Redox-triggered switching of helical chirality of poly(phenylacetylene)s bearing riboflavin pendants†
Hiroki
Iida
,
Tomohisa
Mizoguchi
,
Seong-Dae
Oh
and
Eiji
Yashima
*
Department of Molecular Design and Engineering, Graduate School of Engineering, Nagoya University, Chikusa-ku, Nagoya, 464-8603, Japan. E-mail: yashima@apchem.nagoya-u.ac.jp; Fax: +81-52-789-3185; Tel: +81-52-789-4495
First published on 20th April 2010
Abstract
Two novel optically active poly(phenylacetylene)s bearing riboflavin (vitamin B2) residues as the pendant through different covalent linkages (poly-1 and poly-2) were prepared by the polymerization of the corresponding monomers (1 and 2) with a rhodium catalyst, and their chiroptical properties were investigated by UV-visible and circular dichroism (CD) spectroscopies. Riboflavin-linked poly-2 through the acetal linkage with the ribityl group to the phenylacetylene exhibited a relatively intense induced CD (ICD) in the polymer backbone region. The ICD was temperature-dependent, indicating that the main chain of poly-2 adopts a dynamic preferred-handed helical conformation induced by the optically active ribityl group. In contrast, poly-1 having the riboflavin pendant via the N3-methylene linker showed almost no apparent ICD due to the main-chain helical conformation, probably because the optically active ribityl group of poly-1 is located relatively far from the polymer backbone compared with poly-2. The cyclic voltammetric measurements of poly-1 and poly-2 revealed that these polymers possess reversible redox properties originating from the electron transfer process of the riboflavin pendants. The switching of the chiroptical properties in response to the redox stimuli was also investigated using a chemical reductant (Na2S2O4) and oxidant (O2).
Introduction
Riboflavin (vitamin B2), a redox active unit of cofactors, such as flavin mononucleotide and flavin adenine dinucleotide, is one of the most important bioactive compounds in enzymatic functions.1,2 A commercially produced riboflavin is used as a yellow pigment in many food products daily,3 and its derivatives show unique and diverse functions, such as redox activity, organocatalysis and photosensitivity, arising from the conjugated isoalloxazine ring with ribityl units.1,3,4 As evidenced by the sophisticated functions of flavoproteins,2c,5 the artificial riboflavin-containing optically active polymers with a well-defined structure may be applied to a wide range of fields including optoelectronic materials and biological sciences. However, to the best of our knowledge, riboflavin-containing polymers, in particular, helical polymers, have not yet been reported, although very few polymers bearing riboflavin units as the pendant groups are known.6On the other hand, the synthesis of optically active helical polymers with a controlled helical sense has attracted significant attention during the past decades not only to mimic biological helices and functions, such as DNA and proteins, but also for their possible applications in chiroptical devices and chiral materials as enantioselective absorbents, catalysts and sensors.7 Among the synthetic helical polymers prepared so far, polyacetylenes are one of the most intensively studied helical polymers because of their unique dynamic macromolecular helicity consisting of interconverting right- and left-handed helical segments separated by the rarely occurring helical reversals that have been experimentally and theoretically revealed7f–m like those in polyisocyanates.7a,8 The introduction of optically active substituents to the pendant groups can induce an excess of one-handed helical conformation in the main chain, which further results in a helical array of the pendant groups with a predominant screw-sense along the polymer chain. Therefore, induced helical polyacetylenes exhibit a characteristic induced circular dichroism (ICD) in the polymer backbone regions as well as in the pendant chromophoric regions.9 This unique property of helical polyacetylenes prompted us to synthesize poly(phenylacetylene)s bearing an optically active riboflavin as the pendant group with the expectation that the covalent-bonded chiral riboflavin might induce a preferred-handed helix in the polymer main-chain accompanied by a one-handed helical array of the riboflavin pendants. We report here the design and synthesis of two novel optically active poly(phenylacetylene)s (poly-1 and poly-2 in Chart 1) bearing an optically active riboflavin unit as the pendant groups covalently bonded through different positions. These polymers showed a reversible but different redox behavior depending on the position of the optically active riboflavin pendants accompanied by a reversible change in the chiroptical properties arising from the redox activity of the riboflavin units. Switching of molecular chirality by redox processes has been extensively studied.10–12 However, to the best of our knowledge, only a few examples have been reported regarding the redox-triggered chirality switching of synthetic optically active polymers with a controlled helical sense.10e,g,i
 | ||
| Chart 1 | ||
Results and discussion
Synthesis and polymerization of riboflavin-bound phenyl acetylenes
The optically active poly(phenylacetylene)s, poly-1 and poly-2, were synthesized according to Schemes 1 and 2, respectively. The nucleophilic substitution reaction of riboflavin tetrabutylate 3 with 4 in toluene in the presence of 1,8-diazabicyclo[5.4.0]-7-undecene (DBU) yielded the phenylacetylene monomer 1 bearing the riboflavin pendant via an N3-methylene phenylene linkage at the para position. The monomer 2, in which the riboflavin unit is linked to the para position of the phenyl group through the acetal linkage with the ribityl group, was synthesized from 5′-O-trityl riboflavin 5 and 4-ethynylbenzaldehyde dimethyl acetal 6 according to the reported procedure for analogous compounds.13 The stereoregular (cis-transoidal) poly(phenylacetylene)s, poly-1 and poly-2, were prepared by the polymerization of monomers 1 and 2 using [Rh(nbd)Cl]2 (nbd: norbornadiene) as a catalyst in CHCl3 in the presence of NEt3 in accordance with a previously reported method.9b The number-average molecular weights (Mn) of poly-1 and poly-2 were estimated to be 5.89 × 103 (Mw/Mn = 3.24) and 1.41 × 104 (Mw/Mn = 1.94), respectively, as determined by size exclusion chromatography (SEC) using poly(ethylene oxide) and poly(ethylene glycol) standards in dimethylformamide (DMF) containing 10 mM LiCl as the eluent. The stereoregularities of the obtained polymers were investigated by NMR and Raman spectroscopy. However, we could not estimate the stereoregularity of the polymers by their 1H NMR spectra because of the broadening of the main-chain protons and the overlapping with some flavin protons (Fig. S1†). The Raman spectra of the polymers in the solid state gave useful information and showed weak peaks due to the fluorescence of the riboflavin pendants,14 but apparent peaks resulting from the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–C bond vibrations in the cis polyacetylenes were observed (Fig. S2†).15 Although those in the trans polyacetylene were observed at a lower wavelength (ca. 1200 cm−1), the peaks were weaker than those in the cis polyacetylenes, indicating that the polymers are rich in cis-transoid.
C and C–C bond vibrations in the cis polyacetylenes were observed (Fig. S2†).15 Although those in the trans polyacetylene were observed at a lower wavelength (ca. 1200 cm−1), the peaks were weaker than those in the cis polyacetylenes, indicating that the polymers are rich in cis-transoid.
 | ||
| Scheme 1 Synthesis of poly-1. | ||
 | ||
| Scheme 2 Synthesis of poly-2. | ||
The monomers, 1 and 2, exhibited strong emissions at 511 nm with a similar pattern arising from the conjugated flavin ring upon excitation at 445 and 439 nm, respectively, in diluted DMF (Fig. S3†). Poly-1 and poly-2 also displayed similar emissions with peaks of 514 and 511 nm, respectively, under identical conditions, but the emission intensities were over 50 times smaller than those of the corresponding monomers. Because the main chains of poly-1 and poly-2 consist of π-conjugated double bonds, these fluorescence quenching phenomena may indicate a possible intramolecular energy transfer or electron transfer from the flavin pendants to the polymer backbones. A similar fluorescence quenching was also reported for analogous helical poly(phenylacetylene)s bearing fluorescent pendant groups.16
Chiroptical properties of riboflavin-bound monomers and polymers
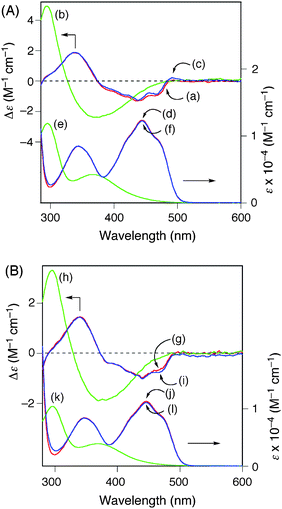
The circular dichroism (CD) and absorption spectra of monomers and polymers were then measured in DMF in order to explore their chiroptical properties (Fig. 1). The monomers 1 (a and c) and 2 (e and g in Fig. 1) gave rise to the typical absorption signals centered at around 350 and 440 nm assigned to the riboflavin moieties and characteristic Cotton effects in the regions,17 although their CD spectral patterns in the longer wavelength regions were different. Poly-1 and poly-2 were expected to adopt a preferred-handed helical conformation biased by the optically active ribityl groups, resulting in induced Cotton effects in the long wavelength regions (300–500 nm) due to the helically twisted π-conjugated polymer backbones.7e–g,i,j,l,m However, poly-1 exhibited a CD similar in its pattern to that of the monomer 1 except for the positive CD at lower wavelengths (< 300 nm). | ||
| Fig. 1 CD (a, b, e, f) and absorption (c, d, g, h) spectra of 1 (a, c) and poly-1 (b, d) (A), and 2 (e, g) and poly-2 (f, h) (B) in DMF (0.5 mg mL−1) at ca. 25 °C. | ||
On the other hand, poly-2 showed the apparent Cotton effects in the flavin chromophore and π-conjugated main-chain regions (290–530 nm), whose CD spectral patterns were considerably different from those of the monomer 2. These drastic CD changes were accompanied by a remarkable decrease in the absorption intensities (hypochromic effect). In addition, the CD intensity of poly-2, in particular, the split-type Cotton effect intensity at 290–400 nm, increased with decreasing temperature, while maintaining the absorption intensity (Fig. 2B). In contrast, the CD and absorption spectra of poly-1 hardly changed even at low temperature (Fig. 2A). These results suggest that poly-2 appeared to possess an excess of a dynamic one-handed helical conformation induced by the optically active ribityl group chemically bonded through the acetal linkage, and the helical sense excess of poly-2 became greater at lower temperature, but the helical structure including the helical pitch most likely remained intact at the temperature ranges (−10–75 °C). The hypochromic effect observed in the poly-2 provided evidence of the π–π stacking between the neighboring conjugated flavin pendant units, which may further arrange in a preferred-handed helical array along the helical main chain. Poly-1, however, may not have a helical sense bias because the optically active ribityl groups are located away from the polymer backbone and the linker is more flexible compared with poly-2.18,19
 | ||
| Fig. 2 CD and absorption spectral changes of poly-1 (A) and poly-2 (B) in DMF (0.5 mg mL−1) at various temperatures. | ||
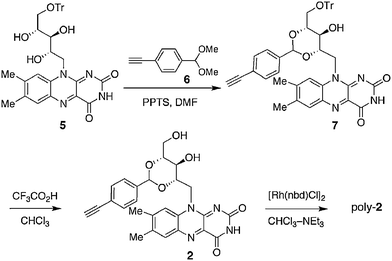
Redox properties of helical polymers and their model compounds
It is well-known that flavins are redox active and occupy three redox states (Scheme 3); in aprotic media, one-electron reduction of oxidized flavin (Flox) occurs to give a radical anion (Flrad−), while in protic or aqueous media, Flox produces the reduced flavin (Flred) via Flrad− by two-electron reduction and protonation.4a,b,20 Quite interestingly, these changes in the redox states of flavins are accompanied by a unique structural change of the flavin skeleton from a planar (Flox and Flrad−) to a bent form (Flred).4a,21 Such an electron-induced structural change of the flavin units would enable flavin-bound helical polymers to act as a switch for their helical conformations by responding to an electrical stimulus. With this in mind, we first investigated the electrochemical properties of poly-1 and poly-2 and the corresponding model compounds 8 and 3, respectively, by cyclic voltammetry (CV) in CH3CN with Bu4NClO4 (0.01 M) as the supporting electrolyte (Fig. 3). The CV profile of poly-1 was reversible with a reduction wave at −1.23 V similar to that of its model compound 8 (Fig. 3 and S7†).
 | ||
| Fig. 3 Cyclic voltammograms of poly-1 (2 mM) (a) and poly-2 film (b) in 0.01 M Bu4NClO4/CH3CN solution at a scan rate of 100 mV s−1 (A). Oxidation (Epa) and reduction (Epc) peaks of poly-1, poly-2, 8, 3, 3 with acetic acid (1 equiv), and ferrocene are shown in (B). | ||
On the contrary, poly-2, measured in the film state because of poor solubility in CH3CN, showed a reversible but broad cyclic voltammogram with an apparent reduction wave at −1.12 V, being different from that of its model compound 3 (−1.21 V) and poly-1 (−1.23 V) (Fig. 3 and S7†). The fact that a poly(phenylacetylene) film bearing no riboflavin pendant groups displayed almost no obvious redox waves under the same conditions (Fig. S6†) indicates that the redox properties of poly-1 and poly-2 totally originate from the riboflavin pendant units. In addition, the large differences in peak shape in CVs and reduction waves observed between poly-1 and poly-2 can be ascribed to the difference in the redox processes of the riboflavin units attached on helical poly(phenylacetylene)s (poly-1 and poly-2) through different linkages. It is likely that a reversible one-electron transfer process between Flox and Flrad− may mainly take place for poly-1 as well as 8 and 3, whereas a two-electron reduction process coupled with protonation of Flrad− from the other Flox pendants may be predominant for poly-2 (Scheme 3).20,22,23 This speculation was supported by the CV profile of poly-2 that exhibited a significant positive shift of the oxidation peak (Epa = −0.77 V) due to the generation of Flred, compared with those of poly-1, 8 and 3 (Epa = −1.12, −1.05 and −1.02 V, respectively). A similar positive shift of the oxidation peak (Epa = −0.80 V) was observed in the CV profile of 3 in the presence of acetic acid (1 equiv.), in which 3 underwent a two-electron reduction coupled with protonation from acetic acid (Fig. 3 and S7†).20
In addition to the electrochemical means, various chemical reductants, such as sodium dithionite (Na2S2O4) and sodium borohydride, can be used for reduction of Flox to generate Flred, which easily regenerates Flox through oxidation with O2 (Scheme 3).1,2a,c The switching of the chiroptical properties of poly-1 and poly-2 as well as the model compounds 3 and 8 in response to the redox stimuli was then investigated using a chemical reductant (Na2S2O4) and oxidant (O2) by following the CD and absorption spectral changes during the reduction and oxidation processes. As shown in Fig. 4, the model compounds 3 and 8 in DMF were reduced to give the reduced form (3red and 8red) upon the addition of aqueous Na2S2O4 solution (20 equiv.) under argon (DMF–water (99/1, v/v)), resulting in the disappearance of the absorption peak at 450 nm accompanied by a drastic change in their CD spectral pattern.24 Each solution was then exposed to air for 2 h, which regenerated the original oxidized 3 and 8 as revealed by the complete recovery of the characteristic absorption and CD peaks.25
 | ||
| Fig. 4 CD (a–c, g–i) and absorption (d–f, j–l) spectra of 3 (A) and 8 (B) (0.5 mg mL−1) in DMF–water (99/1, v/v) before (a, d, g, j) and after (b, e, h, k) reduction with Na2S2O4 (20 equiv.), and further oxidation with air (c, f, i, l) at ca. 25 °C. | ||
In the same way, the reversible absorption and CD spectral changes of poly-1 and poly-2 were clearly observed with the addition of aqueous Na2S2O4 under argon followed by oxidation with air (Fig. 5). Judging from the characteristic changes in the absorption spectra of poly-1 and poly-2 as observed for the model compounds 3 and 8, the riboflavin pendant units appeared to be reduced to give the reduced forms (poly-1red and poly-2red, respectively) in the presence of aqueous Na2S2O4 under argon.26 Subsequent oxidation of poly-1red and poly-2red with air successfully reproduced the original oxidized forms (poly-1 and poly-2, respectively) as supported by the reappearance of the original absorption signals.
 | ||
| Fig. 5 CD (a–c, g–i) and absorption (d–f, j–l) spectra of poly-1 (0.5 mg mL−1) (A) and poly-2 (0.05 mg mL−1) (B) in DMF–water (99/1 and 99.9/0.1, respectively, v/v) before (a, d, g, j) and after (b, e, h, k) reduction with Na2S2O4 (20 and 5 equiv., respectively), and further oxidation with air (c, f, i, l) at ca. 25 °C. | ||
Interestingly, the CD spectral patterns of poly-1 and poly-2 were drastically changed upon reduction with Na2S2O4 but were almost completely recovered to the original CDs after oxidation with air. Thus, the “redox-triggered switching of chirality” on helical polymers has been achieved. The observed CD pattern of poly-1red was quite different from that of the model compound 8red, whereas the poly-2red exhibited a CD pattern similar to that of the reduced model compound 3red, although the CD spectral pattern of the original (oxidized) poly-2 was significantly different from those of the model compound 3 and the monomer 2.
It has been reported that oxidized flavins (Flox) have a tendency to form a face-to-face π-stacked complex either in solution or in the solid state.27 In comparison, reduced flavins (Flred) cannot form such intermolecular contacts because of their bent structure (Scheme 3).4a,21 The same structural features may be the case for flavin-functionalized polymers including poly-1 and poly-2, so that such structural changes in the flavin pendants might be closely related to the significant changes in their CD spectra after reduction with Na2S2O4. In particular, the drastic change in the CD spectral pattern observed in poly-2 before and after reduction suggests that the π-stacked pendant flavin units are arranged in either a right- or left-handed screw-sense along the preferred-handed helical poly-2 backbone may be transformed into a non-helical disordered array after reduction, because of the steric repulsion between the resultant bent-structured, reduced flavin units (Fig. 6A). This structural change in the pendant groups may bring about a further conformational change in the polymer backbone from a predominantly one-handed helical conformation to an almost racemic one. This speculation is supported by the facts that the CD spectral pattern of poly-2red was similar to that of the reduced 8 and that complete recovery of the CD was attained through oxidation with air.
 | ||
| Fig. 6 (A) A schematic illustration of the redox-triggered switching of the helical structure of poly-2. (B) A possible helical structure of poly-2 (30-mer). The structure is shown using the cylinder model. The flavin rings and the main-chain atoms are shown using the space-filling model for clarity. | ||
Fig. 6B shows a possible helical structure of poly-2 calculated by molecular mechanics calculations based on a 23 unit/10 turn (23/10) helical poly(phenylacetylene) whose helical structure was determined by X-ray (see ESI†).28 The calculated structure revealed that the neighboring flavin pendant units29 are overlapped via π–π stacking, resulting in formation of a one-handed helical array along the helical main chain.
Conclusions
We synthesized two novel optically active poly(phenylacetylene)s (poly-1 and poly-2) bearing a naturally occurring riboflavin derivative as the pendant groups. Poly-2 possesses an excess of a dynamic one-handed helical conformation induced by the optically active ribityl pendants, as revealed by the temperature-dependent CD changes in the π-conjugated main-chain chromophore regions. The electrochemical analysis of poly-1 and poly-2 showed reversible but different redox behavior accompanied by a reversible change in the chiroptical properties arising from the redox activity of the riboflavin units. The riboflavin units of poly-1 and poly-2 could also be reversibly reduced by chemical reduction with Na2S2O4, resulting in the reduced forms (poly-1red and poly-2red), which could reproduce the original poly-1 and poly-2 through subsequent oxidation with air. The CD spectral patterns of poly-1 and poly-2 were drastically changed upon reduction with Na2S2O4 but were almost completely recovered to the original CDs after aerobic oxidation. The changes in their CD spectra might be closely related to the structural changes in the flavin pendants from a planar (Flox) to a bent form (Flred); the latter cannot form a face-to-face π-stacked complex with the neighboring riboflavin pendants due to steric repulsion. The redox-triggered switching of chirality, in particular, that observed in poly-2, suggests that the π-stacked pendant flavin units arranging in either a right- or left-handed screw-sense induced by the preferred-handed helical poly-2 backbone may be transformed into a disordered array stimulated by the main-chain conformational change from an excess one-handed helix to an almost racemic one. Flavin derivatives are known to work as an organocatalyst,30 which suggests that the present flavin-bound helical polymers may be promising chiral materials for developing novel asymmetric organocatalysts for enantioselective oxidation; work along this line is now in progress in our laboratory.Experimental section
Synthesis of 3-p-ethynylbenzyl riboflavin tetrabutylate (1)
DBU (0.30 mL, 2.0 mmol) was added to a solution of 4 (0.93 mL, 0.79 mmol) and riboflavin tetrabutylate (3) (0.43 g, 0.70 mmol) in anhydrous toluene (85 mL), and the reaction mixture was stirred at 55 °C for 24 h. Water (3000 mL) was added to this solution and the mixture was extracted with diethyl ether (2000 mL). The organic extracts were dried over MgSO4. After filtration, the solvent was evaporated to dryness. The residue was then purified by silica gel chromatography with hexane–CHCl3 (60/40, v/v) to give 1 (100 mg, 20%) as a yellow powder. Mp: 116–117 °C. [α]20D + 62.96 (c 0.1, CHCl3). Anal. Calcd (%) for C42H59N4: C, 65.44; H, 6.54; N, 7.27. Found: C, 65.45; H, 6.54; N, 7.36. IR (KBr, cm−1): 3263 (ν–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H), 2108 (νCC), 1662, 1588, 1550. 1H NMR (500 MHz, CDCl3): δ 8.00 (s, 1H, ArH), 7.55 (s, 1H, ArH), 7.53 (d, J = 8.5 Hz, 2H, ArH), 7.40 (d, J = 8.5 Hz, 2H, ArH), 5.66 (br s, 1H, ribityl–CH), 5.47 (br s, 1H, ribityl–CH), 5.44–5.40 (m, 1H, ribityl–CH), 5.25 (d, J = 3.0 Hz, 2H, NCH2Ar), 4.88 (br, 2H, ribityl–CH), 4.67 (dd, J = 12.3, 4.4 Hz, 1H, ribityl–CH), 4.22 (dd, J = 12.2, 6.1 Hz, 1H, ribityl–CH), 3.02 (s, 1H, HC), 2.62–1.18 (m, 22H, CH3Ar and COCH2CH2), 1.00 (t, J = 7.3 Hz, 3H, CH2CH3), 0.98 (t, J = 7.3 Hz, 3H, CH2CH3), 0.93 (t, J = 7.4 Hz, 3H, CH2CH3), 0.55 (t, J = 7.5 Hz, 3H, CH2CH3). 13C NMR (125 MHz, CDCl3, rt): δ 173.52, 173.18, 172.73, 160.02, 154.98, 149.51, 147.99, 138.01, 136.95, 136.01, 135.06, 133.24, 132.43, 131.53, 129.75, 121.63, 115.80, 83.93, 77.40, 70.66, 69.31, 62.05, 51.19, 45.09, 44.81, 41.28, 36.29, 36.26, 36.15, 35.76, 30.03, 21.65, 19.73, 18.71, 18.58, 17.95, 13.92, 13.87, 13.53.
C–H), 2108 (νCC), 1662, 1588, 1550. 1H NMR (500 MHz, CDCl3): δ 8.00 (s, 1H, ArH), 7.55 (s, 1H, ArH), 7.53 (d, J = 8.5 Hz, 2H, ArH), 7.40 (d, J = 8.5 Hz, 2H, ArH), 5.66 (br s, 1H, ribityl–CH), 5.47 (br s, 1H, ribityl–CH), 5.44–5.40 (m, 1H, ribityl–CH), 5.25 (d, J = 3.0 Hz, 2H, NCH2Ar), 4.88 (br, 2H, ribityl–CH), 4.67 (dd, J = 12.3, 4.4 Hz, 1H, ribityl–CH), 4.22 (dd, J = 12.2, 6.1 Hz, 1H, ribityl–CH), 3.02 (s, 1H, HC), 2.62–1.18 (m, 22H, CH3Ar and COCH2CH2), 1.00 (t, J = 7.3 Hz, 3H, CH2CH3), 0.98 (t, J = 7.3 Hz, 3H, CH2CH3), 0.93 (t, J = 7.4 Hz, 3H, CH2CH3), 0.55 (t, J = 7.5 Hz, 3H, CH2CH3). 13C NMR (125 MHz, CDCl3, rt): δ 173.52, 173.18, 172.73, 160.02, 154.98, 149.51, 147.99, 138.01, 136.95, 136.01, 135.06, 133.24, 132.43, 131.53, 129.75, 121.63, 115.80, 83.93, 77.40, 70.66, 69.31, 62.05, 51.19, 45.09, 44.81, 41.28, 36.29, 36.26, 36.15, 35.76, 30.03, 21.65, 19.73, 18.71, 18.58, 17.95, 13.92, 13.87, 13.53.
Polymerization: synthesis of poly-1
The polymerization was carried out according to Scheme 1. Monomer 1 (60 mg, 0.080 mmol) was placed in a dry ampule, which was then evacuated on a vacuum line and flushed with dry nitrogen. After this evacuation-flush procedure was repeated three times, a three-way stopcock was attached to the ampule, and anhydrous CHCl3 (0.36 mL) was added with a syringe. To this was added a solution of [Rh(nbd)Cl]2 (0.10 M) containing Et3N ([Et3N]/[Rh] = 20) in CHCl3 at 30 °C. The concentrations of the monomer and the rhodium complex were 0.2 and 0.01 M, respectively. After 36 h, the resulting polymer was precipitated into a large amount of MeOH, washed with diethyl ether and hexane, and collected by centrifugation. The product was purified by reprecipitation from CHCl3 to diethyl ether, washed with diethyl ether and dried in vacuo at room temperature for 24 h (37 mg, 62% yield). The Mn and Mw/Mn values were 5.89 × 103 and 3.24, respectively, as determined by SEC using poly(ethylene oxide) and poly(ethylene glycol) standards in DMF containing 10 mM LiCl as the eluent. Anal. Calcd (%) for (C42H59N4)n C, 65.44; H, 6.54; N, 7.27. Found: C, 65.25; H, 6.50; N, 7.15. IR (KBr, cm−1): 1664, 1587, 1550. 1H NMR (500 MHz, CDCl3, 60 °C): δ 7.97– 6.39 (br m, 6H, ArH), 5.66–4.21 (m, 10H, CHO, NCH2 and HC = CAr), 2.60–1.07 (m, 22H, CH3Ar and COCH2CH2), 1.07−0.56 (m, 12H, CH3).Synthesis of 2′,4′-O-p-ethynylbenzylidene riboflavin (2)
Pyridinium p-toluenesulfonate (PPTS, 80.4 mg, 0.32 mmol) was added to a solution of 5 (1.00 g, 1.62 mmol) and 6 (3.10 g, 17.6 mmol) in DMF (40 mL), and the mixture was stirred at 55 °C for one day. An ice-cold solution of saturated NaHCO3 (100 mL) was added to this solution, and the mixture was extracted with CHCl3 (300 mL). The organic extracts were dried over anhydrous MgSO4. After filtration, the solvent was evaporated to dryness and the crude product containing an intermediate 7 was subjected to the next reaction without further purification. The crude product (1.0 g) was dissolved in CHCl3 (30 mL), and to this was added trifluoroacetic acid (0.6 mL). The reaction mixture was stirred at room temperature for 4 h. An ice-cold solution of saturated NaHCO3 (50 mL) was added to this solution, and the mixture was extracted with CHCl3 (150 mL). The organic extracts were dried over anhydrous MgSO4. After filtration, the solvent was evaporated to dryness. The residue was then purified by silica gel chromatography with MeOH–CHCl3 (0%–5%, v/v) as the eluent and the obtained product was further subjected to SEC fractionation, giving 2 (84.0 mg) as a yellow powder in 11% yield. Mp: 186–188 °C. [α]20D + 76.4 (c 0.1, CHCl3). Anal. Calcd (%) for C26H24N4: C, 63.93; H, 4.95; N, 11.47. Found: C, 63.67; H, 4.71; N, 11.39. IR (KBr, cm−1): 3278 (ν–CC–H), 2103 (νCC), 1664, 1578, 1541. 1H NMR (500 MHz, DMSO-d6): δ 11.35 (br s, 1H, NH), 8.02 (s, 1H, ArH), 7.85 (s, 1H, ArH), 7.38 (d, J = 8.5 Hz, 2H, ArH), 7.25 (d, J = 8.0 Hz, 2H, ArH), 5.67 (d, J = 5.5 Hz, 1H, CHO), 5.53 (s, 1H, ArCH), 5.21 (br s, 1H, OH), 4.86 (br s, 1H, OH), 4.71 (t, 1H, ribityl–CH), 4.19 (s, 1H, HC), 4.18–4.15 (m, 1H, ribityl–CH), 3.77–3.74 (m, 1H, ribityl–CH), 3.66–3.63 (m, 1H, ribityl–CH), 3.56–3.50 (m, 2H, ribityl–CH), 2.46 (s, 3H, CH3), 2.40 (s, 3H, CH3). 13C NMR (125 MHz, DMSO-d6): δ 159.93, 155.46, 150.68, 145.65, 138.49, 137.19, 135.82, 133.76, 132.04, 131.17, 130.52, 126.35, 121.89, 118.19, 98.61, 83.18, 81.89, 81.27, 78.57, 63.28, 60.69, 46.19, 20.62, 18.76.
Polymerization: synthesis of poly-2
The polymerization of 2 (80 mg, 0.16 mmol) was carried out according to Scheme 2 in the same manner for that of 1 in anhydrous CHCl3 (0.72 mL) with [Rh(nbd)Cl]2 (0.02 M) in the presence of Et3N ([Et3N]/[Rh] = 100) at 30 °C. The concentrations of the monomer and the rhodium complex were 0.2 and 0.002 M, respectively. After 24 h, the resulting polymer was precipitated into a large amount of MeOH, washed with hexane, and collected by centrifugation. The product was purified by reprecipitation from DMF to MeOH and from DMF to diethyl ether, and the precipitated polymer was washed with diethyl ether, MeOH and hexane, and dried in vacuo at room temperature for 24 h (67.6 mg, 85% yield). The Mn and Mw/Mn values were 1.41 × 104 and 1.94, respectively, as determined by SEC using poly(ethylene oxide) and poly(ethylene glycol) standards in DMF containing 10 mM LiCl as the eluent. Anal. Calcd (%) for (C26H24N4·3/2H2O)n: C, 60.58; H, 5.28; N, 10.87. Found: C, 60.66; H, 5.45; N, 10.70. IR (KBr, cm−1): 1664, 1580, 1545. 1H NMR (500 MHz, DMSO-d6, 60 °C): δ 11.21 (br s, 1H, NH), 8.00–7.36 (br m, 6H, ArH), 5.53–3.52 (m, 9H, ribityl, ArCH and HCC), 2.44–2.08 (br m, 6H, CH3).
CD and absorption measurements of poly-1, poly-2, 3 and 8 through redox reaction
A typical experimental procedure is described below. Because the reduced forms of riboflavin derivatives are air sensitive, all procedures except for the re-oxidation process using air were carried out under an argon atmosphere. Stock solutions of poly-2 (0.05 mg mL−1, 0.1 mM monomer units) in DMF and 0.5 M Na2S2O4 in water were prepared. A 10 mL portion of the poly-2 solution was transferred to a 10 mL flask equipped with a stopcock. The Na2S2O4 aqueous solution (10 μL, 5 equiv.) was added to this using a Hamilton microsyringe. After standing at room temperature for 1 h, the solution was filtered through a membrane filter (0.45 μm) to remove the precipitated Na2S2O4, and the CD and absorption spectra of the filtrate containing poly-2red were taken (Fig. 5B). The filtrate was then exposed to air in order to re-oxidize poly-2red, and the CD and absorption spectra of the resultant poly-2 solution were recorded after standing it at room temperature for 2 h.Acknowledgements
This work was supported in part by a Grant-in-Aid for Scientific Research (S) from the Japan Society for the Promotion of Science (JSPS) and the Global COE Program “Elucidation and Design of Materials and Molecular Functions” of the Ministry of Education, Culture, Sports, Science, and Technology, Japan.Notes and references
- F. Müller, Chemistry and Biochemistry of Flavoenzymes, CRC Press, Boston, 1991 Search PubMed.
- (a) T. C. Bruice, Acc. Chem. Res., 1980, 13, 256–262 CrossRef CAS; (b) C. Walsh, Acc. Chem. Res., 1980, 13, 148–155 CrossRef CAS; (c) V. Massey, Biochem. Soc. Trans., 2000, 28, 283–296 CrossRef CAS; (d) S. O. Mansoorabadi, C. J. Thibodeaux and H.-w. Liu, J. Org. Chem., 2007, 72, 6329–6342 CrossRef CAS.
- E. Choe, R. Huang and D. B. Min, J. Food Sci., 2005, 70, R28–R36 CrossRef CAS.
- (a) J. J. Hasford, K. W. and C. J. Rizzo, J. Org. Chem., 1997, 62, 5244–5245 CrossRef CAS; (b) J. J. Hasford and C. J. Rizzo, J. Am. Chem. Soc., 1998, 120, 2251–2255 CrossRef CAS; (c) S. G. Bertolotti, C. M. Previtali, A. M. Rufs and M. V. Encinas, Macromolecules, 1999, 32, 2920–2924 CrossRef CAS; (d) S. Fukuzumi, K. Yasui, T. Suenobu, K. Ohkubo, M. Fujitsuka and O. Ito, J. Phys. Chem. A, 2001, 105, 10501–10510 CrossRef CAS; (e) Y. Imada, H. Iida, S.-I. Murahashi and T. Naota, Angew. Chem., Int. Ed., 2005, 44, 1704–1706 CrossRef CAS; (f) C. Smit, M. W. Fraaije and A. J. Minnaard, J. Org. Chem., 2008, 73, 9482–9485 CrossRef CAS; (g) S.-H. Kim and C.-C. Chu, J. Biomed. Mater. Res., Part B, 2009, 91b, 390–400 CrossRef CAS; (h) H. Schmaderer, P. Hilgers, R. Lechner and B. Koenig, Adv. Synth. Catal., 2009, 351, 163–174 CrossRef CAS; (i) Y. Imada, T. Kitagawa, T. Ohno, H. Iida and T. Naota, Org. Lett., 2010, 12, 32–35 CrossRef.
- (a) A. Sancar, Chem. Rev., 2003, 103, 2203–2237 CrossRef CAS; (b) J. T. M. Kennis and M.-L. Groot, Curr. Opin. Struct. Biol., 2007, 17, 623–630 CrossRef CAS.
- (a) H. Kamogawa, J. Polym. Sci., Part A-1, 1969, 7, 409–413 CrossRef CAS; (b) S. Shinkai, S. Yamada and T. Kunitake, Macromolecules, 1978, 11, 65–68 CrossRef CAS; (c) W. J. Spetnagel and I. M. Klotz, Biopolymers, 1978, 17, 1657–1668 CrossRef CAS.
- (a) M. M. Green, J.-W. Park, T. Sato, A. Teramoto, S. Lifson, R. L. B. Selinger and J. V. Selinger, Angew. Chem., Int. Ed., 1999, 38, 3138–3154 CrossRef; (b) T. Nakano and Y. Okamoto, Chem. Rev., 2001, 101, 4013–4038 CrossRef CAS; (c) J. J. L. M. Cornelissen, A. E. Rowan, R. J. M. Nolte and N. A. J. M. Sommerdijk, Chem. Rev., 2001, 101, 4039–4070 CrossRef CAS; (d) M. Fujiki, J. R. Koe, K. Terao, T. Sato, A. Teramoto and J. Watanabe, Polym. J., 2003, 35, 297–344 CrossRef CAS; (e) J. W. Y. Lam and B. Z. Tang, Acc. Chem. Res., 2005, 38, 745–754 CrossRef CAS; (f) K. Maeda and E. Yashima, Top. Curr. Chem., 2006, 265, 47–88 CAS; (g) T. Aoki, T. Kaneko and M. Teraguchi, Polymer, 2006, 47, 4867–4892 CrossRef CAS; (h) T. Masuda, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 165–180 CrossRef CAS; (i) E. Yashima and K. Maeda, Macromolecules, 2008, 41, 3–12 CrossRef CAS; (j) E. Yashima, K. Maeda and Y. Furusho, Acc. Chem. Res., 2008, 41, 1166–1180 CrossRef CAS; (k) K. Akagi, Chem. Rev., 2009, 109, 5354–5401 CrossRef CAS; (l) J. Liu, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2009, 109, 5799–5867 CrossRef CAS; (m) E. Yashima, K. Maeda, H. Iida, Y. Furusho and K. Nagai, Chem. Rev., 2009, 109, 6102–6211 CrossRef CAS.
- M. M. Green, N. C. Peterson, T. Sato, A. Teramoto, R. Cook and S. Lifson, Science, 1995, 268, 1860–1866 CrossRef CAS.
- (a) E. Yashima, S. Huang, T. Matsushima and Y. Okamoto, Macromolecules, 1995, 28, 4184–4193 CrossRef CAS; (b) K. Morino, K. Maeda, Y. Okamoto, E. Yashima and T. Sato, Chem.–Eur. J., 2002, 8, 5112–5120 CrossRef CAS; (c) B. S. Li, K. K. L. Cheuk, L. Ling, J. Chen, X. Xiao, C. Bai and B. Z. Tang, Macromolecules, 2003, 36, 77–85 CrossRef CAS; (d) J. Tabei, M. Shiotsuki, T. Sato, F. Sanda and T. Masuda, Chem.–Eur. J., 2005, 11, 3591–3598 CrossRef CAS; (e) T. Kaneko, Y. Umeda, T. Yamamoto, M. Teraguchi and T. Aoki, Macromolecules, 2005, 38, 9420–9426 CrossRef CAS; (f) V. Percec, J. G. Rudick, M. Peterca, M. Wagner, M. Obata, C. M. Mitchell, W.-D. Cho, V. S. K. Balagurusamy and P. A. Heiney, J. Am. Chem. Soc., 2005, 127, 15257–15264 CrossRef CAS; (g) S.-i. Sakurai, K. Okoshi, J. Kumaki and E. Yashima, Angew. Chem., Int. Ed., 2006, 45, 1245–1248 CrossRef CAS; (h) S. Ohsawa, K. Maeda and E. Yashima, Macromolecules, 2007, 40, 9244–9251 CrossRef CAS.
- For examples of chiroptical switch of optically active polymers, see: (a) M. Porsch, G. Sigl-Seifert and J. Daub, Adv. Mater., 1997, 9, 635–639 CAS; (b) L. A. P. Kane-Maguire, I. D. Norris and G. G. Wallace, Synth. Met., 1999, 101, 817–818 CrossRef CAS; (c) F. X. Redl, M. Lutz and J. Daub, Chem.–Eur. J., 2001, 7, 5350–5358 CrossRef CAS; (d) H. Goto and E. Yashima, J. Am. Chem. Soc., 2002, 124, 7943–7949 CrossRef CAS; (e) N. Hida, F. Takei, K. Onitsuka, K. Shiga, S. Asaoka, T. Iyoda and S. Takahashi, Angew. Chem., Int. Ed., 2003, 42, 4349–4352 CrossRef CAS; (f) G.-L. Yuan and N. Kuramoto, Macromolecules, 2003, 36, 7939–7945 CrossRef CAS; (g) E. Gomar-Nadal, J. Veciana, C. Rovira and D. B. Amabilino, Adv. Mater., 2005, 17, 2095–2098 CrossRef CAS; (h) H. Goto and K. Akagi, Chem. Mater., 2006, 18, 255–262 CrossRef CAS; (i) J. Deng, C. Zhou and N. Song, Macromolecules, 2009, 42, 6865–6872 CrossRef CAS.
- For examples of chiroptical switch of optically active small molecules, see: (a) L. Zelikovich, J. Libman and A. Shanzer, Nature, 1995, 374, 790–792 CrossRef CAS; (b) S. Zahn and J. W. Canary, Angew. Chem., Int. Ed., 1998, 37, 305–307 CrossRef CAS; (c) C. Westermeier, H.-C. Gallmeier, M. Komma and J. Daub, Chem. Commun., 1999, 2427–2428 RSC; (d) G. Beer, C. Niederalt, S. Grimme and J. Daub, Angew. Chem., Int. Ed., 2000, 39, 3252–3255 CrossRef CAS; (e) S. Zahn and J. W. Canary, Science, 2000, 288, 1404–1406 CrossRef CAS; (f) K. Tashiro, K. Konishi and T. Aida, J. Am. Chem. Soc., 2000, 122, 7921–7926 CrossRef CAS; (g) J.-I. Nishida, T. Suzuki, M. Ohkita and T. Tsuji, Angew. Chem., Int. Ed., 2001, 40, 3251–3254 CrossRef CAS; (h) T. Mori and Y. Inoue, J. Phys. Chem. A, 2005, 109, 2728–2740 CrossRef CAS; (i) C. Gautier and T. Bürgi, Chem. Commun., 2005, 5393–5395 RSC; (j) A. E. Holmes, D. Das and J. W. Canary, J. Am. Chem. Soc., 2007, 129, 1506–1507 CrossRef CAS; (k) J. Zheng, W. Qiao, X. Wan, J. P. Gao and Z. Y. Wang, Chem. Mater., 2008, 20, 6163–6168 CrossRef CAS; (l) D. Li, Z. Y. Wang and D. Ma, Chem. Commun., 2009, 1529–1531 RSC.
- For reviews, see: (a) B. L. Feringa, R. A. van Delden, N. Koumura and E. M. Geertsema, Chem. Rev., 2000, 100, 1789–1816 CrossRef CAS; (b) J. W. Canary, Chem. Soc. Rev., 2009, 38, 747–756 RSC.
- H. Takahashi, M. Isobe and T. Goto, Tetrahedron, 1991, 47, 6215–6222 CrossRef CAS.
- M. Sun, T. A. Moore and P.-S. Song, J. Am. Chem. Soc., 1972, 94, 1730–1740 CrossRef CAS.
- (a) H. Shirakawa, T. Ito and S. Ikeda, Polym. J., 1972, 4, 460–462 CrossRef; (b) M. Tabata, Y. Tanaka, Y. Sadahiro, T. Sone, K. Yokota and I. Miura, Macromolecules, 1997, 30, 5200–5204 CrossRef CAS; (c) M. Tabata, T. Sone and Y. Sadahiro, Macromol. Chem. Phys., 1999, 200, 265–282 CrossRef CAS.
- (a) S.-i. Sakurai, A. Ohira, Y. Suzuki, R. Fujito, T. Nishimura, M. Kunitake and E. Yashima, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4621–4640 CrossRef CAS; (b) H. Lin, K. Morino and E. Yashima, Chirality, 2008, 20, 386–392 CrossRef CAS.
- (a) H. Harders, S. Foerster, W. Voelter and A. Bacher, Biochemistry, 1974, 13, 3360–3364 CrossRef CAS; (b) X. Liu, Y. Zhu, G. Cheng and S. Dong, Electroanalysis, 2001, 13, 1071–1075 CrossRef CAS.
- The absorption intensity in the flavin chromophore region of 3 slightly decreased (8%) with an increase in the concentration of 3 (0.01–0.5 mg mL−1), whereas that of 8 hardly changed (Fig. S4†). The absorption intensity change observed for 3 is not significant when compared with that for poly-2 (Fig. 1B), indicating that the π–π stacking between the neighboring conjugated flavin pendant units of poly-2 plays a key role in transferring the preferred-handed helicity to the polymer backbone.
- The thermal stabilities of the polymers were also investigated by measuring the CD before and after annealing at 75 °C for 1 h (Fig. S5†). Poly-2 showed a slight decrease in the CD intensity of ca. 30%, probably due to the cis-to-trans isomerization of the main chain at high temperature. (a) X. Kong, J. W. Y. Lam and B. Z. Tang, Macromolecules, 1999, 32, 1722–1730 CrossRef CAS; (b) S. M. A. Karim, R. Nomura and T. Masuda, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3130–3136 CrossRef CAS; (c) V. Percec, J. G. Rudick, P. Nombel and W. Buchowicz, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3212–3220 CrossRef CAS; (d) K. Morino, T. Asari, K. Maeda and E. Yashima, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4711–4722 CrossRef CAS.
- A. Niemz, J. Imbriglio and V. M. Rotello, J. Am. Chem. Soc., 1997, 119, 887–892 CrossRef CAS.
- (a) Y.-J. Zheng and R. L. Ornstein, J. Am. Chem. Soc., 1996, 118, 9402–9408 CrossRef CAS; (b) J. Rodriguez-Otero, E. Martinez-Nunez, A. Pena-Gallego and S. A. Vazquez, J. Org. Chem., 2002, 67, 6347–6352 CrossRef CAS; (c) Y.-K. Choe, S. Nagase and K. Nishimoto, J. Comput. Chem., 2007, 28, 727–739 CrossRef CAS.
- J. B. Carroll, B. J. Jordan, H. Xu, B. Erdogan, L. Lee, L. Cheng, C. Tiernan, G. Cooke and V. M. Rotello, Org. Lett., 2005, 7, 2551–2554 CrossRef CAS.
- No appreciable changes were observed in the cyclic voltammograms for poly-1 and poly-2 after three redox cycles, revealing that the redox processes were fully reversible.
- S. Ghisla, V. Massey, J. M. Lhoste and S. G. Mayhew, Biochemistry, 1974, 13, 589–597 CrossRef CAS.
- C. Kemal, T. W. Chan and T. C. Bruice, J. Am. Chem. Soc., 1977, 99, 7272–7286 CrossRef CAS.
- Because of poor solubility of the resultant poly-2red in DMF–water, 5 equiv. of aqueous Na2S2O4 was added to a DMF solution of poly-2 (DMF–water (99.9/0.1 v/v)). This caused incomplete reduction of the flavin units of poly-2, as evidenced by a relatively strong absorption peak at around 440 nm (k in Fig. 5B).
- (a) M. Kainosho and Y. Kyogoku, Biochemistry, 1972, 11, 741–752 CrossRef CAS; (b) S. Shinkai, A. Harada, Y.-i. Ishikawa, O. Manabe and F. Yoneda, Chem. Lett., 1981, 10, 479–482 CrossRef; (c) M. F. Zipplies and H. A. Staab, Tetrahedron Lett., 1984, 25, 1035–1038 CrossRef CAS; (d) Y. Yano, E. Ohya and Y. Kuwabara, Chem. Lett., 1984, 1009–1012 CrossRef CAS; (e) M. Ebitani, Y. In, T. Ishida, K. Sakaguchi, J. L. Flippen-Anderson and I. L. Karle, Acta Crystallogr., Sect. B: Struct. Sci., 1993, 49, 136–144 CrossRef; (f) M. Insinska-Rak, E. Sikorska, J. L. Bourdelande, I. V. Khmelinskii, W. Prukala, K. Dobek, J. Karolczak, I. F. Machado, L. F. V. Ferreira, E. Dulewicz, A. Komasa, D. R. Worrall, M. Kubicki and M. Sikorski, J. Photochem. Photobiol., A, 2007, 186, 14–23 CrossRef CAS.
- K. Nagai, K. Sakajiri, K. Maeda, K. Okoshi, T. Sato and E. Yashima, Macromolecules, 2006, 39, 5371–5380 CrossRef CAS.
- The absolute configuration at the C(6′) position of the monomer 2 was assigned to be (S) based on ROESY spectra of 2 (Fig. S8 and S9†). See: (a) A. Mees, C. Behrens, A. Schwögler, M. Ober and T. Carell, Eur. J. Org. Chem., 2003, 2670–2677 CrossRef CAS; (b) A. Schwogler and T. Carell, Org. Lett., 2000, 2, 1415–1418 CrossRef CAS.
- (a) F. G. Gelalcha, Chem. Rev., 2007, 107, 3338–3361 CrossRef CAS; (b) Y. Imada and T. Naota, Chem. Rec., 2007, 7, 354–361 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, molecular modeling and calculations for poly-2, and characterization data for 2, 6, 8, poly-1 and poly-2. See DOI: 10.1039/c0py00044b |
| This journal is © The Royal Society of Chemistry 2010 |