High Tg aliphatic polyesters by the polymerization of spirolactide derivatives†
Gina L.
Fiore
,
Feng
Jing
,
Victor G.
Young, Jr.
,
Christopher J.
Cramer
and
Marc A.
Hillmyer
*
Department of Chemistry, University of Minnesota, Minneapolis, Minnesota 55455-0431, USA. E-mail: hillmyer@umn.edu
First published on 9th April 2010
Abstract
A series of bicyclic and tricyclic lactide derivatives was prepared using L-lactide as the starting material. Exomethylene-lactide served as a platform for the Diels–Alder addition of various dienes to afford norbornene-, cyclohexadiene-, and isoprene-lactide derivatives. Norbornene-lactide (NL) was also hydrogenated to the saturated norbornane analog. These new lactide derivatives were subjected to ring-opening polymerization using 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) as the catalyst. Homopolymers of poly(cyclohexadiene-lactide) (Mn = 6.4 kg mol−1), poly(isoprene-lactide) (Mn = 15.3 kg mol−1), and poly(norbornane-lactide) (Mn = 34.4 kg mol−1) exhibited glass transition temperatures (Tg's) of 119 °C, 77 °C, and 107 °C, respectively. The decomposition temperature (Td) of unsaturated PLA derivatives was ≤280 °C, whereas saturated poly(norbornane-lactide) has a Td of 300 °C. Additionally, a series of poly(norbornene-lactide-s-L-lactide) (P(NL-s-L)) copolymers with varying ratios of NL and L-lactide were prepared. The Tg's of the P(NL-s-L) samples ranged from 61 to 100 °C, and the values were well-described by the Fox equation.
Introduction
Poly(lactide) (PLA) is a biorenewable polymer derived from carbohydrate feedstocks, and its renewable origins and biocompatible properties make it attractive for a host of applications. For instance, PLA has been used as resorbable sutures1,2 and surgical fixtures,1,2 packaging materials,3–6 plastic cups,4–6 and clothing.4–6 However, PLA's broad applicability is often limited by its relatively low glass transition temperature (Tg) (∼50 to 60 °C),1,4,7,8 and consequently, developing PLA-based materials with higher Tg's is of keen interest. Several approaches can be used to tune the thermal properties of polymers, including molecular weight, tacticity, polymer architecture, and plasticization.7,8 Additionally, the Tg of a polymer can be increased by limiting the rotational freedom along a polymer backbone. For PLA, this has been achieved by replacing the methyl group on the lactide ring with bulky substituents, such as branched or cyclic groups.9–11 Baker and co-workers generated a series of lactide derivatives exploring monomer structural effects on the thermal properties of PLA based materials.7 The substituents range from phenyl to alkyl groups of varying length and structure.7,9,10,12–16 When the 3- and 6-positions of the lactide ring contain phenyl10 or cyclohexyl9 substituents, the Tg values increased to 100 and 98 °C, respectively. Conversely, when the methyl was replaced with an n-hexyl substituent, the Tg decreased to −37 °C.12 Möller et al. observed similar trends except with monosubstituted-lactide derivatives.17,18PLA derivatives have also been tailored to create materials with fast degradation profiles and a room temperature rubbery state (low Tg) for biomedical applications.2,7,8 Monomers are often decorated with protecting groups that can be removed postpolymerization to afford hydroxyl, amino, and carboxyl PLA based materials.19–23 For instance, Weck et al. prepared a benzyl protected lactide monomer starting from an amino acid derivative to afford a PLA-based material with a Tg of 18 °C.23 The benzyl groups were removed postpolymerization to afford a hydroxyl functionalized PLA material which was converted to a carboxylic acid and then coupled to RGD peptide sequence to form a bioconjugate.24
The above examples all involve the preparation of α-hydroxy acid derivatives followed by cyclization to produce the cyclic ester needed for ring-opening polymerization (ROP). The polymerization of lactide proceeds due to the release of ring strain. Therefore, this approach requires energy input to form the lactide derivative, and then release of energy upon polymerization. When lactide is used as the starting material the ring-closing step can be avoided provided the ring structure remains upon functionalization. However, very few examples exist where chemical modification of lactide does not lead to opening of the ring.25,26 To our knowledge, Scheibelhoffer et al.26 were the first to report the synthesis of an exomethylene-lactide (EML) derivative by first brominating L-lactide with N-bromosuccinimide (NBS) followed by elimination with triethylamine (Et3N) to afford the desired product. Interestingly, this approach also breaks the symmetry of the lactide ring since the second bromination to give the dibromo intermediate is slowed by the presence of the first bromo substituent. We recently reported the preparation of a tricyclic lactide derivative (norbornene-lactide) where EML was reacted with cyclopentadiene in a Diels–Alder addition.11 Norbornene-lactide was polymerized to afford a PLA-based material with a Tg of 113 °C.11 Here we expand on this work and explored the effects of other cyclic substituents on the Tg of PLA.
Experimental section
Materials
(3R,6S)-3-Bromo-3,6-dimethyl-1,4-dioxane-2,5-dione,11,26 (6S)-3-methylene-6-methyl-1,4-dioxane-2,5-dione (exomethylene-lactide),11,26 and (6S)-spiro[6-methyl-1,4-dioxane-2,5-dione-3,2′-bicyclo[2.2.1]hept[5]ene] (norbornene-lactide)11 were prepared as previously reported. Exomethylene-lactide was stored in a freezer in a dry box prior to use.27 Bromo-lactide was stored at room temperature under a nitrogen atmosphere prior to use.27 (3S)-cis-3,6-Dimethyl-1,4-dioxane-2,5-dione (L-lactide, Purac) was recrystallized from ethyl acetate (2×) and stored in a dry box under a nitrogen atmosphere. Dichloromethane was purified by passage through alumina columns using an MBraun solvent purification system. 1,5,7-Triazabicyclo[4.4.0]dec-5-ene (TBD, Aldrich) and anhydrous benzyl alcohol (Aldrich) were stored in a dry box prior to use. All other reagents were used as received.Methods
1H and 13C NMR spectra were recorded on a Varian INOVA-500 spectrometer at 500 and 125 MHz, respectively. 1H NMR spectra were referenced to the signal for residual protiochloroform at 7.260 ppm and 13C NMR spectra were referenced to the chloroform signal at 77.00 ppm. 1H NMR coupling constants are given in Hz. IR spectra were acquired using a MIDAC M4000 FT-IR spectrometer. Elemental analysis was performed at Atlantic Microlab, In., Norcross, GA. Polymer molecular weights and polydispersity indices were determined by size exclusion chromatography (SEC) using three Polymer Laboratories 5 µm mixed-C columns and a guard column and a HP1047A differential refractometer detector at 40 °C. Chloroform was used as the eluting solvent at a flow rate of 1 mL min−1, and monodisperse polystyrene standards were used to calibrate the molecular weights. Thermogravimetric analysis (TGA) was conducted using a Perkin Elmer Diamond TG/DTA Thermogravimetric/Differential Thermal Analyzer from 50 to 550 °C with a heating/cooling rate of 10 °C min−1 under N2 (Td = onset point of decomposition). Differential scanning calorimetry (DSC) measurements were acquired using a TA DSC Q1000. Samples were run under a nitrogen atmosphere from 0 to 180 °C with a heating/cooling rate of 10 °C min−1. Reported values of thermal events are from the second heating cycle unless indicated otherwise (Tg = the midpoint of the change in the heat capacity; Tm reported as the peak maximum).(6S)-Spiro[6-methyl-1,4-dioxane-2,5-dione-3,2′-bicyclo[2.2.1]hept[5]ane] (norbornane-lactide, 1-L)
Norbornene-lactide (0.500 g), 10% Pd/C (0.031 g) and THF (40 mL) were combined and sealed in an autoclave, purged with hydrogen, and then filled with 500 psi hydrogen gas. The hydrogenation was stirred at room temperature for 18 h. The crude product was filtered to remove the catalyst. The THF was removed in vacuo and the crude product was then purified by sublimation at 50 °C under vacuum. The white solid was recrystallized from cyclohexane to afford a white crystalline product: 0.473 g (94%). 1H NMR (CDCl3): δ 5.17–4.85 (m, 1H), 2.86–2.03 (m, 3H), 2.01–1.78 (m, 2H), 1.75–1.61 (m, 4H), 1.60–1.32 (m, 4H). 13C NMR (CDCl3, only the major isomer shown): δ 168.79, 167.83, 87.44, 72.64, 44.91, 41.46, 37.73, 35.44, 27.73, 21.55, 16.61. Anal. Calcd for C11H14O4: C, 62.85; H, 6.71. Found: C, 62.80; H, 6.83%. HRMS-EI (m/z): [M + H]+ calcd for C11H14O4, 210.0892; found, 210.0886. mp = 89–94 °C.(6S)-Spiro[6-methyl-1,4-dioxane-2,5-dione-3,2′-bicyclo[2.2.2]oct[5]ene] (cyclohexadiene-lactide, 2-L)
Under a nitrogen atmosphere, a 50 mL flask was charged with exomethylene-lactide (1.42 g, 9.97 mmol), 1,3-cyclohexadiene (1.60 g, 19.9 mmol), and benzene (20 mL). The reaction was stirred at reflux for 22 h. The reaction mixture was cooled down to room temperature, benzene and excess cyclohexadiene were removed in vacuo. The crude product was purified by column chromatography on silica gel, first by hexanes to remove residual cyclohexadiene, and then CH2Cl2 to elute the product. The white solid was further purified by sublimation at 50 °C under vacuum to afford white crystals: 1.13 g (51%). 1H NMR (CDCl3): δ 6.54–6.04 (m, 2H), 5.33–4.93 (m, 1H), 3.06–2.73 (m, 2H), 2.61–2.00 (m, 1H), 1.81–1.46 (m, 6H), 1.42–1.13 (m, 2H). 13C NMR (CDCl3, only the major isomer shown): δ 167.74, 167.24, 136.59, 127.48, 85.77, 72.87, 39.65, 35.97, 29.38, 22.25, 20.25, 17.20. Anal. Calcd. for C12H14O4: C, 64.85; H, 6.35. Found: C, 64.76; H, 6.30%. HRMS-EI (m/z): [M + H]+ calcd for C12H14O4, 222.0892; found, 222.0906. mp = 70–75 °C.(6S)-Spiro[6-methyl-1,4-dioxane-2,5-dione-3,4′-(1-methyl)cyclohex-1-ene] (isoprene-lactide, 3-L)
A 25 mL pressure vessel was charged with exomethylene-lactide (1.35 g, 9.54 mmol), isoprene (1.30 g, 19.1 mmol), and benzene (10 mL). Under a nitrogen atmosphere, the reaction mixture was stirred at 80 °C for 20 h. The reaction was cooled down to room temperature, benzene and excess isoprene were removed in vacuo. The crude product was purified by column chromatography on silica gel (CH2Cl2 eluent). The white solid was further purified by sublimation at 50 °C under vacuum to afford a white crystalline product: 1.27 g (63%). 1H NMR (CDCl3): δ 5.58–5.19 (m, 1H), 5.14–4.91 (m, 1H), 2.98–2.56 (m, 1H), 2.48–1.94 (m, 5H), 1.74–1.65 (m, 6H). 13C NMR (CDCl3, only the major isomer shown): δ 168.86, 166.89, 134.89, 113.12, 80.05, 72.62, 33.37, 30.54, 25.36, 23.19, 17.32. Anal. Calcd. for C11H14O4: C, 62.85; H, 6.71. Found: C, 62.89; H, 6.62%. HRMS-EI (m/z): [M + H]+ calcd for C11H14O4, 210.0892; found, 210.0896. mp = 75–79 °C.Poly(norbornene-lactide-s-L-lactide) (P(NL-s-L))
A representative procedure is provided. In a dry box, a catalyst/initiator solution was prepared by adding TBD (0.011 g, 0.078 mmol), benzyl alcohol (16 µL, 0.155 mmol), and CH2Cl2 (5.0 mL) to a 20 mL vial. A 5 mL vial was charged with L-lactide (0.031 g, 0.215 mmol), norbornene-lactide (0.175 g, 0.840 mmol), and CH2Cl2 (890 µL). An aliquot of the freshly prepared catalyst/initiator solution (170 µL) was then added to the lactide solution. The vial was sealed with a Teflon coated cap and the reaction was placed in the freezer (−20 °C). After 14 d, the reaction was precipitated into cold MeOH; the solid collected by vacuum filtration was washed with cold MeOH. The polymer product was vacuum dried overnight to afford a white solid: 0.140 g (68%, XNL = 0.58). 1H NMR (CDCl3): δ 6.51–5.76 (m), 5.16 (q, J = 7.1 Hz), 5.07–4.83 (m), 3.58–2.28 (m), 1.94–1.12 (m). Mn = 26 kg mol−1, PDI = 1.58. Tg = 87 °C.General polymerization of lactide derivatives
A representative procedure is provided. In a dry box, a catalyst/initiator solution was prepared by adding TBD (0.010 g, 0.073 mmol), benzyl alcohol (15 µL, 0.147 mmol), and CH2Cl2 (5 mL) to a 20 mL vial. A 5 mL vial was charged with 1-L (0.101 g, 0.481 mmol), and CH2Cl2 (400 µL). An aliquot of the freshly prepared catalyst/initiator solution (82 µL) was then added to the lactide solution. The vial was sealed with a Teflon coated cap and the reaction was placed in the freezer (−20 °C). After 8 d, the reaction was precipitated into cold MeOH; the solid collected by vacuum filtration was washed with cold MeOH. The white solid polymer product was vacuum dried overnight and analyzed by NMR spectroscopy, SEC, and DSC.X-Ray crystallography
A crystal of (3R,6S)-3-bromo-3,6-dimethyl-1,4-dioxane-2,5-dione and (6S)-3-methylene-6-methyl-1,4-dioxane-2,5-dione was placed onto the tip of a 0.1 mm diameter glass capillary and mounted on a CCD area detector diffractometer of a Bruker SMART system for data collection at 173 ± 2 K. A preliminary set of cell constants was calculated from reflections harvested from three sets of 20 frames. These initial sets of frames were oriented such that orthogonal wedges of reciprocal space were surveyed. This produced initial orientation matrices determined from 33 reflections. The data collection was carried out using MoKα radiation (graphite monochromator) with a frame time of 15 seconds and a detector distance of 4.9 cm. A randomly oriented region of reciprocal space was surveyed to the extent of one sphere and to a resolution of 0.77 Å. Four major sections of frames were collected with 0.30° steps in ω at four different ϕ settings and a detector position of −28° in 2θ. The intensity data were corrected for absorption and decay (SADABS).28 Final cell constants were calculated from 2432 strong reflections from the actual data collection after integration (SAINT).29 The structure was solved using SIR-97 and refined using Bruker SHELXTL.30 The space group P21 was determined based on systematic absences and intensity statistics. A direct-methods solution was calculated which provided most non-hydrogen atoms from the E-map. Full-matrix least squares/difference Fourier cycles were performed which located the remaining non-hydrogen atoms. All non-hydrogen atoms were refined with anisotropic displacement parameters. All hydrogen atoms were placed in ideal positions and refined as riding atoms with relative isotropic displacement parameters. The CIF files for the two structures are included in the ESI†.Results and discussion
Lactide derivatives
Exomethylene-lactide (EML) can serve as a platform for the addition of various dienes using a Diels–Alder approach (Scheme 1). EML was prepared by first brominating L-lactide at the 3-position using NBS, followed by elimination with Et3N to afford the captodative alkene (Scheme 1).11,26 The configurations of the bromo-lactide and EML were confirmed by X-ray crystallography (3S,6S-stereocenters see Fig. 1 and S1†) and supported by computation. Specifically, the observed stereoselectivity in the bromination is consistent with reduced strain in the transition-state (TS) (Fig. 2) and product (Fig. S2†) structures predicted for the (3R,6S) isomer. Calculations at the M06-2X/6-311G(2df,p)//M06-L/6-31G(d) level31,32 indicate that at 298 K the difference in activation free energies33 is 2.3 kcal mol−1 between the two TS structures for the reaction of Br2 with the educt radical to generate the preferred (3R,6S) and non-preferred (3S,6S) isomers. This difference is readily attributed to the reduced 1,4-diaxial strain between the reacting Br2 molecule and a hydrogen atom in (3R,6S) compared to a methyl group in (3S,6S); such strain is imposed by the boat-like TS structures preferred by the six-membered rings incorporating two esters (Fig. 2). That strain persists in the products (ΔG = 2.2 kcal mol−1 at the same theoretical level; see Fig. S2†), suggesting that kinetic control is not necessary for stereoselection in the bromination reaction. While the TS structures were computed for Br2 as a brominating agent, the greater steric demands of an NBS molecule suggest that stereoselection would be enhanced if NBS was the active brominating agent. | ||
| Scheme 1 | ||
 | ||
| Fig. 1 Molecular drawing of exomethylene-lactide (EML) with 50% probability ellipsoids. | ||
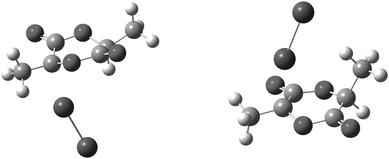 | ||
| Fig. 2 Stereostructures computed at the M06-L/6-31G(d) level for bromination TS structures (Br–Br approaching the intermediate lactide radical) leading to the preferred (3R,6S)- (left) and unpreferred (3S,6S)-3-bromo-3,6-dimethyl-1,4-dioxane-2,5-dione (right). | ||
The synthesis of norbornene-lactide (NL) was recently reported,11 where cyclopentadiene was added to EML in a Diels–Alder reaction to afford NL as a mixture of isomers with the stereoisomer shown in Scheme 1 as the preferred cycloadduct. The distribution of isomers is largely determined by the position of the 6-methyl group. When the methyl (S-stereocenter) is positioned opposite the approaching diene, this leads to the least sterically hindered transition state and thus the preferred reaction pathway to afford the major isomer (74%; 85 : 15 exo : endo selectivity).11 These conclusions were supported by 1D NOE, 1H–1H COSY, and 1H NMR analyses.
To explore the effects of cyclic substituents on the thermal properties of the substituted PLA, three new bicyclic and tricyclic lactide derivatives (1-L, 2-L, 3-L) were prepared and subsequently polymerized to afford the respective homopolymers (Scheme 1). A saturated norbornane-lactide derivative (1-L) was prepared by hydrogenation of norbornene-lactide (Scheme 1). The 1H NMR spectra of NL and 1-L are shown in Fig. S3a†. The disappearance of the olefin chemical shifts at 6.56–6.34 and 6.32–5.87 ppm (CH, CH) of NL suggests that the double bond was effectively hydrogenated. The distribution of isomers was determined by the relative integrations of the lactide ring methine resonances (CH, 5.17–4.85 ppm). From that analysis we concluded that hydrogenation reaction conditions did not affect the stereochemistry of the monomer products.
The size of the cyclic substituent was altered by using 1,3-cyclohexadiene and isoprene as reagents to create the tricyclic and bicyclic structures, respectively (Scheme 1). In comparison to NL, a cyclohexadiene derivative has an additional methylene unit in the bridge and therefore leads to a bulkier bicyclic structure on the lactide ring. The 1H NMR of cyclohexadiene-lactide (2-L) is shown in Fig. S4a†. Here too, a mixture of isomers is produced as evident by the chemical shift of the lactide ring methine proton (CH). The major isomer is located at 5.16 ppm while the minor isomers are shifted both upfield (5.04 and 4.97 ppm) and downfield (5.30 ppm) (see Fig. S4a† for assignments). Using 1H NMR, HMQC, 13C NMR, 1H–1H COSY and 1D NOE spectroscopy (Fig. S4a–e†), the distribution and assignment of each isomer were determined (the major (73%) isomer is depicted in Scheme 1; 3 : 1 exo : endo selectivity). These results are comparable to norbornene-lactide,11 suggesting that a similar transition state occurs in this reaction where the methyl group on the dienophile is opposite to the approaching diene.
Isoprene was combined with EML to form a monocyclic spiro-derivative (Scheme 1). To prevent the loss of isoprene during the reaction, the reaction mixture was sealed in a pressure vessel and stirred at 80 °C for 20 h (Scheme 1). Unlike cyclopentadiene and cyclohexadiene, isoprene is an asymmetric diene, and therefore can result in a mixture of stereo- and regioisomers. The preferred cycloadduct is formed when isoprene adds to EML with “para” substitution and the dipole–dipole of the diene and dienophile is minimized (Scheme S1†).34 Using 1H NMR, HMQC, 13C NMR, 1H–1H COSY and 1D NOE spectroscopy (Fig. S5a–e†), the distribution and assignments of the isomers were determined. The major isomer is depicted in Scheme 1, and was found to make up 48% of the mixture with 65 : 35 para selectivity.
NL-L statistical copolymers
A series of statistical copolymers with varying amounts of L-lactide (L) and NL was prepared by ROP using TBD (Scheme 2). The polymer products were isolated by precipitation and analyzed by NMR spectroscopy, SEC, and DSC. A representative 1H NMR spectrum of P(NL-s-L) is shown in Fig. S6†. The mole ratios of L and NL were calculated by the relative integrations of the olefin of norbornene (CH, CH, 6.49–5.93 ppm) and the methine of PLA (CH, 5.16 ppm). The mole ratio of NL (XNL) ranged between 0.07 and 0.72 (Table 1) and was lower than the corresponding feed composition. This is likely a result of the differences in the inherent reactivity of L-lactide and NL, where PLLA polymerization reaches completion in 1 min with TBD catalyst at room temperature,35–37 and PNL in 5–8 d at −20 °C.11 The reaction times for P(NL-s-L) varied depending on the mole ratios of the monomers in the feed. When a XNL < 0.5 was targeted, reaction times were typically 5 d, while higher mole ratios of NL required longer reaction times of 9–14 d. The product molecular weights, mole ratios, and Tg values are listed in Table 1. For each sample in the copolymer series, a single Tg was obtained by DSC suggesting that the polymer products are statistical in nature (Fig. 3). PLA is known to undergo transesterification when reacted with TBD for extended periods of time.35 It is possible that the copolymers are initially blocky and that transesterification plays a role in the incorporation of NL to afford a statistical polymer. The Fox equation can be used to predict the Tg of a statistical copolymer based on the weight fractions of the A and B monomers, and the Tg values of the respective homopolymers. The inverse Tg's of the copolymers as a function of the NL weight fractions are given in Fig. 4. The experimental values are consistent with theory suggesting that the Tg of a P(NL-s-L) copolymer can be tuned by varying the molar ratio of NL and L monomers in the feed. | ||
| Scheme 2 | ||
| Polymer | X NL (feed)b | X NL (NMR)c | M n /kg mol−1 | PDI | Time/d | Yield (%) | T g/°C | T m/°C |
|---|---|---|---|---|---|---|---|---|
| a Polymerization conditions: [M]0/[I]0/[cat] = 200/1/0.5, [M]0 = 1 M in CH2Cl2, −20 °C. b Mole ratio of norbornene-lactide added to the feed. c Determined by the relative integration of methine of PLA (CH at 5.16 ppm) vs. the double bond of PNL (CH, CH at 6.49–5.93 ppm). d Molecular weights determined by SEC calibrated with PS standards. e Determined during the first heating cycle. | ||||||||
| PNL | 1 | 1 | 26 | 1.4 | 8 | 81 | 105 | |
| P(NL-s-L) | 0.9 | 0.72 | 16 | 1.3 | 9 | 29 | 100 | |
| 0.8 | 0.58 | 26 | 1.6 | 14 | 68 | 87 | ||
| 0.7 | 0.34 | 17 | 1.5 | 14 | 28 | 76 | ||
| 0.6 | 0.37 | 25 | 1.7 | 14 | 73 | 78 | ||
| 0.5 | 0.19 | 20 | 1.7 | 8 | 50 | 67 | ||
| 0.3 | 0.17 | 28 | 1.6 | 5 | 73 | 66 | ||
| 0.2 | 0.07 | 22 | 1.8 | 5 | 78 | 61 | 150e | |
| PLLA | 0 | 0 | 31 | 1.1 | 0.2 | 83 | 58 | 170 |
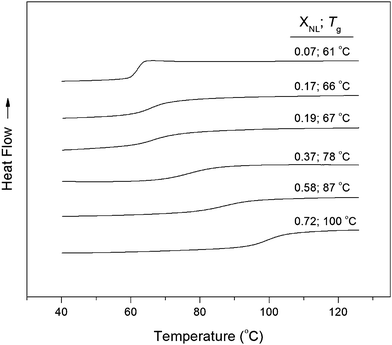 | ||
| Fig. 3 DSC traces of P(NL-s-L) copolymers of varying norbornene-lactide mole fractions (XNL). Plotted curves are of the second heating cycle. | ||
 | ||
| Fig. 4 Plot of 1/Tgversus weight percent of NL for P(NL-s-L) (■). The theoretical line (—) was generated using the Flory–Fox relationship and the Tg values of PLLA (58 °C) and PNL (105 °C) homopolymers. | ||
Other cyclic PLA derivatives
The structure–property relationship of the cyclic substituents on the lactide monomer structure of 1-L, 2-L, 3-L and the thermal properties of their respective homopolymers were explored. Analogous to the polymerization of NL, lactide derivatives were combined with a benzyl alcohol initiator and TBD catalyst in CH2Cl2, sealed under nitrogen, and placed in a freezer (−20 °C). Polymer products were isolated by precipitation and analyzed by NMR spectroscopy (Fig. S7–S9†) and SEC. A summary of the molecular weights and polydispersities (PDI) is listed in Table 2 (both determined using SEC calibrated by polystyrene standards).| Polymer | M n (calcd)a/kg mol−1 | M n /kg mol−1 | PDI | Time/d | Yield (%) | T g/°C |
|---|---|---|---|---|---|---|
| a Calculated molecular weights based on percent yield and initial monomer/initiator loadings. b Molecular weights determined by SEC calibrated with PS standards. c Polymerization conditions: [M]0/[I]0/[cat] = 200/1/0.5, [M]0 = 1 M. d Polymerization conditions: [M]0/[I]0/[cat] = 200/1/1, [M]0 = 1 M. | ||||||
| P(1-L) | 28.2 | 24.9c | 1.20 | 8 | 67 | 95 |
| 36.3 | 34.4c | 1.41 | 14 | 86 | 107 | |
| P(2-L) | 26.7 | 5.6c | 1.28 | 15 | 60 | 113 |
| 27.6 | 6.4d | 1.13 | 8 | 62 | 119 | |
| 32.9 | 8.2d | 1.37 | 18 | 74 | 118 | |
| P(3-L) | 8.0 | 9.5d | 1.22 | 2 | 19 | 76 |
| 29.9 | 15.3d | 1.38 | 8 | 71 | 77 |
The polymerization of lactide proceeds by the release of ring-strain upon opening of the lactide ring,38 and the bulky nature of the side group can limit the exothermicity of this process.11,38 When the polymerization of lactide derivatives is performed at low temperatures, this shifts the equilibrium towards polymer formation; this was evident with the 3-L lactide derivative. When the polymerization of isoprene-lactide (P(3-L)) was attempted at room temperature for 14 d, polymer formation was not observed. However, when the polymerization was performed at −20 °C for 8 d a polymer product was formed in 71% yield (Mn = 15.3 kg mol−1, PDI = 1.38).
The polymerization of the saturated version of NL, 1-L, was performed at −20 °C for 8 d to afford a white polymer product in 67% yield (Mn = 24.9 kg mol−1, PDI = 1.20). The structure of the 2-L derivative is similar to NL, but is slightly bulkier due to the additional CH2 in the bridge. The reaction time for poly(cyclohexadiene-lactide) (P(2-L)) was longer in comparison to PNL. Specifically, the polymerization of NL required 0.5 equiv of TBD catalyst and a 5–8 d reaction time to afford a polymer with Mn of ∼24 kg mol−1.11 However, under the same reaction conditions and an increased reaction time of 15 d, 2-L affords a polymer product of 5.6 kg mol−1. Upon an increase in catalyst loading to 1 equiv of TBD, a reaction time of 18 d yielded a polymer of 8.2 kg mol−1. It was previously reported that the polymerization of lactide derivatives proceeds by preferential cleavage of the ester at the 1,2-position of the lactide ring.11 As seen with P(2-L), the steric bulk of the bicyclic spiro-group can hinder nucleophilic substitution at the carbonyl and thus slow the reaction rate. Similar trends have been noted in alkyl substituted lactide derivatives. For instance, Yin and Baker12 reported the synthesis and polymerization of a disubstituted glycolide series with ethyl, hexyl, and isobutyl substituents. It was shown that when the size of the substituent was increased, the polymerization rate decreased. Hall and Schneider39 also demonstrated that a tetramethylglycolide monomer will not polymerize to a high molecular weight due to steric hindrance at the α-carbon.
Thermal analysis
The PNL, P(1-L), P(2-L), and P(3-L) homopolymers were analyzed by DSC and TGA under nitrogen to determine their Tg and Td, respectively. The DSC traces of P(1-L), P(2-L), and P(3-L) are represented in Fig. 5. Incorporation of bulky substituents along a polyester backbone can increase the barrier to segmental motion, and thus increase the Tg of the materials.7 The P(3-L) introduced a monocyclic structure and increased the Tg to 76–77 °C (Table 2). As previously demonstrated,11 a bicyclic spiro structure resulted in a significant increase in Tg (PNLTg = 113 °C; Mn = 30 kg mol−1).11 The saturated P(1-L) gave a Tg of 95 °C (Mn = 24.9 kg mol−1) and 107 °C (Mn = 34.4 kg mol−1) (Table 2). These values are somewhat lower than the Tg of unsaturated PNL of comparable molecular weight (Tg = 105 °C, Mn = 26 kg mol−1; Tg = 113 °C, Mn = 30 kg mol−1),11 and these results follow a similar trend with work presented in the literature.7,9,40 Increasing the bulkiness further in P(2-L) derivative gave rise to a Tg of 119 °C (Mn = 6.4 kg mol−1) and 118 °C (Mn = 8.2 kg mol−1), resulting in a 11 °C increase in comparison to PNL of slightly higher molecular weight (Tg = 108 °C; Mn = 12.2 kg mol−1).11 The cyclohexadiene-lactide, P(2-L), has the highest Tg reported to date among PLA derivatives, and we anticipate that increasing the molecular weight further would lead to even higher values.7,9–11,19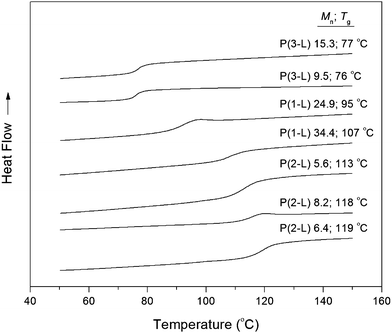 | ||
| Fig. 5 DSC curves of P(1-L), P(2-L) and P(3-L) homopolymers. Plotted curves are of the second heating cycle. Molecular weights are listed in kg mol−1. | ||
The onset of decomposition temperatures (Td) of P(1-L), P(2-L), and P(3-L) was 300, 280, and 280 °C, respectively (Fig. 6). PNL had an onset of the decomposition at 248 °C with a second transition at ∼310 °C, this is likely due to a retro-Diels–Alder reaction where cyclopentadiene is thermally eliminated41,42 followed by decomposition of the polyester backbone. The mass loss of the first transition was ∼20%, and cyclopentadiene makes up 30% weight of the polymer. However, the saturated P(1-L) derivative had a single transition at 300 °C suggesting that hydrogenation increased the stability of the material likely due to the absence of a retro-Diels–Alder reaction.
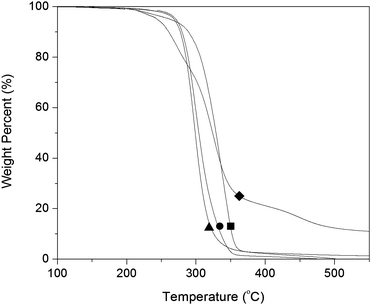 | ||
| Fig. 6 TGA data for P(3-L) (▲, Mn = 15.3 kg mol−1), P(2-L) (●, Mn = 6.4 kg mol−1), P(1-L) (■, Mn = 25 kg mol−1), and PNL (◆, Mn = 26.4 kg mol−1). | ||
Summary
A series of lactide derivatives and PLA based materials was prepared using L-lactide, a biorenewable resource, as the starting material. Exomethylene-lactide served as a template and dienophile in subsequent Diels–Alder reactions with cyclopentadiene, 1,3-cyclohexadiene and isoprene to produce the corresponding tricyclic and bicyclic lactide derivatives. Additionally, norbornene-lactide was hydrogenated to afford a saturated norbornane-lactide derivative. A series of statistical copolymers of P(NL-s-L) were prepared with NL mole ratios ranging from 0.07–0.72. The Tg values for P(NL-s-L) samples (61–100 °C) matched well with predicted values from the Fox equation based on the Tg's of the corresponding homopolymers. Homopolymers of P(1-L), P(2-L), and P(3-L) exhibited Tg values of 107 °C, 119 °C, and 77 °C, respectively. Decomposition temperatures of these materials were ≤280 °C, except for P(1-L) (Td = 300 °C). Here, saturation increases the stability of the polymer likely due to the absence of a retro-Diels–Alder reaction. These results demonstrate that the direct attachment of cyclic structures to the lactide ring can significantly increase the Tg of materials, and that even the smallest changes (e.g., saturation and insertion of CH2) can have a measurable impact. This work serves as a model for the tuning of thermal properties of lactide-based materials, important for understanding new materials from biorenewable resources. In addition, this work illustrates how EML serves as a versatile platform for the preparation of new lactide derivatives that can be converted into aliphatic polyesters with tunable properties. We envision that EML can be used for the synthesis of various cyclic esters that will increase the diversity of polymers that can be derived from renewable materials.Acknowledgements
We thank generous financial support from NatureWorks. Jennifer Lowe is acknowledged for her advice and technical assistance.References and notes
- A. P. Gupta and V. Kumar, Eur. Polym. J., 2007, 43, 4053–4074 CrossRef CAS.
- A.-C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS.
- R. Auras, B. Harte and S. Selke, Macromol. Biosci., 2004, 4, 835–864 CrossRef CAS.
- S. Mecking, Angew. Chem., Int. Ed., 2004, 43, 1078–1085 CrossRef CAS.
- E. T. H. Vink, K. R. Rábago, D. A. Glassner and P. R. Gruber, Polym. Degrad. Stab., 2003, 80, 403–419 CrossRef CAS.
- R. Datta and M. Henry, J. Chem. Technol. Biotechnol., 2006, 81, 1119–1129 CrossRef CAS.
- G. L. Baker, B. M. Vogel and M. R. Smith, III, Polym. Rev., 2008, 48, 64–84 Search PubMed.
- K. M. Benabdillah, M. Boustta, J. Coudane and M. Vert, in Polymers from Renewable Resources: Biopolyesters and Biocatalysis, ed. C. Scholz and R. A. Gross, American Chemical Society, Washington DC, 2000, vol. 764, pp. 200–220 Search PubMed.
- F. Jing, M. R. Smith and G. L. Baker, Macromolecules, 2007, 40, 9304–9312 CrossRef CAS.
- T. Liu, T. L. Simmons, D. A. Bohnsack, M. E. Mackay, M. R. Smith and G. L. Baker, Macromolecules, 2007, 40, 6040–6047 CrossRef CAS.
- F. Jing and M. A. Hillmyer, J. Am. Chem. Soc., 2008, 130, 13826–13827 CrossRef CAS.
- M. Yin and G. L. Baker, Macromolecules, 1999, 32, 7711–7718 CrossRef CAS.
- T. L. Simmons and G. L. Baker, Biomacromolecules, 2001, 2, 658–663 CrossRef CAS.
- X. Jiang, M. R. Smith and G. L. Baker, Macromolecules, 2008, 41, 318–324 CrossRef CAS.
- X. Jiang, E. B. Vogel, M. R. Smith and G. L. Baker, Macromolecules, 2008, 41, 1937–1944 CrossRef CAS.
- X. Jiang, E. B. Vogel, M. R. Smith and G. L. Baker, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5227–5236 CrossRef CAS.
- T. Trimaille, M. Möller and R. Gurny, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4379–4391 CrossRef CAS.
- T. Trimaille, K. Mondon, R. Gurny and M. Möller, Int. J. Pharm., 2006, 319, 147–154 CrossRef CAS.
- K. Marcincinova-Benabdillah, M. Boustta, J. Coudane and M. Vert, Biomacromolecules, 2001, 2, 1279–1284 CrossRef CAS.
- M. Leemhuis, C. F. van Nostrum, J. A. W. Kruijtzer, Z. Y. Zhong, M. R. ten Breteler, P. J. Dijkstra, J. Feijen and W. E. Hennink, Macromolecules, 2006, 39, 3500–3508 CrossRef CAS.
- B. Saulnier, S. Ponsart, J. Coudane, H. Garreau and M. Vert, Macromol. Biosci., 2004, 4, 232–237 CrossRef CAS.
- K. Marcincinova-Benabdillah, J. Coudane, M. Boustta, R. Engel and M. Vert, Macromolecules, 1999, 32, 8774–8780 CrossRef CAS.
- W. W. Gerhardt, D. E. Noga, K. I. Hardcastle, A. J. Garcia, D. M. Collard and M. Weck, Biomacromolecules, 2006, 7, 1735–1742 CrossRef CAS.
- D. E. Noga, T. A. Petrie, A. Kumar, M. Weck, A. J. Garcia and D. M. Collard, Biomacromolecules, 2008, 9, 2056–2062 CrossRef CAS.
- N. K. Abayasinghe and D. W. Smith, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2001, 42, 485 CAS.
- A. S. Scheibelhoffer, W. A. Blose and H. J. Harwood, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 1969, 10, 1375–1380 CAS.
- Exomethylene-lactide and bromo-lactide are susceptible to degradation when stored under ambient conditions. The storage of lactide derivatives under a dry inert atmosphere increased the shelf-life of these materials from a few days or weeks to several months.
- R. Blessing, An empirical correction for absorption anisotropy, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 33–38 CrossRef.
- SAINT+ V6.45, Bruker Analytical X-Ray Systems, Madison, WI Search PubMed.
- SHELXTL V6.14, Bruker Analytical X-Ray Systems, Madison, WI Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–246 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS.
- Computed using the standard idea-gas, rigid-rotator, harmonic oscillator approximations; see: C. J. Cramer, Essentials of Computational Chemistry: Theories and Models, John Wiley & Sons, Chichester, 2nd edn, 2004 Search PubMed.
- W. R. Roush and B. B. Brown, J. Org. Chem., 1992, 57, 3380–3387 CrossRef CAS.
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2006, 128, 4556–4557 CrossRef CAS.
- B. G. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 8574–8583 CrossRef CAS.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS.
- A. Duda, A. Kowalski, J. Libiszowski and S. Penczek, Macromol. Symp., 2005, 224, 71–84 CrossRef CAS.
- H. K. Hall and A. K. Schneider, J. Am. Chem. Soc., 1958, 80, 6409–6412 CrossRef.
-
G. L. Baker and M. R. Smith, III, Process for the preparation of polymers of dimeric cyclic esters, PCT Int. Appl., WO 2
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 001042333, 2001.
001042333, 2001. - J. L. Ripoll, A. Rouessac and F. Rouessac, Tetrahedron, 1978, 34, 19–40 CrossRef CAS.
- W. Carruthers, Cycloaddition Reactions in Organic Synthesis, Pergamon Press, Oxford, 1990, p. 373 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization of polymers (1H NMR spectroscopy) and monomers. X-Ray structural information, CIF files for crystal structures, computed energies and stereostructures and associated Cartesian coordinates, 1H, 13C, 1H-1H COSY, HMQC, 1D NOE NMR spectra. CCDC reference numbers 772542 and 772543. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0py00029a |
| This journal is © The Royal Society of Chemistry 2010 |