Novel liquid crystalline hyperbranched polyesters with interior 4,7-diphenyl-2,1,3-benzothiadiazole units
Moriyuki
Sato
*,
Yoko
Kobayashi
,
Toshihiro
Shimizu
and
Isao
Yamaguchi
Department of Materials Science, Faculty of Science and Engineering, Shimane University, Matsue, Shimane, 690-8504, Japan. E-mail: msato@riko.shimane-u.ac.jp
First published on 5th March 2010
Abstract
Novel hyperbranched (HB) polyesters with interior 4,7-diphenyl-2,1,3-benzothiadiazole (DBT) units were prepared by melt polycondensation of a dimethyl ester derivative of DBT (BMBT) (A2 monomer) with 2-hydroxymethyl-2-methyl-1,3-propanediol (HMP) (B3 monomer) at various mole ratios (A2/B3) via an A2 + B3 approach. The liquid crystalline (LC) and optical properties were investigated. FT-IR and 1H NMR spectroscopies revealed that the HB polymers have good solubility in chloroform, and can be synthesized without gelation during the polymerization with a degree of branching (DB) of 36–94%. The branched structures of HB polymers are dependent on the feed mole ratio. The HB polymer prepared from a mole ratio of A2/B3 = 1/3 had the highest number average molecular weight (Mn) and a more linear structure than the other HB polymers (A2/B3 = 3/1, 2/1, 3/1 and 1/2), which had the lowest DB values. DSC measurements, polarizing microscopy observations and X-ray analyses suggested that the HB polymer (A2/B3 = 1/3) forms the smectic A phase, whereas the others formed the smectic C phase. UV-vis and photoluminescent (PL) spectra in the solutions and in films indicated that the HB polymers show maximum absorbances and yellow-light emission attributable to the DBT unit. The PL spectra of HB polymers in film showed another broad peak, which was likely to be due to interchain interactions or aggregations, at longer wavelengths. Relative quantum yields (Φ) of the HB polymers in the chloroform solutions were lower than those of HB polymers comprising the 2-phenylbenzothiazole unit. Polarized absorption and fluorescent spectra revealed that the HB polymers have poor orientation properties and the luminescent properties are independent of the orientation structures of HB polymers.
Introduction
Dendritic polymers such as hyperbranched (HB) polymers have attracted much attention as materials having higher potential for various applications and dendrimers.1–10 HB polymers are macromolecules with an irregular and low degree of branching (DB), which can be produced by a simple and low-cost method compared to dendrimers. Yet the polymers have very similar properties to dendrimers, including the presence of functional groups at the chain ends and low viscosity due to lack of interchain entanglement.1–10 A large amount of work on HB polymers has been reported and the physical and biological properties have been well-characterized.11,12 Typically, the HB polymers have been prepared using AB2 monomers or from a combination of A2 and B3 monomers (A2 + B3 method) via a one-pot method.11,12 The latter method is of interest as a simple and cost-effective synthetic strategy for the HB polymers. Traditional polymerization techniques have been useful for the synthesis, interpretation of the molecular structures and polymerization kinetics in the A2 + B3 type HB polymers.12cOn the other hand, low molecular weight compounds, linear polymers and HB polymers composed of calamitic five-membered heterocyclic mesogens such as aromatic benzothiazole13 and 1,3,4-thiadiazole moieties14 are known to have photoluminescent (PL) properties due to the π–π* electron transition of the heterocyclic compounds, as well as thermotropic liquid crystalline (LC) properties. Previously, thermotropic LC HB polymers containing heterocyclic calamitic mesogens (2-phenyl-5-phenylethenyl-1,3,4-thiadiazole,15 2,5-diphenyl-1,3,4-thiadiazole14 (DTD) and 2-phenylbenzothiazole13 (PBT)) in the interiors were prepared under various polymerization conditions via the A2 + B3 approach, and their LC and optical properties were determined to be dependent on the polymerization methods and conditions (mole ratios of A2/B3). Unfortunately, the quantum yields were low, with the exception of those for the HB polymers containing the PBT moiety.13
4,7-Diphenyl-2,1,3-benzothiadiazoles (DBT), which are analogous heterocyclic compounds containing a 2,1,3-thiadiazole unit lateral to terphenyl, are used as good emitting and electron-transporting materials for organic electroluminescent devices, with strong PL intensities.16 2,1,3-Benzothiadiazole (BT) dyes containing linear π-conjugated aromatic compounds emit strong fluorescent light due to the π–π* electron transition, with high relative quantum yields (Φ).16a,16b Until recently, there have been no reports that the DBT derivatives form LC phases. This is thought to be because the aspect ratio of the DBT unit is low. Kelly and co-workers synthesized red-light emitting photopolymerizable smectic LC DBT compounds for use in organic light-emitting diodes and found that the resulting polymer networks show not only PL properties based on the emission of BT unit, but also thermotropic LC properties.13a, 16c
In this work, HB polymers 4 bearing a DBT unit in the interiors were synthesized by melt polycondensation of 4,7-bis[(4-methoxycarbonyloxydecyloxy)phenyl]-2,1,3-benzothiadiazole (BMBT) 2 (A2 monomer), with an aliphatic triol, 2-hydroxymethyl-2-methyl-1,3-propanediol (HMP) 3 (B3 monomer) at various mole ratios of A2/B3 (Scheme 1). The resultant HB polymers 4 were examined to determine whether or not LC phases form. This would suggest that the materials could show thermotropic LC properties based on intermolecular interactions between the branched polymer chains in spite of the low aspect ratio of the DBT unit. In addition, the relationship between the LC and optical properties of the resulting HB polymers 4 was evaluated, because the DBT moiety emits strong fluorescent light based on the π–π* electron transition. On the other hand, the orientation of the LC polymer chains may prevent PL properties. The resulting HB polymers 4 with DBT units in the interiors are also expected to show more interesting and better emitting properties compared with the analogous LC HB polymers containing DTD and PBT units.
 | ||
| Scheme 1 | ||
Results and discussion
Synthesis of A2 monomer 2 and HB polymers 4
Dioxydiundecanoic acid dimethyl ester derivative of DBT (BMBT) 2 (A2 monomer) was prepared from a bisphenol derivative of DBT, 4,7-bis(4-hydroxypheny)-2,1,3-benzothiadiazole (BHBT) 1, and 11-bromoundecanoic acid dimethyl ester (BADE) in the presence of potassium carbonate in dimethylsulfoxide (DMSO) and analyzed by FT-IR and 1H NMR spectroscopies and elemental analyses. Polarizing microscopy observation, differential scanning calorimetry (DSC) measurement and powder X-ray diffraction (XRD) analysis suggested that the A2 monomer 2 forms the enantiotropic thermotropic smectic C phase (broken fan texture, 2θ = 2.46°, 4.92° and 25.2°, d spacing value: 35.9 Å, calculated molecular length: 48.0 Å). Compound 2 emits strong fluorescent light at room temperature, which can be observed under solar light. The relative Φ value is 20.9% using coumarin 311 as a standard.HB polymers 4 with DBT units in the interiors were synthesized by melt polycondensation of the A2 monomer 2 (BMBT) with aliphatic triol (B3 monomer) 3 (HMP) in the presence of p-toluene sulfonic acid monohydrate (p-TsOH) as a catalyst.17,18 The polymerization conditions were carefully controlled, since the reaction in this monomer system (A2 + B3) is theoretically known to form gelated polymers beyond the critical point of gelation.19 Optimum conditions for the synthesis of HB polymers 4via the A2 + B3 approach were investigated under the following conditions, reaction temperature: 110 °C, 160 °C and 180 °C, reaction time: 20 min, 4 h and 7 h, and mole ratio: A2/B3 = 3/2. During the polymerization, methanol was removed from the reaction system in vacuo every 5 or 15 min. The polycondensation proceeded gradually at 180 °C and the reaction mixture became viscous after 3 h. Under other conditions (shorter reaction times and lower temperatures) the reaction proceeded more slowly and the system did not become viscous. As listed in Table 1, HB polymer 4–4 (soluble in chloroform) with maximum inherent viscosity (ηinh = 0.32 dL g−1) is successfully prepared without gelation under the conditions (reaction temperature: 180 °C, reaction time: 7 h). Therefore, the synthesis of expected HB polymers 4 from 2 (A2 monomer) and 3 (B3 monomer) was carried out at various mole ratios of A2/B3 = 3/1, 2/1, 3/2, 1/2 and 1/3 under the following conditions, reaction time: 7 h, reaction temperature: 180 °C. The preparative data for HB polymers 4 are summarized in Table 2. The table shows that, in HB polymers 4a–4b with higher A2 monomer content and HB polymer 4c with an equimolar ratio of A2/B3, the Mns vary and increase with the feed mole ratios of A2/B3. The HB polymers 4d–4e, which have higher B3 monomer content, show Mns equal to HB polymer 4c or higher Mns than HB polymers 4a–4b. The reactions for HB polymers 4d and 4e were complete in 2.5 h and 30 min, respectively, because the reaction proceeded more rapidly and afforded gelated polymers at longer reaction times. These results are inconsistent with Flory's theory,19 which describes that in preparing HB polymers via the A2 + B3 approach, the HB polymer prepared in the mole ratio of A2/B3 = 3/2 (equal numbers of functional groups in the reaction system) will have the highest Mn in the HB polymers prepared at various mole ratios. This is thought to be due to the fact that HB polymer 4e prepared under the reaction conditions in this work has a more linear and rigid structure than the others 4a–4d. In general, linear polymers have higher Mns than analogous branched polymers. This was also observed in the preparation of HB polymers from the diester of DTD and HMP by the A2 + B3 type melt polycondensation.18
| Run No. | Temp./°C | Time/h | Yield (%) | ηinhb/dL g−1 |
|---|---|---|---|---|
| a Melt polycondensation was carried out at a feed mole ratio of A2/B3 = 3/2. b Measured at a concentration of 0.2 g dL−1 in DCAA at 30 °C. | ||||
| 4–1 | 110 | 1/3 | 33 | 0.18 |
| 4–2 | 160 | 1/3 | 30 | 0.18 |
| 4–3 | 180 | 4 | 29 | 0.24 |
| 4–4 | 180 | 7 | 30 | 0.32 |
| No. | Mole ratio (A2/B3) | Time/h | Yield/% | DBb/% | M n | M w/Mnc |
|---|---|---|---|---|---|---|
| a Melt polycondensation was carried out at 180 °C in the presence of p-TsOH (7 mg). A2 monomer of 0.225 g (3.0 × 10−4 mol) was used. b Degree of branching: (D + T) / (D + T + L). c Measured in chloroform using polystyrene as a standard by SEC. d L unit %: 5.8% (4c); 55% (4d); 63% (4e). | ||||||
| 4a | 3/1 | 1.0 | 49 | 94 | 7300 | 3.94 |
| 4b | 2/1 | 7.0 | 78 | 92 | 6700 | 3.67 |
| 4c | 3/2 | 7.0 | 85 | 94d | 11900 | 3.96 |
| 4d | 1/2 | 2.5 | 61 | 45d | 11900 | 3.54 |
| 4e | 1/3 | 0.5 | 67 | 36d | 12100 | 4.13 |
The structures of the HB polymers 4 were confirmed by FT-IR and 1H NMR spectroscopies. The FT-IR spectra of HB polymers 4 depicted absorption bands based on CH stretching at 2921 and 2853 cm−1, ester C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 1740 cm−1, CC and CN at 1608 and 1557 cm−1, and C–O–C at 1251 and 1180 cm−1. In HB polymers 4c–4e, the OH absorption bands of terminal alcoholic group based on the B3 monomer (HMP) were detected at around 3300–3500 cm−1. The 1H NMR spectra of HB polymers 4 in CDCl3 showed proton signals for aromatic rings of the DBT unit at 7.06–7.91 ppm, OCH2 and C(O)OCH2 at 4.03 ppm, CH2C(O)O at 2.33 ppm and aliphatic CH2 chains at 1.29–1.85 ppm. In addition, the terminal C(O)OCH3 was observed at 3.67 ppm and CH3 proton signals attributed to the HMP (B3 monomer) at 0.93–1.02 ppm. Proton signals for the CH2 chain next to the OH group (–CH2OH) were observed at 3.4 ppm in the HB polymers 4a and 4b, but not in the HB polymers 4c–4e. The CH3 proton signals at 0.93–1.02 ppm vary systematically with the mole ratios of A2/B3 and the branched structures of the HB polymers 4 as shown in Fig. 1 and Table 2. The DB values of HB polymers 4 were estimated from the extended 1H NMR spectra (the CH3 proton signals at 0.93–1.02 ppm in Fig. 1) in CDCl3. The DBs1a were calculated using eqn (1) and are presented in Table 2.
O at 1740 cm−1, CC and CN at 1608 and 1557 cm−1, and C–O–C at 1251 and 1180 cm−1. In HB polymers 4c–4e, the OH absorption bands of terminal alcoholic group based on the B3 monomer (HMP) were detected at around 3300–3500 cm−1. The 1H NMR spectra of HB polymers 4 in CDCl3 showed proton signals for aromatic rings of the DBT unit at 7.06–7.91 ppm, OCH2 and C(O)OCH2 at 4.03 ppm, CH2C(O)O at 2.33 ppm and aliphatic CH2 chains at 1.29–1.85 ppm. In addition, the terminal C(O)OCH3 was observed at 3.67 ppm and CH3 proton signals attributed to the HMP (B3 monomer) at 0.93–1.02 ppm. Proton signals for the CH2 chain next to the OH group (–CH2OH) were observed at 3.4 ppm in the HB polymers 4a and 4b, but not in the HB polymers 4c–4e. The CH3 proton signals at 0.93–1.02 ppm vary systematically with the mole ratios of A2/B3 and the branched structures of the HB polymers 4 as shown in Fig. 1 and Table 2. The DB values of HB polymers 4 were estimated from the extended 1H NMR spectra (the CH3 proton signals at 0.93–1.02 ppm in Fig. 1) in CDCl3. The DBs1a were calculated using eqn (1) and are presented in Table 2.
| DB = (D + T)/(D + T + L) | (1) |
 | ||
| Fig. 1 Extended 1H NMR spectra of HB polymers 4 in CDCl3. | ||
These spectral data support production of the expected HB polymers 4 bearing the interior DBT unit via the A2 + B3 approach and the branched structures of obtained HB polymers 4 depend on the feed mole ratio.
Optical properties of HB polymers 4
Solution and solid-state UV-vis absorption and PL spectra of HB polymers 4 were measured. Fig. 2 shows UV-vis absorption and PL spectra of HB polymer 4c in chloroform solution and in film. Table 3 lists the UV-vis and PL spectral data of A2 monomer 2 and HB polymers 4. The UV-vis absorption spectra of HB polymers 4 in the chloroform solutions were normalized to the absorption peak maxima at 412–413 nm on the basis of a π–π* electron transition for the DBT unit as well as the BMBT (A2 monomer). The PL spectra of HB polymers 4 in chloroform solutions when excited at 412–413 nm showed peak maxima at 540–542 nm with a yellow emission. The Stokes shifts were 126.6–130 nm, which are considerably higher than those for previously-reported HB polymers with DTD14 and PBT units.13 The relative quantum yields (Φ) of HB polymers 4 measured in the solution using coumarin 311 as a standard are lower than those for the A2 monomer and our previously-reported HB polymers with an internal PBT unit13 (25–40%).| No. | In solution | In film | |||
|---|---|---|---|---|---|
| λ max, abs/nm | λ max, PL /nm | Φ /% | λ max, abs/nm | λ max, PL /nm | |
| a Excited at maximum absorption wavelength. b Coumarin 311 used as standard. | |||||
| 2 | 412 | 541 | 20.9 | 411 | 545 |
| 4a | 413 | 541 | 17.6 | 405 | 540, 597 |
| 4b | 413.5 | 540 | 17.1 | 403 | 540, 590 |
| 4c | 412 | 542 | 17.0 | 400.5 | 538, 597 |
| 4d | 412 | 541 | 18.6 | 398 | 537, 596 |
| 4e | 413 | 541 | 17.1 | 400.5 | 538, 595 |
 | ||
| Fig. 2 UV-vis and PL spectra of HB polymer 4c in chloroform solutions and in film. | ||
The UV-vis absorption spectra of HB polymers 4 in film showed similar absorption curves, with maximum wavelengths at 398–405 nm, but were blue-shifted, similar to our previously reported HB polymers.13f The absorption maxima for 4 were at a lower wavelength by about 8–14 nm compared with those in chloroform solutions and were blue-shifted with an increase in the B3 content. This was thought to be due to the intermolecular interactions between the OH groups and the HB polymer backbones at the termini. In BMBT 2 (A2 monomer), the absorption maximum of the solid-state UV-vis spectra was observed at the same wavelength as that in the chloroform solution. The PL spectra of HB polymers 4a–4b with higher A2 content in the film showed emission maxima at almost the same wavelengths as those in the solutions. Furthermore, the HB polymers 4c–4e, which have equal or higher B3 contents and terminal OH groups, have emission maxima that are blue-shifted by 3–4 nm compared with those in the solutions. On the other hand, the HB polymers with a PBT moiety bear red-shifted emission maxima in film.13 The HB polymers 4 displayed another broad emission maxima at longer wavelengths. This was thought to be due to interchain interactions between polymer backbones or aggregations based on the DBT unit. Similar behaviours were reported in dendronized polymers20 and polymers containing the DTD unit.14 The different terminal groups and the heterocyclic units (DBT or DTD and PBT) in the HB polymer backbones will likely affect their PL behaviours. This is because PL behaviour is related to changes in the HOMO–LUMO band gap energy levels due to the intermolecular interactions arising from the electron-donor or electron-withdrawing properties. The HB polymers 4 emit yellow light, which has a larger Stoke shifts than those in chloroform solutions. Furthermore, for BMBT 2 (A2 monomer) the emission maximum in the solid state was detected at a longer wavelength than those of the HB polymers 4.
LC properties of HB polymers 4
The LC properties of HB polymers 4 were examined using a polarizing microscope to observe the optical textures. DSC measurements and powder XRD analyses of polymer samples quenched from the LC states were also undertaken. These measurements suggested that all HB polymers 4 form thermotropic LC mesophases.The DSC curves of HB polymers 4 on the first cooling runs are illustrated in Fig. 3. Table 4 shows the phase transition temperatures and thermodynamic data for the HB polymers 4 on the first cooling and the second heating runs. The DSC curves of the HB polymers 4 on the heating runs, two or three endothermal peaks on the basis of Tk, Tm and Ti transitions are observed. On the cooling runs, the corresponding two or three exothermal peaks are detected. The HB polymers 4c–4e with mole ratios of A2/B3 = 1/1, 1/2 and 1/3 tend to form more stable LC phases compared to the HB polymers 4a–4b, where the ΔTs depend on their Mn values. Melting enthalpies (ΔHm) of the HB polymers 4 change and decrease with the change in mole ratio from A2/B3 = 3/1 to 1/3. Polarizing microscopy observations indicate that all the HB polymers 4 form enantiotropic thermotropic LC phases (broken fan or schlieren for 4a–4d and grainy texture for 4e) between Tm and Ti. This suggests that the HB polymers 4 form the smectic phase. The polarizing microphotograph for the HB polymer 4a is presented in Fig. 4.
| No. | T k | T m | ΔHm | T i | ΔHi | ΔTb |
|---|---|---|---|---|---|---|
| /°C | /°C | /Jg−1 | /°C | /Jg−1 | /°C | |
| a Observed on the second heating runs. Data in the parentheses on the first cooling scans. Tk: solid-to-solid transition temperature; Tm: solid-to-LC phase transition temperature; Ti: LC phase-to-isotropization transition temperature. b ΔT: Temperature range of LC phase (ΔT = Ti-Tm). | ||||||
| 4a | 76 (56) | 90 (69) | 49.6 (50.9) | 128 (124) | 8.94 (8.35) | 38 (55) |
| 4b | 81 | 95 (69) | 34.9 (40.5) | 126 (119) | 6.73 (7.28) | 31 (50) |
| 4c | 82 (56) | 91 (71) | 32.2 (26.7) | 135 (129) | 8.23 (7.88) | 44 (58) |
| 4d | 68 | 91 (72) | 18.0 (25.5) | 134 (129) | 7.20 (7.59) | 43 (57) |
| 4e | 65 | 89 (60) | 11.4 (21.9) | 133 (128) | 8.22 (6.94) | 44 (68) |
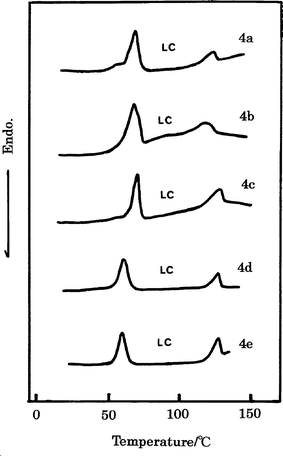 | ||
| Fig. 3 DSC curves for HB polymers 4 on the first cooling runs. | ||
 | ||
| Fig. 4 A polarizing microphotograph of HB polymer 4a at 123 °C on the first cooling (magnification: ×200). A broken fan texture can be observed. | ||
The LC phases of HB polymers 4 were identified by powder XRD analyses of samples quenched from the LC states between Tm and Ti. Fig. 5 shows the XRD patterns for HB polymers 4, which indicates that the HB polymers 4a–4d have two sharp and one broad reflection at middle angles. In HB polymer 4e one sharp and one broad reflection are observed at the middle angles. These XRD patterns suggest that the HB polymers 4 form either the smectic A or the smectic C phase. The d spacing values estimated from the reflections at the middle angles for the HB polymers 4a–4d and the HB polymer 4e are 38.5–39.8 Å and 52.0 Å, respectively. These values depict that the d spacing values for the HB polymers 4a–4d are considerably shorter than molecular length of polymer repeating unit. On the other hand, that for HB polymer 4e is about the same as the repeating unit length as shown in Table 5 and Fig. 6. Therefore, the LC phases of HB polymers 4a–4d and 4e are suggested to be the smectic C and smectic A phases, respectively, based on the polarizing microscope observations and the XRD data.
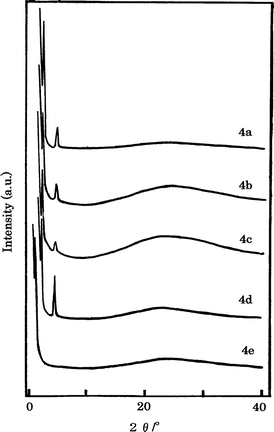 | ||
| Fig. 5 Powder XRD patterns of HB polymers 4 quenched from the LC states. | ||
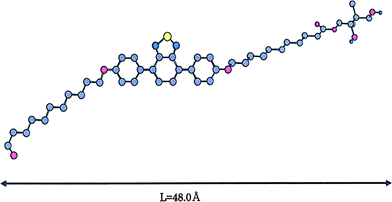 | ||
| Fig. 6 A possible molecular model and the length of the repeating unit of HB polymers 4. | ||
These results show that all the HB polymers 4 composed of the DBT unit exhibit smectic phases. Of them, HB polymers 4d–4e with the higher B3 content and with DB values lower than those of the HB polymers 4a–4c, form more stable LC phases compared to HB polymers 4a–4b, which have higher A2 content. This is thought to be due to intermolecular interaction between the terminal OH groups and the branched polymer chains or the effect of the Mn values. HB polymer 4e, which has the lowest DB value and the most linear structure (highest L unit %) shows smectic A phase, whereas the others 4a–4d are smectic C phase.
Polarized absorption and fluorescence in LC states of HB polymers 4
Polarized absorption and fluorescent spectra of the sheared HB polymers 4 with frozen LC phases retained by quenching from the LC states below room temperature were examined and the relationship between the optical properties and the liquid crystallinity was investigated. Molecular orientations of the HB polymers 4 quenched from the LC states were evaluated from the polarized absorption spectra. Table 6 lists the dichroic ratio (RA) and order parameter (S) values for the HB polymers 4 and the BMBT 2 (A2 monomer) in the sheared state. The RA and S values of HB polymers 4b–4c are higher than those of the others 4a, 4d and 4e, and the A2 monomer 2 (BMBT), but are very low compared with those of our previously-reported block copolymers composed of DTD units16f and side-chain LC polymers bearing 1,3,4-oxadiazole units.21 This suggests that the orientation properties of HB polymers 4 in the LC states are poor. This observation is likely due to a low aspect ratio of the DBT unit, in spite of emergence of the smectic A or C phase in the HB polymers 4.| No. | Dichroic ratioa | Order parameterb | PL dichroic ratioc |
|---|---|---|---|
| (RA) | (S) | (RI) | |
| a R A = A∥/A⊥, where A∥is the absorbance parallel to the shearing direction and A⊥ is the absorbance perpendicular to the shearing direction. b S = (RA−1) / (RA + 2). c R I = I∥/I⊥, where I∥ is the intensity parallel to the shearing direction and I⊥ is the intensity perpendicular to the shearing direction. | |||
| 2 | 1.36 | 0.11 | 1.11 |
| 4a | 1.39 | 0.11 | 1.14 |
| 4b | 1.59 | 0.16 | 1.07 |
| 4c | 1.41 | 0.12 | 1.90 |
| 4d | 1.25 | 0.08 | 1.62 |
| 4e | 1.45 | 0.13 | 1.26 |
The polarized fluorescent spectra of HB polymers 4 with frozen LC phases are shown in Fig. 7. Photoluminescence dichroic ratios (RI) calculated from the spectral data are tabulated in Table 6. The data indicate that the HB polymers 4 emit polarized fluorescent yellow-light at room temperature and the RI values are also lower than those in the block copolymers composed of the DTD unit.14f The HB polymers 4c–4d have higher RI values than the others 4a, 4b and 4e and A2 monomer 2 (BMBT). The RI values of HB polymers 4 are as low as the RA and S values. The HB polymers 4b–4d in the vicinity of the mole ratio of A2/B3 = 3/2 have higher RI values than the others (4a and 4e). These data suggest that the luminescent properties are independent of the orientation structures of A2 monomer 2 (BMBT) and HB polymers 4. This may be due to little or no ground-state aggregation effect.22 The detailed mechanism remains unclear at the present time. Thus, no clear relationship between the optical and LC properties can be established for the HB polymers 4.
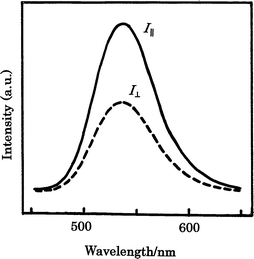 | ||
| Fig. 7 Polarized PL spectra of HB polymer 4c quenched from the LC state. | ||
Conclusions
HB polymers 4 including those with the interior DBT unit were successfully prepared by melt polycondensation of a dimethyl ester derivative of DBT (BMBT) (A2 monomer) with HMP (B3 monomer) at various mole ratios (A2/B3) via the A2 + B3 approach. The HB polymer 4e achieved from a mole ratio of A2/B3 = 1/3 formed the smectic A phase and the others formed the smectic C phase in spite of low aspect ratio of the DBT unit. The HB polymers 4 exhibited absorption and PL peak maxima with yellow emission, which was attributed to the DBT unit in both chloroform solutions and in film. The PL spectra in film showed emission maxima at longer wavelengths, which was likely to be due to interchain interactions or aggregations. The Φ values for the HB polymers 4 were lower than those of the HB polymers comprising the PBT unit. The HB polymers 4b–4d in the vicinity of the mole ratio of A2/B3 = 3/2 displayed better orientation properties, however, their RA, S and RI values were low. The luminescent properties are independent of the orientation structures of the HB polymers 4. Unfortunately, there is no clear relationship between the optical and the LC properties.Experimental
Materials
Compound 1 (BHBT)13a was prepared from 4,7-bis(4-methoxy- phenyl)-2,1,3-benzothiadiazole (BMPBT) by refluxing in a mixture of hydrobromic acid and acetic acid. The BMPBT was synthesized according to the method previously described.16a Hydrobromic acid, acetic acid, potassium carbonate, BADE, HMP 3 and p-TsOH were purchased from Kanto Chemical Co., Inc. and Tokyo Chemical Industry Co., Ltd. and used as received. HMP 3 was used after recrystallization. Acetone, chloroform and DCAA were purified by distillation before use. DMSO was distilled under vacuum and stored over molecular sieves (4 Å).O), 1608 and 1557 (CN and CC), 1254 and 1017 (C–O–C). Elem. Anal. Calcd for C42H56N2O6S (717.1): C, 70.34; H, 7.89; N, 3.91. Found: C, 69.98; H, 7.86; N, 3.90%.
The other HB polymers 4a–4b, 4d–4e were synthesized using the same procedure under the conditions shown in Table 2.
Measurements
The 1H NMR spectra were recorded using a JEOL JNM-AL400 spectrometer in CDCl3 and DMSO-d6. The FT-IR spectra were obtained on a Jasco FT/IR-660 Plus15 spectrometer using a KBr disc. The UV and the PL spectra in chloroform and in the solid phase were measured on a Jasco V-630 UV-vis spectrophotometer and FP-6200 fluorescence spectrophotometer, respectively. The polarizing microscope observations of the texture were achieved using a polarizing microscope (Nikon) equipped with a hot stage (magnification: ×200). The DSC measurements were carried out with a Shimadzu DSC-60 calorimeter at a heating and a cooling rate of 10 °C min−1 in nitrogen. The powder diffraction X-ray analyses of polymers quenched from the LC state were performed using a Rigaku Denki RINT 2500 generator with Cu Kα irradiation. The size exclusion chromatography (SEC) measurements were undertaken using a Jasco 830-RI refractometer and a column combination (K-803/K-804) (Shodex), with a polystyrene standard using chloroform as the eluent. The inherent viscosity measurements were carried out at a concentration of 0.2 g dL−1 in chloroform at 30 °C using an Ostwald-type viscometer. The order parameters (S) were evaluated from the polarized absorption spectra of the HB polymer samples measured using a polarizer. Oriented samples for solid-state polarized absorption and fluorescent spectroscopy were prepared by quenching the sheared compound and HB polymers from the LC states. In the samples, the measuring directions were determined as follows. The polarized film was fitted to the UV-vis spectrometer and the sites where the maximum absorptions of samples were observed were found. These are the absorbances parallel to the direction (A∥). The polarized film was then rotated 90° and the absorbances of samples in the sites were those perpendicular to that direction (A⊥). In the polarized fluorescence spectrum measurements, the measuring directions were determined using the same procedure as those for the polarized absorbance.Acknowledgements
The authors thank Ms. Michiko Egawa for her help in obtaining the elemental analysis data.References
- (a) C. J. Hawker and J. M. J. Fréchet, J. Am. Chem. Soc., 1990, 112, 7638 CrossRef CAS; (b) J. M. J. Fréchet and D. A. Tomalia, Dendrimers and Other Dendritic Polymers, Chichester, UK, 2001, Wiley Search PubMed.
- F. Vögtle, S. Gestermann, R. Hesse, H. Schwierz and B. Windisch, Prog. Polym. Sci., 2000, 25, 987 CrossRef CAS.
- D. K. Smith and F. Diederich, Top. Curr. Chem., 2002, 12, 767.
- J. M. J. Fréchet, Science, 1994, 263, 1710 CrossRef CAS.
- D. A. Tomalia and J. M. J. Fréchet, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2719 CrossRef CAS.
- C. Gao and D. Yan, Prog. Polym. Sci., 2004, 29, 183 CrossRef CAS.
- C. R. Yates and W. Hayes, Eur. Polym. J., 2004, 40, 1257 CrossRef CAS.
- A. Sunder, J. Heinmann and H. Frey, Chem.–Eur. J., 2000, 6, 2499 CrossRef CAS.
- B. Voit, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 2679 CrossRef CAS.
- B. Voit, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2505 CrossRef CAS.
- (a) Y. Chen, J. Loccufier, L. Vanmaele, E. Barriau and H. Frey, Macromol. Chem. Phys., 2007, 208, 1694 CrossRef CAS; (b) L. Ding, Z. Bo, Q. Chu, J. Li, L. Dai, Y. Pamg, F. E. Karasz and M. F. Durstock, Macromol. Chem. Phys., 2006, 207, 870 CrossRef CAS; (c) L. Tian, X. Shu and J. Zhu, Adv. Mater., 2007, 19, 4548 CrossRef CAS; (d) C. Gottschalk, F. Wolf and H. Frey, Macromol. Chem. Phys., 2007, 208, 1657 CrossRef CAS; (e) Z. Shi, Y. Zhou and D. Yan, Macromol. Rapid Commun., 2008, 29, 412 CrossRef CAS; (f) J. Shi, B. Tong, Z. Li, J. Shen, W. Zhao, H. Fu, J. Zhi, Y. Dong, M. Häussler, J. W. Y. Lam and B. Z. Tang, Macromolecules, 2007, 40, 8195 CrossRef CAS; (g) P. Wang, X. Wang, K. Meng, S. Hong, X. Liu, H. Cheng and C. C. Han, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3424 CrossRef CAS; (h) S. Hecht and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2001, 40, 74 CrossRef CAS; (i) P. Ceroni, G. Bergarnini, F. Marchioni and V. Balzani, Prog. Polym. Sci., 2005, 30, 453 CrossRef CAS; (j) A. T. Lebedev, A. V. Cheprakov, S. Sakadžić, D. A. Boas, D. F. Wilson and S. A. Vinogradov, ACS Appl. Mater. Interfaces, 2009, 1, 1292 Search PubMed; (k) A. Scarpaci, E. Blart, V. Montembault, L. Fontaine, V. Rodriguez and F. Odobel, ACS Appl. Mater. Interfaces, 2009, 1, 1799 Search PubMed; (l) R. Guan, Y. Xu, L. Ying, W. Yang, H. Wu, Q. Chen and Y. Cao, J. Mater. Chem., 2009, 19, 531 RSC; (m) T. Satoh, Soft Matter, 2009, 5, 1972 RSC.
- (a) A. Reisch, H. Komber and B. Voit, Macromolecules, 2007, 40, 6846 CrossRef CAS; (b) C. Oguz, S. Unal, T. E. Long and M. A. Gallivan, Macromolecules, 2007, 40, 6529 CrossRef CAS; (c) S. Unal and T. E. Long, Macromolecules, 2006, 39, 2788 CrossRef CAS; (d) Q. Lin and T. E. Long, Macromolecules, 2003, 36, 9809 CrossRef CAS.
- (a) M. P. Aldred, P. Vlachos, D. Dong, S. P. Kitney, W. C. Tsoi, M. O'Neill and S. M. Kelly, Liq. Cryst., 2005, 32, 951 CrossRef CAS; (b) M. Funahashi and J. Hanna, Appl. Phys. Lett., 1998, 73, 3733 CrossRef CAS; (c) J. Belmar, M. Parra, C. Zũñniga, C. Pérez and C. Muñoz, Liq. Cryst., 1999, 26, 389 CrossRef CAS; (d) T. M. H. Costa, V. Stefani, M. R. Gallas, N. M. Balzaretti and J. A. H. da Jornada, J. Mater. Chem., 2001, 11, 3377 RSC; (e) K. Tanaka, T. Kumagai, H. Aoki, N. Deguchi and S. Iwata, J. Org. Chem., 2001, 66, 7328 CrossRef CAS; (f) M. Sato, A. Nakashima, Y. Sato and I. Yamaguchi, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6688 CrossRef CAS; (g) J.-B. Baek and L.-C. Chien, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 3587 CrossRef CAS.
- (a) M. Sato, R. Ishii, S. Nakashima, K. Yonetake and K. Kido, Liq. Cryst., 2001, 28, 1211 CrossRef CAS; (b) M. Sato, S. Nakashima and Y. Uemoto, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 2676 CrossRef CAS; (c) M. Sato, M. Notsu and S. Nakashima, Liq. Cryst., 2004, 31, 1195 CrossRef CAS; (d) M. Sato, R. Ohta, M. Handa and K. Kasuga, Liq. Cryst., 2002, 29, 1441 CrossRef CAS; (e) M. Sato, Y. Matsuoka and I. Yamaguchi, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2998 CrossRef CAS; (f) S. Nakashima, M. Sato and I. Yamaguchi, Polym. Int., 2008, 57, 39 CrossRef CAS.
- (a) M. Sato and R. Ohta, Liq. Cryst., 2007, 34, 295 CrossRef CAS; (b) M. Sato, A. Nakashima and I. Yamaguchi, Kobunshi Ronbunshu, 2007, 64, 929 CrossRef CAS.
- (a) X. Zhang, H. Gorohmaru, M. Kadowaki, T. Kobayashi, T. Ishii, T. Thiemann and S. Makata, J. Mater. Chem., 2004, 14, 1901 RSC; (b) X. Zhang, R. Yamaguchi, K. Moriyama, M. Kadowaki, T. Kobayashi, T. Ishii, T. Thiemann and S. Makata, J. Mater. Chem., 2006, 16, 736 RSC; (c) M. P. Aldred, M. Carrasco-Orozco, A. E. A. Contoret, D. Dong, S. R. Farrar, S. M. Kelly, S. P. Kitney, D. Mathieson, M. O'Neill, W. C. Tsoi and P. Vlachos, Liq. Cryst., 2006, 33, 459 CrossRef CAS; (d) H. Yan, P. Lee, N. R. Armstrong, A. Graham, G. A. Evmenenko, P. Dutta and T. J. Marks, J. Am. Chem. Soc., 2005, 127, 3172 CrossRef CAS.
- M. A. Ogliaruso and J. F. Wolfe, The Chemistry of Functional Groups, Supplement B: The Chemistry of Acid Derivatives Part 1, ed. S. Patai, John Wiley & Sons, Chichester, 1979, p. 419 Search PubMed.
- Our unpublished data.
- P. J. Flory, J. Am. Chem. Soc., 1952, 74, 2718 CrossRef CAS.
- X. Xiao, Y. Wu, M. Sun, J. Zhou, Z. Bo, L. Li and C. Chan, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 574 CrossRef CAS.
- M. Millaruelo, L. S. Chinellato, Jr., J. L. Serrano, L. Oriol and M. Piñol, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4804 CrossRef CAS.
- P. K. Tsolakis and J. K. Kallitsis, Chem.–Eur. J., 2003, 9, 936 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |