimuli were studied, such as, temperature, pH, ionic strength, or light. Many studies focus on the design of temperature-responsive materials able to respond in a physiological environment. Small organic compounds as well as polymers may exhibit a phase transition from soluble to insoluble upon heating. The lower critical solution temperature (LCST) is the critical temperature below which a mixture is miscible in all proportion. This behaviour is based on hydrogen bonding between water molecules and groups of the polymer chain. Weakening of the hydrogen bonds at higher temperatures causes entropy-driven phase transition of the polymer to a hydrophobic collapsed state.4 The polymers with LCST behaviour show a sudden and mostly reversible change from hydrophilic to hydrophobic that makes them attractive for application in biotechnology, drug delivery systems,5 tissue engineering6 and biomolecule separation7,8 but are also of interest for catalysis.9The most widely studied thermoresponsive polymer is poly(N-isopropylacrylamide) (P(NIPAM)) due to its LCST of 32 °C close to body temperature and the fact that its phase transition temperature is relatively insensitive to the environmental conditions like pH and concentration.10 However, in recent years more polymers have been introduced as potential alternatives to P(NIPAM) including poly(oligoethylene glycol methacrylate)s11,12 and poly(2-oxazoline)s.13–15 An interesting polymer that surprisingly received only little attention is poly(2-hydroxypropyl acrylate) (P(2HPA)), with a cloud point of 16 °C at a concentration of 10 wt%.4 Although the thermosensitivity of P(2HPA) was already reported in 1975,4 only a few studies refer to 2HPA as comonomer for the preparation of thermoresponsive hydrogels.16,17 Recently the copolymerization of 2HPA with N,N-dimethylacrylamide, N-acryloyl morpholine, aminoethyl methacrylate hydrochloride and 2-hydroxyethyl acrylate was reported with the goal to prepare well-defined copolymers with tuneable LCST.18–20 Another polymer with a low LCST is poly(diethylene glycol ethyl ether acrylate) (P(DEGEA)). Recently, Boyer et al. determined the cloud point of P(DEGEA) to be 15 °C at a concentration of 0.1 wt%,21 while Skrabania et al. observed the cloud point at 9 °C also at the same concentration.22 Another recent paper has reported the synthesis of P(DEGEA) by RAFT polymerization, with the homopolymer presenting also a cloud point (CP) of 9 °C.23 Because of its low LCST, copolymerization of DEGEA with more hydrophilic monomers should lead to a library covering a broad range of transition temperatures. Thus, copolymers of DEGEA with oligoethylene glycol acrylate revealed copolymers with LCSTs ranging from 15 °C till 92 °C.21 A similar study was reported by Lutz for methacrylate monomers.12,24
Because the LCST is strongly dependent on parameters, such as the molecular weight and polydispersity index,25 controlled radical polymerizations are mostly used to prepare well-defined polymers. In recent years, significant advances have been made in the field of controlled radical polymerization (CRP). A variety of well-defined polymers with complex architectures have been obtained by atom transfer radical polymerization (ATRP),26,27 reversible addition–fragmentation chain transfer polymerization (RAFT)28,29 and nitroxide mediated polymerization (NMP).30,31 Radical polymerizations are particularly interesting for the preparation of functional polymers, due to the high compatibility with many functional groups and polar solvents, including water. This is in contrast to ionic polymerizations, which require strictly inert conditions and cannot tolerate protic solvents. The major drawback of free radical polymerization is related to the lack of control over the polymer structure,32,33 which could be important for the preparation of polymers with sharp LCST transitions.25
Besides FRP, NMP was applied in the current study inspired by the progress in the development of highly reactive alkoxyamine initiators (N-tert-butyl-N-(1′-diethylphosphono-2,2′-dimethylpropyl)-O-(2-carboxyl-prop-2-yl); Blocbuilder™)34 and nitroxides (N-tert-butyl-N-(1′-diethylphosphono-2,2′-dimethylpropyl); SG-1)18,19,35,36 (Scheme 1). The initiating system—Blocbuilder™/SG-1, greatly improved the efficiency of acrylate polymerizations.37–40
 | ||
| Scheme 1 Chemical structures of Blocbuilder™ alkoxyamine initiator, SG-1 free nitroxide and the monomers. Boxes indicate structural isomers that are obtained via enzymatic acylation of the diol/triol substrates. | ||
In the present work we report the nitroxide mediated polymerization as well as the free radical polymerization of DEGEA, 2HPA and dihydroxyhexyl acrylate, DHHA (Scheme 1), obtained via enzymatic-catalyzed transacylation.41,42 In addition, the copolymerization of DEGEA with various amounts of HEA and copolymerization of DHHA with various amounts of MA was studied in order to prepare a library of copolymers with tuneable LCST. The effect of the polymerization procedure on the LCST was investigated as well.
2. Experimental part
2.1. Materials
Methyl acrylate (MA, 99%, Acros Organics), methyl methacrylate (MMA, 99%, Fluka), diethylene glycol monoethyl ether (DEGEE, 99%, Sigma-Aldrich), 2-hydroxyethyl acrylate (HEA, 96%, Sigma-Aldrich), 1,2-propanediol (1,2Pdiol, 99.5+%, Sigma-Aldrich), 1,2,6-hexanetriol (1,2,6Htriol, 97%, Acros Organics), azobisisobutyronitrile (AIBN, 98+%, Fluka), 2-methyl-2-butanol (2Me2BuOH, 96+%, Fluka), N,N-dimethylformamide (DMF, 98%, Fluka) and all solvents were used as received. MA was passed through a neutral aluminium oxide column before use to remove the inhibitor. A commercial lipase, Novozyme 435 (Lipase B from Candida antarctica immobilized on a macroporous acrylic resin, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 U g−1 Novo Nordisk) was dried under reduced pressure at room temperature for 24 h and stored under nitrogen before it was used as a biocatalyst for the transacylation reactions. Blocbuilder™ initiator and SG-1 (85%) free nitroxide were kindly provided by Arkema and used as received.
000 U g−1 Novo Nordisk) was dried under reduced pressure at room temperature for 24 h and stored under nitrogen before it was used as a biocatalyst for the transacylation reactions. Blocbuilder™ initiator and SG-1 (85%) free nitroxide were kindly provided by Arkema and used as received.
All reactions were carried out under nitrogen atmosphere. Nitrogen (Linde, 5.0) was passed over molecular sieves (4 Å) and finely distributed potassium on aluminium oxide before use.
2.2. Measurements
1H NMR spectra were recorded on a Bruker DPX-400 FT-NMR spectrometer at 400 MHz, using deuterated dimethyl sulfoxide (DMSO-d6) as solvent. Chemical shifts are reported relative to the residual solvent signals.Size exclusion chromatography (SEC) was carried out using DMF (containing 1.0 g LiBr per L) or DMAc (containing 2.1 g LiCl per L) as eluting solvents. For DMF as eluting solvent a high pressure liquid chromatography pump (Bischoff HPLC), a Jasco 2035-plus RI detector and four MZ-DVB gel columns (30 Å, 100 Å, 1000 Å and 3000 Å) were used in series at 30 °C with a flow rate of 1.0 mL min−1. For DMAc as eluting solvent a Shimadzu system with a SCL-10A system controller, a LC-10AD pump, a RID-10A refractive index detector and PSS gram 30 (pore size 30 Å; bead size 10 µm) and PSS gram 1000 (pore size 1000 Å; bead size 10 µm) columns were used in series at 60 °C with a flow rate of 1.0 mL min−1. The molecular weights and polydispersities were calculated using a poly(methyl methacrylate) (PMMA) calibration.
Cloud points of aqueous polymer solutions were determined by turbidity measurements in a Crystal 16™ by Avantium Technologies or by measuring the absorbance in a Varian Cary 100 spectrophotometer for the copolymers P[(DHHA)-co-(MA)]. In the first case, four blocks of four parallel temperature controlled sample holders are connected to a Julabo FP40 cryostat allowing 16 simultaneous measurements. Turbidity of the solutions was measured by the transmission of red light through the sample vial as a function of the temperature. In the case of the UV-VIS spectrometer, the absorbance was monitored as a function of temperature at a fixed wavelength of 500 nm and in the end the results were transformed in transmittance. The cloud points are given as the 50% transmittance point during the second heating ramp. Solutions of the polymers were prepared in deionized water and were stirred at room temperature until all polymer was dissolved or dispersed. Two heating/cooling cycles were applied from 0 °C to 105 °C at 1.0 °C min−1 with hold steps of 5 min at the extreme temperatures.
Dynamic light scattering measurements were performed on a Malvern Zetasizer Nano ZS Model ZEN3600 used for the determination of the Rh the cumulants analysis and for the determination of the distribution CONTIN. The back scattered light was determined at 173°.
3. Syntheses
3.1. Representative procedure for lipase-catalyzed transacylation reactions
Methyl acrylate (5 g, 58 mmol), diethylene glycol monoethyl ether (7.78 g, 58 mmol) and Novozyme 435 (500 mg, 10 wt% with respect to MA), were added and stirred for 24 h at ambient temperature. The reaction progress was monitored by 1H NMR spectroscopy. After 24 h the following molar ratio of products was calculated: MA : DEGEA = 70% : 30%. The enzyme was removed by filtration, MA by distillation under reduced pressure at 40 °C and the stock solution was stored at 4 °C up to polymerization (Table 1, entries 1 to 8). All other enzymatic transacylation reactions were performed according to this procedure, Table 1.| No. | MA/g (mmol) | Alcohol | Novozyme 435/g | |
|---|---|---|---|---|
| Name | g (mmol) | |||
| a Reactions performed in bulk for 24 h at ambient temperature. b Reactions performed in 50 vol% 2Me2BuOH at 50 °C, t = 24 h. c Reactions performed in 50 vol% 2Me2BuOH at 50 °C, t = 8 h. d Reactions performed in 50 vol% 2Me2BuOH at 50 °C, t = 4 h. e Reactions performed in 50 vol% 2Me2BuOH at 50 °C, t = 2 h. | ||||
| 1a | 5 (58) | DEGEE | 7.78 (58) | 0.5 |
| 2a | 5 (58) | DEGEE | 7.78 (58) | 0.5 |
| 3a | 15 (174) | DEGEE | 23.38 (174) | 1.5 |
| 4a | 15 (174) | DEGEE | 23.38 (174) | 1.5 |
| 5a | 5 (58) | DEGEE | 7.78 (58) | 0.5 |
| 6a | 5 (58) | DEGEE | 7.78 (58) | 0.5 |
| 7a | 5 (58) | DEGEE | 7.78 (58) | 0.5 |
| 8a | 5 (58) | DEGEE | 7.78 (58) | 0.5 |
| 9b | 10 (116) | 1,2Pdiol | 17.7 (232.3) | 1 |
| 10b | 10 (116) | 1,2,6Htriol | 46.7 (348) | 1 |
| 11c | 10 (116) | 1,2,6Htriol | 46.7 (348) | 1 |
| 12d | 10 (116) | 1,2,6Htriol | 46.7 (348) | 1 |
| 13e | 10 (116) | 1,2,6Htriol | 46.7 (348) | 1 |
3.2. Representative procedure for free radical polymerization (FRP) of the monomers resulting from the enzymatic transacylation reactions
In a Schlenk tube, a mixture of DEGEA stock solution in DEGEE (5.6 mL, 10.08 mmol DEGEA), ethanol (5.6 mL) and AIBN (16.5 mg, 0.10 mmol) was bubbled with nitrogen for at least 30 min to deoxygenate the solution and then immersed into an oil bath, preheated to 70 °C. For analyses, samples (200 µL) were withdrawn from the polymerization mixture after 240 minutes. For SEC analysis, 100 µL of this sample were diluted with 2.0 mL DMAc and for 1H NMR analysis 100 µL of the sample were diluted with 600 µL DMSO-d6. Subsequently, the reaction mixture was cooled to room temperature and the crude polymerization mixture was precipitated in hexane (400 mL) followed by overnight standing at 4 °C. The precipitated polymer was collected by decanting the solvent followed by drying under reduced pressure at 40 °C, yielding the polymer in 70% yield, (entry 1, Table 2). All other FRP reactions were performed according to this procedure and are listed in Table 2.| Namea | Stock solution from transacylation | EtOH/mL | AIBN/mg (mmol) | HEA/g (mmol) | C M /M | ||
|---|---|---|---|---|---|---|---|
| No.b | mL | mmol acrylate | |||||
| a Names indicate the molar ratio of the monomers. b The numbers refer to the stock solutions reported in Table 1. c Reactions performed after removal of MA by distillation under reduced pressure. d Reactions performed without the removal of MA after the enzymatic transacylation. e C M is the monomer molar concentration in the final monomer solution and was determined, knowing the product distribution from 1H NMR spectroscopy and the densities of the compounds in the system. For the density values of the acrylates, which are not reported in the literature we used 1 g L−1. All CM values from the table are determined after addition of EtOH or DMF. | |||||||
| P(DEGEA) | 1c | 5.6 | 10.08 | 5.6 | 16.5 (0.10) | — | 0.9 |
| P(DEGEA75-co-HEA25) | 3c | 6.3 | 12.6 | 6.7 | 28 (0.17) | 0.49 (4.2) | 1.3 |
| P(DEGEA50-co-HEA50) | 5c | 5.3 | 10.6 | 6.4 | 35 (0.21) | 1.23 (10.6) | 1.7 |
| P(DEGEA25-co-HEA75) | 7c | 5.6 | 11.2 | 9.1 | 73 (0.44) | 3.9 (33.6) | 2.5 |
| P(2HPA) | 9c | 6 | 14.4 | 6 | 23.6 (0.14) | — | 1.2 |
| P(DHHA) | 10c | 10 | 14.27 | 10 | 23.4 (0.14) | — | 0.7 |
| P(DHHA63-co-MA37) | 11d | 10 | 11 | 10 | 18.1 (0.11) | — | 0.55 |
| P(DHHA55-co-MA45) | 12d | 10 | 11 | 10 | 18.1 (0.11) | — | 0.55 |
| P(DHHA38-co-MA62) | 13d | 10 | 11 | 10 | 18.1 (0.11) | — | 0.55 |
The copolymers P[(DEGEA)-co-(HEA)] were isolated by precipitation in a mixture of diethyl ether/hexane, while P(2HPA), P(DHHA) as well as the P[(DHHA)-co-(MA)] were purified via dialysis against water, using a cellulose ester membrane with 100–500 Da molecular weight cut off (MWCO) and isolated by lyophilization. The polymers were obtained in 50–80% yield.
3.3. Representative procedure for nitroxide mediated polymerization (NMP) of the monomers resulting from the enzymatic transacylation reactions
In a Schlenk tube, a mixture of DEGEA stock solution in DEGEE (6 mL, 12 mmol DEGEA), Blocbuilder™ initiator (45 mg, 0.12 mmol) and SG-1 free nitroxide (3.5 mg, 0.012 mmol) was bubbled with nitrogen for at least 30 minutes to deoxygenate the solution and then the tube was immersed into an oil bath, preheated to 110 °C. For the kinetic investigations, samples (600 µL) were withdrawn from the polymerization mixture after 7, 15, 30, 60, 120, 240 and 480 minutes. For SEC analysis, 100–500 µL of this sample were diluted with 2.0 mL DMF and 100 µL of the sample were diluted with 600 µL DMSO-d6 for 1H NMR analysis. After 8 hours polymerization time, the reaction mixture was cooled to room temperature and the crude polymerization mixture was precipitated in hexane (400 mL) followed by overnight standing at 4 °C. The precipitated polymer was collected by decanting the solvent followed by drying under reduced pressure at 40 °C, yielding the polymer in 47% yield, entry 1, Table 3.| Namea | Stock solution from transacylation | Observations | C M /M | ||
|---|---|---|---|---|---|
| No.b | mL | mmol acrylate | |||
| a Numbers in the name indicate the molar ratio of the monomers. b The numbers refer to the stock solution reported in Table 1. c Reactions performed after removal of MA by distillation under reduced pressure. d Reactions performed without the removal of MA after the enzymatic transacylation. e C M is the monomer molar concentration in the final monomer solution and was determined, knowing the product distribution from 1H NMR spectroscopy and the densities of the compounds in the system. For the density values of the acrylates, which are not reported in the literature we used 1 g L−1. All CM values from the table are determined after addition of EtOH or DMF. | |||||
| P(DEGEA) | 2c | 6 | 12 | Polymerization in bulk | 2 |
| P(DEGEA75-co-HEA25) | 4c | 6 | 11.66 | Add HEA (0.451 g, 3.88 mmol) | 2.4 |
| P(DEGEA50-co-HEA50) | 6c | 5 | 9.85 | Add HEA (1.14 g, 9.85 mmol) | 3.2 |
| P(DEGEA25-co-HEA75) | 8c | 4 | 7.9 | Add HEA (2.74 g, 23.64 mmol) | 4.8 |
| P(2HPA) | 9c | 7 | 16.8 | Add DMF (1 mL) | 2.1 |
| P(DHHA) | 10c | 7 | 10 | Add DMF (3 mL) | 1 |
| P(DHHA63-co-MA37) | 11d | 7 | 7.7 | Add DMF (3 mL) | 0.77 |
| P(DHHA55-co-MA45) | 12d | 7 | 7.7 | Add DMF (3 mL) | 0.77 |
| P(DHHA38-co-MA62) | 13d | 7 | 7.7 | Add DMF (3 mL) | 0.77 |
Same procedure was applied for the homopolymerization of HEA (CM = 3 M) in DEGEE, the homopolymer was precipitated in diethyl ether. All other NMP reactions were performed according to this procedure and are presented in Table 3. The copolymers P(DEGEA-co-HEA) were isolated by precipitation in a mixture of diethyl ether/hexane, while P(2HPA), P(DHHA) as well as the P(DHHA-co-MA) were purified via dialysis against water, using a cellulose ester membrane with 100–500 Da molecular weight cut off (MWCO), and isolated by lyophilization. Yields were not determined.
4. Results and discussions
Our previous research focused on developing a simple route for preparing highly functional poly[(meth)acrylate]s via a cascade reaction of enzymatic transacylation and free radical or nitroxide mediated polymerization.41,43 In the present work, this previous approach is expanded to the more challenging syntheses of hydroxy functional poly(acrylate)s via enzymatic transacylation of methyl acrylate with different functional alcohols: a functional monoalcohol, DEGEE; a functional diol, 1,2Pdiol and a functional triol, 1,2,6Htriol (Scheme 2, 1st step). In a subsequent step, after removal of the enzyme by filtration and removal of the MA by distillation, the monomer mixture was copolymerized via free radical polymerization and nitroxide mediated polymerization. In some cases different amounts of HEA were added as comonomer to control the phase transition temperature of the thermoresponsive polymers (Scheme 2, 2nd step).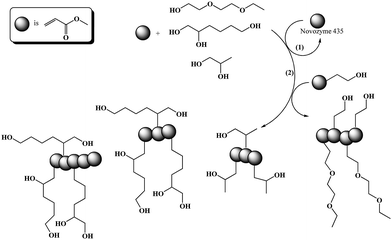 | ||
| Scheme 2 Cascade reaction comprising (1) an enzymatic transacylation and (2) a radical polymerization. | ||
4.1. Lipase-catalyzed transacylation
In order to prepare functional acrylates or methacrylates, the transacylation of MA and MMA as substrates was first investigated with a monofunctional alcohol, DEGEE, as reagent using Novozyme 435 as catalyst. Under the applied conditions, namely ambient temperature and an equimolar ratio between DEGEE and M(M)A in bulk, this transacylation resulted in a mixture of two (meth)acrylates.The product mixture obtained was analyzed by 1H NMR spectroscopy using DMSO-d6 as solvent since it is known that in this solvent coupling between CH2 and the OH protons might be observed.44Fig. 1 shows the signals of the starting materials (t0): characteristic for MA are signal 3 at δ = 3.68 ppm (the methyl ester group) and signals 1 and 2 at δ = 5.9–6.3 ppm (protons at the sp2 hybridised C-atoms). After 24 h (t24) the formation of DEGEA is evidenced by the appearance of signals A (δ = 4.22 ppm) and B (δ = 3.64 ppm) as triplets. The appearance of signals for methanol (δ = 3.17 ppm and δ = 4.11 ppm) further indicates that the transacylation was successful. Since the signals of the double bonds of the product (DEGEA) and MA overlap, the integral of these signals was taken as reference. The ratio of DEGEA/MA was calculated to be 30/70 mol%. This result was confirmed by the ratio of methanol to MA. Similar calculations were performed for all the transacylation reactions. The removal of MA and methanol by distillation was confirmed by the disappearance of the corresponding signals (Fig. 1, tf). In addition, the molar concentration of the DEGEA in the solution was calculated using the ratios between DEGEA and DEGEE determined by 1H NMR spectroscopy and the densities of the compounds (dDEGEA = 1.013 g L−1 and dDEGEE = 0.987 g L−1).
 | ||
| Fig. 1 1H NMR spectra from the enzymatic transacylation reaction between MA and DEGEE at time 0 h (t0), after 24 h (t24) and after removal of MA under reduced pressure (tf). (Reaction conditions: MA : DEGEE = 1 : 1 molar ratio, rt in bulk.) | ||
In order to gain a better understanding of the lipase-catalyzed transacylation, kinetic studies were performed using MA and MMA as acyl donors, DEGEE as a functional monoalcohol and Novozyme 435 as catalyst. All the reactions were performed in bulk using equimolar amounts of alcohol and acyl donor at ambient temperature, proving the simplicity of the procedure. At different reaction times samples were taken for the determination of the product yield. From the time/conversion diagram, (Fig. 2) it was concluded that acrylates react faster than methacrylates, which is in very good agreement with our previous results and is most likely related to the higher sterical demand of MMA than MA in the enzymatic transacylation reaction.41,42 Furthermore, in the case of MA as substrate, the equilibrium is reached after 24 h, while with MMA as substrate even after 4 days, equilibrium is not achieved. Based on this kinetic study, all following transacylations of MA with DEGEE were performed for 24 h at ambient temperature using an equimolar ratio of the reagents.
 | ||
| Fig. 2 Transacylation of M(M)A with DEGEE (molar ratio 1 : 1), in bulk, at ambient temperature: conversion of M(M)A in time. | ||
The enzymatic transacylations of a diol (1,2Pdiol) and triol (1,2,6Htriol) were performed with MA using 2-methyl-2-butanol, a polar tertiary alcohol, as co-solvent to reduce the viscosity of the reaction mixture. The temperature of the reaction was set to 50 °C while the molar ratio of the acyl donor/diol (triol) was set to 1/number of OH groups in the molecule according to a theoretical model presented in the literature.45–47 The equilibrium of these enzymatic transacylations is reached after 24 h.42 For the reaction of MA with 1,2Pdiol, an isomeric mixture of monoacrylates (2HPA 88 mol% and 1MeHEA 11 mol%) containing less than 1 mol% bisacrylate was obtained after 24 hours at a MA conversion of 50%. For the asymmetrical triol, 1,2,6Htriol, an isomeric mixture of three monoacrylates (94 mol%) and a concentration of 6 mol% bisacrylates are obtained after 24 hours at 75% MA conversion. These results are in accordance with those reported previously42 showing the reproducibility of the enzymatic transacylation reactions. After quenching the reactions by cooling to room temperature, filtering the Novozyme 435 and removing the excess of MA by distillation under reduced pressure, the monomers were analyzed by 1H NMR spectroscopy to determine the ratio between hydroxy acrylate, diol (triol) and 2Me2BuOH. The molar concentrations of the acrylates in the remaining solutions were calculated using the densities of the compounds. If no densities were reported in literature, a density of 1.00 g L−1 was assumed for the acrylates. Hence, a molar concentration of 2.4 mol L−1 (HPA) and 1.4 mol L−1 (DHHA) was determined.
In the case of 1,2,6Htriol as asymmetrical triol, a kinetic study was performed in order to determine the conversion of MA in time (Fig. 3). Samples were taken at: 0 h, 2 h, 4 h, 8 h, 16 h and 24 h and were analysed by 1H NMR spectroscopy demonstrating that the equilibrium of this transacylation is reached after 16 hours. Based on this kinetic study, three reaction times were selected for the transacylation reactions, namely 2, 4 and 8 hours, to obtain different DHHA/MA ratios that can be directly copolymerized after removal of the enzyme.
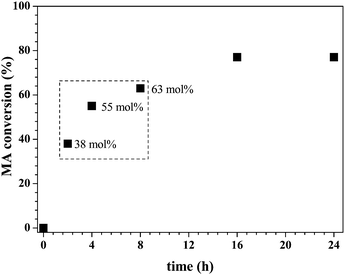 | ||
| Fig. 3 Transacylation of MA with 1,2,6Htriol (molar ratio 1 : 3), in 2Me2BuOH, at T = 50 °C: conversion of MA in time. Dashed box indicates the composition of the stock solutions used for polymerizations. | ||
4.2. Free radical polymerization
The cascade reaction concept41 was applied for the synthesis of hydroxy functional polyacrylates: transacylation of MA with a functional alcohol, diol or triol in the first step resulted in the desired acrylates, which in the second step, after filtration of Novozyme 435 and (eventually) removal of MA by distillation and/or addition of HEA, were polymerized. Free radical polymerization was our first choice since radical polymerization is particularly attractive for industrial applications of acrylates.Thus, after removal of Novozyme 435 and of MA from the reaction mixture of enzymatic transacylation of MA and DEGEE, 1 mol% AIBN was added and the mixture was heated to 70 °C. The monomer conversion was determined via1H NMR spectroscopy by comparison of the integral of the acrylate signals at δ = 6.3–5.9 ppm at time 0 and at the end of polymerization using the signal at δ = 4.36 ppm corresponding to the –OH group of DEGEE, as reference. The final polymer was purified by precipitation in hexane and analysed by 1H NMR spectroscopy and size exclusion chromatography (SEC) using DMF as eluting solvent (Table 4).
| Polymera | Conversionb (%) | M n /Da | PDId | Polymer compositionb (mol%) |
|---|---|---|---|---|
| a Numbers in the name indicate the monomer molar ratio. b Calculated by 1H NMR spectroscopy. c Number average molecular weight (Mn) and polydispersity index (PDI) of isolated polymers determined by size exclusion chromatography (SEC) using N,N-dimethylformamide as eluent. d Total monomer conversion calculated by 1H NMR spectroscopy. | ||||
| P(DEGEA) | 93 | 16600 |
2.1 | P(DEGEA) |
| P(DEGEA75-co-HEA25) | 95/97 | 21000 |
2 | P(DEGEA78-co-HEA22) |
| P(DEGEA50-co-HEA50) | 94/95 | 22200 |
3 | P(DEGEA55-co-HEA45) |
| P(DEGEA25-co-HEA75) | 99/99 | 30900 |
2.7 | P(DEGEA29-co-HEA71) |
| P(DHHA) | 98 | 6100 | 1.3 | P(DHHA) |
| P(DHHA63-co-MA37) | 92d | 8100 | 2.1 | P(DHHA67-co-MA33) |
| P(DHHA55-co-MA45) | 91d | 7200 | 2 | P(DHHA60-co-MA40) |
| P(DHHA38-co-MA62) | 88d | 8000 | 2.2 | P(DHHA43-co-MA57) |
| P(2HPA) | 75 | 11200 |
3.2 | P(2HPA) |
It was reported in the literature that P(DEGEA) exhibits a CP at ∼9 °C.22,23 To increase the cloud point of the polymer, copolymerization with a more hydrophilic monomer is required. Thus, DEGEA was copolymerized with HEA in different molar ratios: 25 mol%, 50 mol% and 75 mol%, under the same conditions. The final copolymers were isolated from the polymerization mixture by precipitation in a mixture of hexane and diethyl ether. The compositions of the isolated copolymers were determined by 1H NMR spectroscopy using the integrals of the signals corresponding to the –OH groups of HEA at δ = 4.66 ppm and those corresponding to the methylene groups of both DEGEA and HEA at δ = 4.11 ppm and δ = 4.00 ppm.
According to the same procedure P(2HPA) and P(DHHA) were prepared starting with the stock solutions obtained after enzymatic transacylation. To reduce the viscosity of the reaction medium, 50 vol% ethanol was added to the stock solution. The characteristics of the polymers are summarized in Table 4.
Finally, in order to increase the hydrophobicity of P(DHHA) direct copolymerization of the MA–DHHA monomer mixture resulting after the enzymatic transacylation was performed. For this purpose three transacylation reactions of MA with 1,2,6Htriol were performed using the same amount of reagents, enzyme and temperature but different reaction times: 2 h, 4 h and 8 h (according to Fig. 3). 1H NMR analysis of the reaction product revealed the composition of the copolymers (Table 4). A representative example for an 1H NMR spectrum is given in Fig. 4. Characteristic signals are observed for the hydroxyl protons (δ = 4.8–4.2 ppm) and the methylene and methine groups adjacent to oxygen atoms (δ = 4.1–3.2 ppm).
![1H NMR spectrum of the isolated copolymer P[(DHHA)67-co-(MA)33].](/image/article/2010/PY/c0py00051e/c0py00051e-f4.gif) | ||
| Fig. 4 1H NMR spectrum of the isolated copolymer P[(DHHA)67-co-(MA)33]. | ||
All hydroxy (co)polymers obtained via free radical polymerization as well as those prepared by nitroxide mediated polymerization, see next section, were purified by dialysis in water, using a cellulose ester membrane with 100–500 Da molecular weight cut off (MWCO). The molecular weight (Mn) and the polydispersity indices (Mn/Mw) of the purified polymers are summarized in Table 4. SEC analyses using DMF as eluent revealed for all the polymers the expected molar mass distribution for free radical polymerization, namely monomodal molecular weight distribution with polydispersity indices around 2 (Table 4). The low Mn and PDI values for P(DHHA) are most likely due to limited solubility of this polymer in DMF, resulting in a smaller hydrodynamic volume during SEC.
4.3. Nitroxide mediated polymerization (NMP)
In order to obtain well-defined polymers, the monomers resulting from the enzymatic transacylation were also polymerized via nitroxide mediated polymerization. In the current work, the nitroxide mediated (co)polymerization of the new functional acrylates was performed using the previously obtained optimal conditions:18,19,43 polymerization temperature T = 110 °C and 10% excess SG-1 free nitroxide relative to the Blocbuilder™ initiator. The polymerizations were performed for eight hours either in the direct stock solution obtained after enzymatic transacylation or by addition of DMF which is known to increase the polymerization rate in NMP.48 Since the monomer solutions are viscous due to the excess of diol/triol remaining from the monomer synthesis, the addition of DMF is also required for decreasing the viscosity. In the following, first the homopolymerization of DEGEA and HEA and their copolymerization are described followed by the copolymerization of DHHA with MA.DEGEA (2 M solution) and HEA (3 M solution) were polymerized directly after enzymatic transacylation in DEGEE stock solution. The reaction progress was monitored by analysing samples withdrawn from the polymerization mixture at different time intervals. The first order kinetic plots are linear indicating a controlled polymerization (Fig. 5, up). From the slope of the first-order kinetic plot the apparent rate constants (kapp) were determined.
![Kinetic plot of ln([M]0/[M]t) versus time (up) and number-average molecular weight (Mn) and polydispersity index (PDI) vs. conversion (down) for the nitroxide mediated radical polymerizations of DEGEA and HEA. (Reaction conditions: monomer/Blocbuilder™/Sg-1 ratio of 100/1/0.1 in DEGEE and T = 110 °C.)](/image/article/2010/PY/c0py00051e/c0py00051e-f5.gif) | ||
| Fig. 5 Kinetic plot of ln([M]0/[M]t) versus time (up) and number-average molecular weight (Mn) and polydispersity index (PDI) vs. conversion (down) for the nitroxide mediated radical polymerizations of DEGEA and HEA. (Reaction conditions: monomer/Blocbuilder™/Sg-1 ratio of 100/1/0.1 in DEGEE and T = 110 °C.) | ||
The linear increase of the molecular weight with conversion as well as the relatively narrow molecular weight distributions (polydispersity index, PDI < 1.4) further demonstrate a controlled polymerization of HEA as well as DEGEA (Fig. 5, down). In addition, the SEC traces of the polymers are monomodal and are shifted to lower elution volumes, with increasing reaction time (Fig. 6, up).
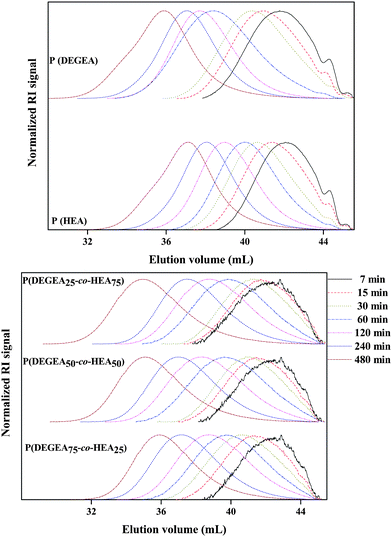 | ||
| Fig. 6 Size exclusion chromatography traces for the homopolymerizations of DEGEA and HEA (up) and the copolymerizations of DEGEA with HEA (down). (Reaction conditions: monomer/Blocbuilder™/SG-1 ratio of 100/1/0.1 in DEGEE and T = 110 °C.) | ||
The copolymerization kinetics of DEGEA with 25, 50 and 75 mol% HEA were investigated too. The molar concentration of the DEGEA solution was determined via1H NMR spectroscopy using the ratios between DEGEA and DEGEE and the densities of the compounds (dDEGEA = 1.013 g L−1 and dDEGEE = 0.987 g L−1). Then, the desired amount of HEA was added. Fig. 7 clearly demonstrates that the monomer composition does not influence the control over the polymerization. The apparent rate constant increases with a factor 1.5 for each 25 mol% of HEA added. An explanation for this experimental result could be the interaction of the hydroxyl groups with the nitroxide via hydrogen bonding which has been reported to accelerate the NMP.49 This accelerating effect of the hydroxyl groups is more pronounced for the copolymerization compared to the HEA homopolymerization due to the higher monomer concentration. Furthermore, all copolymerizations showed a linear increase of molecular weight (Mn) with conversion, almost no deviation from the theoretical values (Mn,th) and relatively low PDI < 1.5 indicative of a controlled polymerization. In addition, all SEC traces are monomodal and shifted to lower elution volumes with increasing monomer conversion (reaction time) (Fig. 6, down).
![Kinetic plot of ln([M]0/[M]t) versus time (up) and number-average molecular weight (Mn) and polydispersity index (PDI) vs. theoretical number average molecular weight (Mn,th, the dashed line represents the ideal situation where Mn = Mn,th) (down) for the nitroxide mediated radical copolymerizations of DEGEA and HEA. (Reaction conditions: monomer/Blocbuilder™/SG-1 ratio of 100/1/0.1 in DEGEE and T = 110 °C.)](/image/article/2010/PY/c0py00051e/c0py00051e-f7.gif) | ||
| Fig. 7 Kinetic plot of ln([M]0/[M]t) versus time (up) and number-average molecular weight (Mn) and polydispersity index (PDI) vs. theoretical number average molecular weight (Mn,th, the dashed line represents the ideal situation where Mn = Mn,th) (down) for the nitroxide mediated radical copolymerizations of DEGEA and HEA. (Reaction conditions: monomer/Blocbuilder™/SG-1 ratio of 100/1/0.1 in DEGEE and T = 110 °C.) | ||
To evaluate the microstructure of the copolymers, i.e. monomer composition and distribution, the fraction of HEA incorporated into the polymer was determined by means of 1H NMR spectroscopy. The incorporated HEA fraction (FHEA) is close to the HEA fraction in the feed (fHEA, dotted lines) for all three copolymerizations indicating a nearly ideal random copolymerization of the two monomers (Fig. 8, up). The small deviations of FHEA from the theoretical composition are within experimental error and are due to overlapping signals in the 1H NMR spectra. The FHEA at ∼8% total monomer conversion is plotted against the HEA fraction in the feed (fHEA) (the Fineman–Ross plot (Fig. 8, down)50) proving nearly ideal random copolymerization (rHEA = 1.07 ± 0.02 and rDEGEA = 1.08 ± 0.02).
 | ||
| Fig. 8 Nitroxide-mediated copolymerization of HEA with DEGEA: fraction of HEA as a function of monomer conversion (FHEA: determined by 1H NMR spectroscopy) (up). Plot of mole fraction of HEA (FHEA) in the copolymer as a function of the mole fraction in the feed (fHEA) (down) (total monomer conversion ≈ 8%). | ||
The NMP of 2HPA and DHHA proceeded in a controlled manner as reported elsewhere,43 however, it is important to mention that due to the relatively high concentration of bisacrylates (6 mol%), in the Mnvs. conversion plots, deviation from linearity was observed at high monomer conversions (for DHHA at conversions >50%).
The copolymerization kinetics of the monomer stock solution resulting from three enzymatic transacylation of MA with 1,2,6Htriol performed at the same reaction conditions but for different times (2 h, 4 h and 8 h) were investigated. As depicted in the Fig. 9 for all three copolymerizations the first order plots revealed a constant concentration of radicals during the polymerization while the linear increase of Mn with conversion and the relatively low molecular weight distributions (PDI < 1.4) confirm the control over the polymerization. The monomodal SEC traces shown in Fig. 10 further illustrate the controlled character of the copolymerizations.
![Kinetic plot of ln([M]0/[M]t) versus time (up) and number-average molecular weight (Mn) and polydispersity index (PDI) vs. theoretical number average molecular weight (Mn,th, the dashed line represents the ideal situation where Mn = Mn,th) (down) for the nitroxide mediated radical copolymerizations of DHHA and MA. (Reaction conditions: monomer/Blocbuilder™/SG-1 ratio of 100/1/0.1 in DEGEE and T = 110 °C.)](/image/article/2010/PY/c0py00051e/c0py00051e-f9.gif) | ||
| Fig. 9 Kinetic plot of ln([M]0/[M]t) versus time (up) and number-average molecular weight (Mn) and polydispersity index (PDI) vs. theoretical number average molecular weight (Mn,th, the dashed line represents the ideal situation where Mn = Mn,th) (down) for the nitroxide mediated radical copolymerizations of DHHA and MA. (Reaction conditions: monomer/Blocbuilder™/SG-1 ratio of 100/1/0.1 in DEGEE and T = 110 °C.) | ||
 | ||
| Fig. 10 Size exclusion chromatography traces for the copolymerizations of DHHA with MA. (Reaction conditions: monomer/Blocbuilder™/SG-1 ratio of 100/1/0.1 in DMF and T = 110 °C.) | ||
The final polymers resulting from NMP were purified by precipitation in diethyl ether for P(DEGEA), in hexane for P(HEA), in mixture of diethyl ether and hexane for P[(DEGEA)-co-(HEA)] or by dialysis in water for P(2HPA), P(DHHA) and P[(DHHA)-co-(MA)] to remove the alcohols and unreacted monomers. The molecular weight and PDI of the purified copolymers are summarized in Table 5. A first conclusion after analysing the purified copolymers by 1H NMR spectroscopy, is that the DHHA has a higher reactivity in NMP than MA since the copolymers contain a higher fraction of DHHA than in the feed (Table 5). Another important observation is the big difference between the theoretical molecular weight and that determined by SEC using DMF as eluting solvent. This might be explained by the unsuitable calibration protocol based on poly(methyl methacrylate) standards, which are likely to have a different hydrodynamic radius in solution compared to these copolymers containing nearly 70 mol% DHHA repeating units.
| Polymera | Conversionb (%) | M n,th | M n /Da | PDId | Polymer compositionb (mol%) |
|---|---|---|---|---|---|
| a Numbers in the name indicate the monomer molar ratio. b Calculated by 1H NMR spectroscopy. c Theoretical number average molecular weight determined by conversion. d Number average molecular weight (Mn) and polydispersity index (PDI) of isolated polymers determined by size exclusion chromatography (SEC) using N,N-dimethylformamide as eluent. e Total monomer conversion calculated by 1H NMR spectroscopy. | |||||
| P(DEGEA) | 73 | 14100 |
9900 | 1.3 | P(DEGEA) |
| P(DEGEA75-co-HEA25) | 47/52 | 8500 | 8400 | 1.3 | P(DEGEA79-co-HEA21) |
| P(DEGEA50-co-HEA50) | 58/65 | 9700 | 12600 |
1.2 | P(DEGEA51-co-HEA49) |
| P(DEGEA25-co-HEA75) | 70/77 | 10300 |
12800 |
1.4 | P(DEGEA29-co-HEA71) |
| P(HEA) | 73 | 8800 | 7700 | 1.2 | P(HEA) |
| P(DHHA) | 63 | 12200 |
6100 | 1.2 | P(DHHA) |
| P(DHHA63-co-MA37) | 61e | 9500 | 5100 | 1.3 | P(DHHA72-co-MA28) |
| P(DHHA55-co-MA45) | 57e | 8500 | 4200 | 1.3 | P(DHHA69-co-MA31) |
| P(DHHA38-co-MA62) | 75e | 9800 | 3700 | 1.3 | P(DHHA62-co-MA38) |
| P(2HPA) | 75 | 10000 |
9000 | 1.4 | P(2HPA) |
4.4. Thermoresponsive properties of the polymers in aqueous solution
The solubility and LCST behaviour of the copolymers were investigated by turbidimetry at a concentration of 5 mg mL−1. Aqueous polymer solutions were heated and cooled in between 0 °C and 105 °C and cloud points were determined at 50% transmittance during polymer precipitation in the second heating run. The results are presented in Table 6 for the P[(DEGEA)-co-(HEA)] and P(2HPA) and in Table 7 for P[(DHHA)-co-(MA)] exhibiting cloud points between 13.2 °C (P(DEGEA)) and 47.8 °C (P[(DEGEA)25-co-(HEA)75]).| Monomer ratio in the feeda | Polymerization method | Polymer compositionb (mol%) | CPc/°C | Width of the CP,d ΔT/°C |
|---|---|---|---|---|
| a Numbers indicate the monomer molar ratio. b Calculated by 1H NMR spectroscopy. c Cloud points were determined at 50% transmittance point in second heating ramp for a 0.5 wt% polymer concentration. d Temperature interval for a complete transition from maximal to minimal transmittance. | ||||
| P(DEGEA) | FRP | P(DEGEA) | 16.5 | 1.1 |
| P(DEGEA75-co-HEA25) | FRP | P(DEGEA78-co-HEA22) | 23.7 | 2 |
| P(DEGEA50-co-HEA50) | FRP | P(DEGEA55-co-HEA45) | 29.4 | 2.4 |
| P(DEGEA25-co-HEA75) | FRP | P(DEGEA29-co-HEA71) | 47.8 | 7.3 |
| P(2HPA) | FRP | P(2HPA) | 31.7 | 6.2 |
| P(DEGEA) | NMP | P(DEGEA) | 13.2 | 1.4 |
| P(DEGEA75-co-HEA25) | NMP | P(DEGEA79-co-HEA21) | 21.3 | 2.3 |
| P(DEGEA50-co-HEA50) | NMP | P(DEGEA51-co-HEA49) | 26.9 | 4.9 |
| P(DEGEA25-co-HEA75) | NMP | P(DEGEA29-co-HEA71) | 38.5 | 10.7 |
| P(2HPA) | NMP | P(2HPA) | 28.7 | 5.8 |
| Monomer ratio in the feeda | Polymerization method | Polymer compositionb (mol%) | CPc/°C | Width of the CP,e ΔT/°C |
|---|---|---|---|---|
| a Numbers indicate the monomer molar ratio. b Calculated by 1H NMR spectroscopy. c Cloud point determined at 50% transmittance point in second heating ramp for a 0.5 wt% polymer concentration. d In the investigated temperature range from 1–105 °C. e Temperature interval for a complete transition from maximal to minimal transmittance. | ||||
| P(DHHA) | FRP | P(DHHA) | Solubled | — |
| P(DHHA63-co-MA37) | FRP | P(DHHA67-co-MA33) | 23 | 13.3 |
| P(DHHA55-co-MA45) | FRP | P(DHHA60-co-MA40) | 22.3 | 11.3 |
| P(DHHA38-co-MA62) | FRP | P(DHHA43-co-MA57) | 17.3 | 13.3 |
| P(DHHA) | NMP | P(DHHA) | Solubled | — |
| P(DHHA63-co-MA37) | NMP | P(DHHA72-co-MA28) | 44.5 | 17.8 |
| P(DHHA55-co-MA45) | NMP | P(DHHA69-co-MA31) | 42 | 20 |
| P(DHHA38-co-MA62) | NMP | P(DHHA62-co-MA38) | 36 | 18.6 |
A first remark with respect to the library of P[(DEGEA)-co-(HEA)] copolymers is that the cloud points increase with increasing FHEA due to the higher hydrophilicity of HEA in comparison with DEGEA (Table 6), which shows that the balance between hydrophilic and hydrophobic moieties in the molecular structure of the polymers is the key-parameter that determines their solution properties. In addition the polymers prepared by FRP showed in general a ∼3 °C higher cloud point in comparison to those prepared by NMP, which might be attributed to the relatively large hydrophobic SG-1 end group of the polymers obtained by NMP (Fig. 11A). However, in the case of the copolymers P[(DEGEA)25-co-(HEA)75] synthesized by FRP and NMP the difference between the cloud points is 9.3 °C. This larger difference in cloud point is most likely due to the fact that in these copolymers the ratio of hydrophilic to hydrophobic repeating units is the highest as is also the difference in molar mass. The more hydrophilic character of the copolymer will enhance the effect of the large hydrophobic SG-1 end-group in the NMP copolymer. Furthermore, this hydrophobic end-group effect is stronger for the shorter polymer chains than the longer chains from FRP.
 | ||
| Fig. 11 Cloud point as a function of polymer composition estimated by 50% transmittance points for the second heating ramp for D100 = P(DEGEA), D.H. = P(DEGEA-co-HEA) the numbers indicate the monomer ratio in the feed, P(2HPA) (A) and P(DHHA-co-MA), the numbers indicating the monomer ratio in the feed (B). | ||
In the case of the dihydroxyhexyl acrylate copolymer library, the P(DHHA) homopolymers prepared both by FRP and NMP were soluble in water because of the hydrophilicity of the hydroxyl groups that form hydrogen bonds with the surrounding water molecules. As a consequence, there was no cloud point observed in between 20 °C and 105 °C; although the solutions became translucent during heating indicative of the formation of larger aggregates, which were investigated by dynamic light scattering of 5 mg mL−1 solutions. For both homopolymers prepared via FRP and NMP the hydrodynamic radius (Rh) obtained from cumulant analysis increased with increasing temperature demonstrating the formation of soluble aggregates due to partial hydrophobic collapse of the P(DHHA), whereby larger aggregates are observed for the less-defined polymer from FRP (Fig. 12). It should be noted that mostly broad size distributions with PDI > 0.2 were obtained indicative for the formation of ill-defined aggregates as expected from the hydrophobic collapse of homopolymers. CONTIN analysis revealed that the P(DHHA) from FRP is mostly present as solubilised aggregates (Rh ≈ 50 nm; 95% intensity) at 20 °C while the more defined P(DHHA) from NMP forms single hydrated polymer chains (Rh ≈ 5 nm; 35% intensity) with a minor fraction of large aggregates (Rh ≈ 300 nm; 65% intensity). These results indicate that the lower molar mass P(DHHA) from NMP has a higher water-solubility compared to the larger polymer chains obtained by FRP. Upon heating the polymer prepared by NMP, the fraction of aggregates continuously increases while the size of the aggregates decreases most likely due to hydrophobic collapse of the aggregates. Increasing the temperature of the aqueous solution of the P(DHHA) from FRP above 20 °C causes further aggregation, surprisingly leading to a single size distribution of well-defined aggregates with Rh ≈ 300 nm and PDI ≈ 0.1 at 50 °C (100% intensity). Further heating yields a bimodal size distribution where the second distribution has a size in the micrometre range due to aggregation of the defined aggregates. It is hypothesized that the large number of hydroxyl groups in the P(DHHA) ensures solubility keeping the aggregated species in solution explaining the absence of macroscopic precipitation during the (partial) hydrophobic collapse that occurs upon heating.
![Hydrodynamic radius (Rh) as a function of temperature. [P(DHHA) aqueous solution (5 mg mL−1).]](/image/article/2010/PY/c0py00051e/c0py00051e-f12.gif) | ||
| Fig. 12 Hydrodynamic radius (Rh) as a function of temperature. [P(DHHA) aqueous solution (5 mg mL−1).] | ||
To decrease the hydrophilicity, copolymers of DHHA and MA were prepared by copolymerization of the monomer solutions resulting from three enzymatic transacylation reactions without the removal of MA. The resulting copolymers revealed a large difference in cloud point of around 19 °C for the copolymers prepared by FRP and those by NMP, (Table 7 and Fig. 11B). However, the cloud point values show the same trend for the two polymerization methods. In this case the FRP copolymers present lower cloud point values. This is rather surprising since both MA-DHHA and DEGEA-HEA copolymers prepared via NMP have the same hydrophobic SG-1 end groups, and thus, a similar slightly lower cloud point is expected. However, due to the higher reactivity of DHHA compared to MA, the composition and the molar mass of the polymers obtained via FRP and NMP are different. (i) The lower conversion in NMP leads to a higher content of hydrophilic repeating units and thus to a higher cloud point. (ii) The higher conversion in FRP leads to higher molar masses, which at the same composition will result in lower cloud points (compare P(DHHA60-co-MA40), Mn = 7200 g mol−1, obtained by FRP with P(DHHA62-co-MA38), Mn = 3700 g mol−1, obtained via NMP).
All observed phase transitions were fully reversible and almost no hysteresis was observed for the copolymers of DEGEA and HEA as well as for the homopolymer P(2HPA), as depicted in Fig. 13A and C. Nonetheless, in the case of the copolymers of DHHA and MA during the cooling process, the rehydration of the polymer chains seems to be hindered by hydrophobic interchain interactions, leading to a marked hysteresis (Fig. 13B and D). The hysteresis depends on the concentration of the polymer in solution as well as on the hydrophilic content in the copolymer. There seems to be a relation of the observed hysteresis and the content of MA in the copolymer, i.e. increasing the MA content and thus the hydrophobicity, decreases the hysteresis.
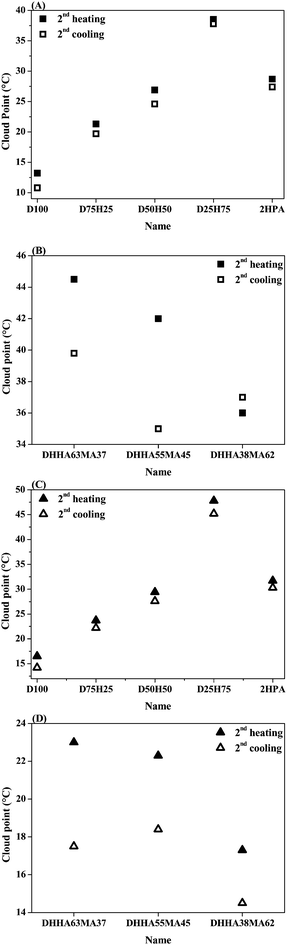 | ||
| Fig. 13 Cloud point as a function of polymer composition estimated by 50% transmittance points for the second heating ramp and for the second cooling ramp for D100 = P(DEGEA), D.H. = P(DEGEA-co-HEA) numbers indicate the monomer ratio in the feed, 2HPA (A and C) and P(DHHA-co-MA) numbers indicate the monomer ratio in the feed (B and D). The squares (■ and □) indicate the polymers synthesized by NMP and the triangles (▲ and △) the polymers synthesized by FRP. | ||
Fig. 14 illustrates the transmittance versus temperature curves and surprisingly the width of the transitions seems not to be strongly affected by the polymerization method as was previously reported in literature11,25 due to the broader molar mass distribution of the polymers resulting from FRP. In contrast to these earlier reports, a slightly broader transition is observed in the current study for the polymers synthesized by NMP for both copolymer libraries (Tables 6 and 7). This unexpected trend is related to the molar mass of the copolymers, which is significantly higher for the polymers from FRP. As such, the molar mass of these FRP polymers is above the threshold where the cloud point depends on the molar mass and, thus, the molar mass distribution does not influence the cloud point. The molar mass of the NMP polymers is below this threshold and, thus, the cloud point depends on the molar mass distribution. Furthermore, the phase transitions observed for random copolymers are slightly broader than those observed for homopolymers which is in agreement with the literature.12 Moreover, in the case of the copolymers P[(DEGEA)-co-(HEA)] an increase in the width of the CP can be observed with increasing of the FHEA. Therefore both methods can be used to prepare polymers with sharp LCST transition, while if more defined structures are targeted NMP is the method of choice.
 | ||
| Fig. 14 Transmittance as a function of temperature for aqueous solutions of (A) homopolymers P(DEGEA) and P(2HPA) obtained through free radical polymerization and nitroxide mediated polymerizations, see the graph legend; (B) library of copolymers P(DEGEA-co-HEA) obtained through free radical polymerization and nitroxide mediated polymerizations, see the graph legend. (Polymer concentration 5 mg mL−1.) | ||
5. Conclusions
Different functional (co)polymers with thermoresponsive properties were prepared following a cascade reaction comprising enzymatically (lipase-catalyzed transacylation) and chemically (FRP and NMP) catalyzed steps. Thus two different libraries of thermoresponsive copolymers were obtained on the basis of DEGEA and HEA as well as on the basis of DHHA and MA. The NMP kinetic investigations revealed that the polymerizations proceeded in a controlled manner. DEGEA and HEA revealed the same polymerization kinetics in DEGEE, while the copolymerization kinetics demonstrated that DEGEA and HEA are polymerized in a nearly ideal random fashion. The NMP copolymerization of DHHA and MA revealed that the catalytic interaction of the hydroxyl groups with the nitroxide via hydrogen bonding plays an important role in the final composition of the copolymers, which can be observed in the determination of the cloud points. The cloud points of aqueous solution of all the copolymers revealed that by increasing the fraction of hydrophilic HEA and DHHA monomers an increase in the cloud points is also observed. In the case of the FRP and NMP P[(DEGEA)-co-(HEA)] copolymer libraries, the polymers prepared by NMP showed in general a lower cloud point with around 3 °C due to the hydrophobic SG-1 end-group. In the case of the FRP and NMP P[(DHHA)-co-(MA)] copolymer libraries, the polymers prepared by FRP showed a lower cloud point with around 19 °C, which is ascribed to the inhomogeneity of the copolymers from FRP. Finally the width of the LCST transitions is not strongly affected by the polymerization method due to the relatively high molar mass of the FRP copolymers that diminishes the effect of end-groups and molar mass distribution on the phase transition.Acknowledgements
This work was carried out in the framework of the IP-project “Sustainable Microbial and Biocatalytic Production of Advanced Functional Materials” (BIOPRODUCTION/NMP-2-T-2007-026515) funded by the European Commission. Richard Hoogenboom is grateful to the Alexander von Humboldt foundation and the Netherlands Scientific Organisation (NWO) for financial support. Arkema is thanked for providing Blocbuilder™ initiator and SG-1 free nitroxide.References
- Y. Osada and J. P. Gong, Adv. Mater., 1998, 10, 827–837 CrossRef CAS.
- I. Y. Galaev and B. Mattiasson, Trends Biotechnol., 1999, 17, 335–340 CrossRef CAS.
- E. R. Gillies and J. M. J. Frechet, Pure Appl. Chem., 2004, 76, 1295–1307 CrossRef CAS.
- L. D. Taylor and L. D. Cerankowski, J. Polym. Sci., Polym. Chem. Ed., 1975, 13, 2551–2570 CrossRef.
- D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS.
- I. Lynch, I. A. Blute, B. Zhmud, P. MacArtain, M. Tosetto, L. T. Allen, H. J. Byrne, G. F. Farrell, A. K. Keenan, W. M. Gallagher and K. A. Dawson, Chem. Mater., 2005, 17, 3889–3898 CrossRef CAS.
- M. D. Costioli, I. Fisch, F. Garret-Flaudy, F. Hilbrig and R. Freitag, Biotechnol. Bioeng., 2003, 81, 535–545 CrossRef CAS.
- A. Kikuchi and T. Okano, Prog. Polym. Sci., 2002, 27, 1165–1193 CrossRef CAS.
- D. E. Bergbreiter, B. L. Case, Y.-S. Liu and J. W. Caraway, Macromolecules, 1998, 31, 6053–6062 CrossRef CAS.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- J.-F. Lutz, O. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS.
- J.-F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- R. Hoogenboom, Angew. Chem., Int. Ed., 2009, 48, 7978–7994 CrossRef CAS.
- C. Diab, Y. Akiyama, K. Kataoka and F. M. Winnik, Macromolecules, 2004, 37, 2556–2562 CrossRef CAS.
- R. Hoogenboom, H. M. L. Thijs, M. Jochems, B. M. van Lankvelt, M. W. M. Fijten and U. S. Schubert, Chem. Commun., 2008, 5758–5760 RSC.
- D. Christova, R. Velichkova, W. Loos, E. J. Goethals and F. Du Prez, Polymer, 2003, 44, 2255–2261 CrossRef CAS.
- D. I. Perera and R. A. Shanks, Polym. Int., 1995, 37, 133–139 CrossRef CAS.
- T. M. Eggenhuisen, C. R. Becer, M. W. M. Fijten, R. Eckardt, R. Hoogenboom and U. S. Schubert, Macromolecules, 2008, 41, 5132–5140 CrossRef CAS.
- R. Hoogenboom, D. Popescu, W. Steinhauer, H. Keul and M. Möller, Macromol. Rapid Commun., 2009, 30, 2042–2048 CrossRef CAS.
- K. L. Deng, H. Tian, P. F. Zhang, X. B. Ren and H. B. Zhong, eXPRESS Polym. Lett., 2009, 3, 97–104 Search PubMed.
- C. Boyer, M. R. Whittaker, K. Chuah, J. Liu and T. P. Davis, Langmuir, 2010, 26, 2721–2730 CrossRef CAS.
- K. Skrabania, J. Kristen, A. Laschewsky, Ö. Akdemir, A. Hoth and J.-F. Lutz, Langmuir, 2007, 23, 84–93 CrossRef CAS.
- A. P. Vogt and B. S. Sumerlin, Soft Matter, 2009, 5, 2347–2351 RSC.
- J.-F. Lutz, A. Hoth and K. Schade, Des. Monomers Polym., 2009, 12, 343–353 Search PubMed.
- H. Mori, H. Iwaya, A. Nagai and T. Endo, Chem. Commun., 2005, 4872–4874 RSC.
- J. S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- K. Matyjaszewski and J. H. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- G. R. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS.
- A. Studer and T. Schulte, Chem. Rec., 2005, 5, 27–35 CrossRef CAS.
- D. H. Solomon and G. Moad, The Chemistry of Free Radical Polymerization, Oxford Pergamon, 1995, p. 315 Search PubMed.
- V. Coessens, T. Pintauer and K. Matyjaszewski, Prog. Polym. Sci., 2001, 26, 337–377 CrossRef CAS.
- J. L. Couturier, O. Guerret, D. Bertin, D. Gigmes, S. Marque, P. Tordo, F. Chauvin, and P. E. Dufils (ATOFINA), Pct WO 2004/014926, 2004.
- S. Grimaldi, F. Lemoigne, J. P. Finet, P. Tordo, P. Nicol and M. Plechot (ATOCHEM ELF SA), Pct WO 96/24620, 1996.
- S. Grimaldi, J.-P. Finet, F. Le Moigne, A. Zeghdaoui, P. Tordo, D. Benoit, M. Fontanille and Y. Gnanou, Macromolecules, 2000, 33, 1141–1147 CrossRef CAS.
- F. Chauvin, P.-E. Dufils, D. Gigmes, Y. Guillaneuf, S. R. A. Marque, P. Tordo and D. Bertin, Macromolecules, 2006, 39, 5238–5250 CrossRef CAS.
- J. Nicolas, B. Charleux, O. Guerret and S. Magnet, Macromolecules, 2004, 37, 4453–4463 CrossRef CAS.
- J. Nicolas, C. Dire, L. Mueller, J. Belleney, B. Charleux, S. R. A. Marque, D. Bertin, S. Magnet and L. Couvreur, Macromolecules, 2006, 39, 8274–8282 CrossRef CAS.
- C. Dire, B. Charleux, S. Magnet and L. Couvreur, Macromolecules, 2007, 40, 1897–1903 CrossRef CAS.
- D. Popescu, H. Keul and M. Möller, Macromol. Chem. Phys., 2009, 210, 123–139 CAS.
- D. Popescu, R. Hoogenboom, H. Keul and M. Möller, J. Mol. Catal. B: Enzym., 2010, 62, 80–90 CrossRef.
- D. Popescu, R. Hoogenboom, H. Keul and M. Möller, J. Polym. Sci., Part A: Polym. Chem., 2010 DOI:10.1002/pola.24041.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS.
- M. T. Iglesias, J. Guzman and E. Riande, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 2565–2576 CrossRef CAS.
- M. T. Iglesias, J. Guzman and E. Riande, J. Polym. Sci., Part A: Polym. Chem., 1995, 33, 2057–2067 CrossRef CAS.
- F. Garcia, J. L. De la Pena, J. J. Delgado, N. Garcia, J. Guzman, E. Riande and P. Calle, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 1843–1853 CrossRef CAS.
- K. Bian and M. F. Cunningham, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 414–426 CrossRef CAS.
- E. Harth, B. V. Horn and C. J. Hawker, Chem. Commun., 2001, 823–824 RSC.
- M. Fineman and S. D. Ross, J. Polym. Sci., 1950, 5, 259–262 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |