DOI:
10.1039/C0PY00042F
(Paper)
Polym. Chem., 2010,
1, 916-921
Synthesis and characterization of functionalized silane-based copolymers for thermally robust polymer–silica hybrids
Received
9th February 2010
, Accepted 6th April 2010
First published on
4th May 2010
Abstract
We report the preparation and characterization of a group of thermally stable silane-based copolymers prepared by radical polymerization of various phenylmaleimides with vinyl- or allyltrialkoxysilanes. These polymers exhibit thermal stability up to 400 °C or better, as determined by thermal analysis. Polymer preparation and stability (resistance to hydrolysis) were confirmed by FTIR, 1H NMR, and 13C NMR spectroscopy. Polymer molecular weights (5000 to 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) and polydispersity indices (1.87–3.06) were determined by gel permeation chromatography (GPC). While controlling the functional groups on the phenylmaleimide affords the tuning of the chemical properties of the resulting polymers, the choice of the alkoxysilane leads to control over the rate and extent of reactivity of the polymers. These materials are being employed in the design of thermally robust polymer–silica materials for use in high-temperature chromatography.
000 g mol−1) and polydispersity indices (1.87–3.06) were determined by gel permeation chromatography (GPC). While controlling the functional groups on the phenylmaleimide affords the tuning of the chemical properties of the resulting polymers, the choice of the alkoxysilane leads to control over the rate and extent of reactivity of the polymers. These materials are being employed in the design of thermally robust polymer–silica materials for use in high-temperature chromatography.
Introduction
Polymer-based hybrid materials have attracted much attention due to their ease of preparation and ability to be tuned towards specific applications through the incorporation of a variety of functionalities in the polymer backbone. These properties facilitate their use as specific and selective sensors where biological moieties such as enzymes, antibodies, and proteins can be entrapped within the polymer matrix. Polymer materials1–3 have also been employed for chemical separations (e.g. chromatography) where alkyl-modified metal oxide stationary phases have dominated for years. Silica based materials have also been extensively used as they can be modified chemically through facile routes. They can also be designed to control the properties including pore size, surface area, and particle diameter.4 While alkyl-modified silica phases have been employed in the separation of a broad range of compounds,5,6 there are some disadvantages associated with these materials, including limited applications at basic pH. Basic compounds or chelating compounds are retained more on silica-based materials at higher pH,7 which is also an issue.
Organic polymer-based materials are another class of materials8,9 that have been used as stationary phases in reverse phase liquid chromatography (RP-LC) separations. These materials have advantages over silica-based materials because of the diversity of chemical modifications.10 Chemical stability11,12 over a wide pH range as well as elimination of secondary effects like surface silanol activity is possible by utilizing organic polymeric material. However, due to their physical nature these materials pose a problem by forming mesopores. Swelling and shrinkage can also occur in these polymeric materials, depending upon the solvents in the mobile phase, which effectively limits the use of many solvents when polymeric stationary phases are used.
Materials based on the sol–gel technology13 provide an alternative to the types of stationary phases mentioned above. Here organic–inorganic hybrid materials are employed as a stationary phase.14,15 In this hybrid material, silica and organosilanes provide material properties which are intermediate between pure organic and inorganic materials. Moreover, materials based on sol–gel technology provide good mechanical strength to the column,16 facile methods of preparation,17 and the resulting materials being thermally and chemically18–20 robust. Many functional moieties can also be incorporated into the sol–gel based materials21,22 which allow the use of an array of solvents as a mobile phase and works over wide range of pH. Since analyte selectivity is a prime requirement for separations, such materials should have the ability to achieve near complete resolution of analyte within shortest possible time. They should also provide predictable response over a wide range of operating conditions.
Considering the prerequisites of a good material for separations, we report the preparation and characterization of a group of stable phenylmaleimide-co-alkoxysilane polymers that have been designed by incorporating silane side-groups directly into the polymer backbone. These polymers are capable of participating directly in the sol–gel formation process in the presence of a catalyst (acid or base). The incorporation of the silane groups into the polymer backbone is expected to minimize or eliminate the occurrence of phase segregation, a common problem with polymer blending. Phase segregation is minimized as the silane side groups are an integral part of the polymer and when these alkoxysilane side groups are hydrolyzed, they can bond covalently within the sol–gel matrix, thereby reducing heterogeneity. These polymers are stable in ambient conditions for extended periods of time in solution with no evidence of cross-linking of the silane groups except in the presence of a catalyst (aqueous base, for example). This is particularly important as this evades special protocols to handle or store these polymers. The polymers are also thermally stable up to 400 °C which might find applications in high temperature chromatography, particularly subcritical water chromatography.
Results and discussion
Through the facile synthesis of the maleimides, a variety of functional groups can be added to modify the chemical properties of the stationary phase. This also allows for the stationary phase to be tuned towards a particular separation application, by changing the functionalized aniline group. When preparing the copolymer, maleimides tend to form alternating copolymers which ensures a regular structure to the copolymers as well as consistent properties. Having consistent properties in a copolymer provides the stationary phase with uniform selectivity leading to a more reproducible separation.
Comonomers
Fig. 1 represents a typical IR spectrum of one of the monomers, N-phenylmaleimide (NPM).31 First are the aromatic methylene stretches associated with the N-phenyl ring between 3000 and 3100 cm−1. There are two carbonyl stretches, the first centered at 1775 cm−1and the second at 1710 cm−1. This spectral signature is characteristic of the presence of the maleimide groups and is evident in all of the polymers we report here. The band at 1775 cm−1 is associated with the symmetric (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretch and the band at 1710 cm−1 is associated with the asymmetric (CO) stretch in the succinimide moiety. We also observe the characteristic (CC) stretching modes of the phenyl ring at 1597, 1502, and 1457 cm−1 and out-of-plane ring bending at 694 cm−1. The band centered at 1381cm−1 has been assigned to the (C–N) bond of the tertiary aromatic amine. This band is centered at 1381 cm−1 rather than the expected 1360 cm−1 due to resonance stabilization of the amine group with the phenyl ring as well as the presence of the succinimide carbonyl functionalities. The 1189 cm−1 band has been assigned to the maleimide (C–N–C) stretch. For the other monomer, vinyl/allyltrimethoxy- or triethoxysilanes were mainly used, although copolymers have been made with different alkoxysilanes as well. The choice of alkoxysilane groups (allyl or vinyl) can affect properties such as chain length which further influences the pore size within the copolymer. The reactivity of the copolymers depends on the choice of alkoxysilane (methoxy or ethoxy). These alkoxysilane groups (SiOCH3 or SiOC2H5) are the most important part of the copolymer, allowing the copolymer to covalently link to a silica surface such as a silica particle or capillary wall. Silica particles and capillary walls contain silanol groups (Si–OH) which can react with the copolymer when its silane groups are hydrolyzed to silanols. Incorporation of these properties provides a copolymer than can be tuned and attached to a silica surface in order to prepare different forms of stationary phases.
O) stretch and the band at 1710 cm−1 is associated with the asymmetric (CO) stretch in the succinimide moiety. We also observe the characteristic (CC) stretching modes of the phenyl ring at 1597, 1502, and 1457 cm−1 and out-of-plane ring bending at 694 cm−1. The band centered at 1381cm−1 has been assigned to the (C–N) bond of the tertiary aromatic amine. This band is centered at 1381 cm−1 rather than the expected 1360 cm−1 due to resonance stabilization of the amine group with the phenyl ring as well as the presence of the succinimide carbonyl functionalities. The 1189 cm−1 band has been assigned to the maleimide (C–N–C) stretch. For the other monomer, vinyl/allyltrimethoxy- or triethoxysilanes were mainly used, although copolymers have been made with different alkoxysilanes as well. The choice of alkoxysilane groups (allyl or vinyl) can affect properties such as chain length which further influences the pore size within the copolymer. The reactivity of the copolymers depends on the choice of alkoxysilane (methoxy or ethoxy). These alkoxysilane groups (SiOCH3 or SiOC2H5) are the most important part of the copolymer, allowing the copolymer to covalently link to a silica surface such as a silica particle or capillary wall. Silica particles and capillary walls contain silanol groups (Si–OH) which can react with the copolymer when its silane groups are hydrolyzed to silanols. Incorporation of these properties provides a copolymer than can be tuned and attached to a silica surface in order to prepare different forms of stationary phases.
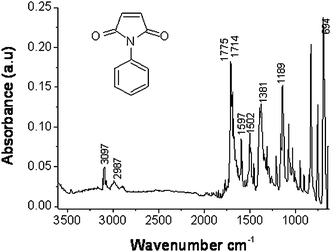 |
| | Fig. 1 FTIR spectrum of N-phenylmaleimide (NPM) with the structure in the inset. | |
Structural characterization of copolymers
Fig. 2 represents the IR spectrum of the copolymers with allyl side chain (allyltrimethoxysilane, NPM–ATMS and allyltriethoxysilane, NPM–ATES). The copolymers were not hydrolyzed under preparation conditions as evidenced by the absence of (OH) stretch in the 3200–3500 cm−1 region or (SiO) stretch from Si–OH at 920 cm−1. This confirms that the copolymer is still terminated in the respective alkoxysilane that are used in their preparation. If hydrolysis had occurred, the copolymer would begin reacting with itself instead of the silica surface. Peaks associated with the carbonyl groups of the phenylmaleimide and the (SiO) from Si–OR23–25 of the alkoxysilane are apparent at ∼1710 cm−1 and 1070 cm−1 respectively. Spectra between these copolymers are very similar as stated earlier. However, there is a strong band at 957 cm−1 that is present only in the ethoxysilane copolymers. This appears to be characteristic of (SiOC2H5) since the methoxysilane copolymers do not have any peaks in that region. The absence of an (OH) stretch around 3500 cm−1 is a strong evidence of the fact that the stretch at 957 cm−1 is not a result of hydrolysis of (SiOR) groups which appears at 920 cm−1. To further show the lack of hydrolysis of the copolymers at ambient conditions, Fig. 3 shows a comparison of the FTIR spectra of the copolymer (NPM–ATES) before and after being treated with a basic catalyst for one hour. Ammonium hydroxide was added as a catalyst to induce hydrolysis of the silane groups within the polymer backbone. A strong (OH) stretch 3464 cm−1 and (SiOH) stretch at about 910 cm−1 (Fig. 3) clearly indicates the difference between the stable copolymer and the hydrolyzed one. The spectra clearly indicate that there is a decrease in the stretch at 1072 cm−1 after hydrolysis due to the loss of −OR groups. The formation of siloxanes Si–O–Si, occurring due to aggregation of the polymer appears as a broad band in the region 1020–1090 cm−1. Further, NMR (both 1H and 13C) spectral analyses were performed to gain better understanding of the chemical structure of the monomer, vinyltrimethoxysilane (VTMS) and the copolymer, NPM–VTMS. 1H and 13C NMR analyses were performed using 300 MHz NMR spectrometer in CDCl3 with 1 v/v% of tetramethylsilane (TMS). Fig. 4a represents the 1H NMR of the alkoxysilane monomer, VTMS (where resonance from the protons on the vinyl (5.7–6.3 ppm) and methoxysilane, Si–OCH3, (3.5 ppm) are observed along with some impurities between 2 and 3 ppm). Fig. 4b is the 13C NMR spectrum of the monomer VTMS with its characteristic peaks from the vinyl (128 and 137 ppm) and methoxysilane (∼51 ppm) carbons. 1H NMR of the copolymer, NPM–VTMS, is presented in Fig. 4c.
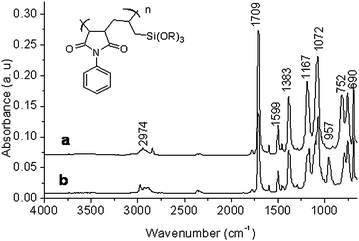 |
| | Fig. 2 FTIR spectra of the copolymers: NPM–ATMS (a), NPM–ATES (b), and the inset of the general structure where R = CH3 or C2H5. | |
A broad peak at 7.1–7.6 ppm26 is representative of aromatic protons from the phenyl ring. Succinimide protons, which are between carbonyl groups on the maleimide ring, were detected at 3.5 ppm,27 where the chemical shift can vary with cis (3.2 ppm) or trans (2.7 ppm),26 proving the disappearance of the alkene through the polymerization process. At the attachment of the alkoxysilane to the maleimide ring there are methylene protons (2.3 ppm)26 along with a methine proton (1.7 ppm)28 showing loss of the vinyl moiety. Terminal –OCH3 groups are intact through resonance at 3.5 ppm28 due to the methyl groups which can also be seen in the proton NMR of VTMS. The silane shifts are also visible in the 13C NMR spectrum of NPM–VTMS (Fig. 4d) at 51.13 ppm.29 Representative peaks from the phenyl ring are presented at 126–129 ppm.30 The carbonyl and succinimide carbons, on the maleimide ring were detected at 176.6226,27 and 39.34 ppm26,30 respectively. Successful preparation of the copolymer is also confirmed through the loss of the vinyl moiety and peaks from the –CH2 and –CH (22–23.5 ppm)26 between the maleimide and alkoxysilane. With the 1H NMR, 13C and the IR data, we have confirmed that the copolymers are stable under normal laboratory conditions with the alkoxysilane side groups intact.
Molecular weight and polydispersity
Table 1 shows the molecular weights and the polydispersity of the copolymers measured by gel permeation chromatography (GPC). On comparing the number average molecular weight (Mn), weight average molecular weight (Mw) and polydispersity of different copolymers with 5 and 10% radical initiator, it has been observed that using a higher initiator concentration leads to an increase in the polydispersity of the copolymers. This is true as a higher initiator concentration will lead to more number of reactive species, thereby leading to higher polydispersity. A trend was also observed in the allyl vs. vinyl side chains and methoxy vs. ethoxy side chains. The allyl (2.11 and 2.46) side chain yields copolymers with a narrower distribution when compared to the vinyl copolymers (2.92 and 3.06). In addition, the allyl copolymers (5215 and 21043 g mol−1) had lower molecular weights than the vinyl copolymers (17747 and 25982 g mol−1). The vinyl group has one less carbon which makes a more reactive radical than the allyl group, leading to a larger distribution in molecular weights. However, analyzing the reaction mechanism requires more comprehensive experiments and is beyond the scope of this paper.
Table 1 Molecular weight data of various NPM copolymers at 5 and 10 mol% AIBN
| Copolymers (mol%) |
M
n/g mol−1 |
M
w/g mol−1 |
PDIa |
|
Polydispersity index: Mw/Mn.
|
| NPM–ATMS (5%) |
2684 |
5043 |
1.87 |
| NPM–ATMS (10%) |
2470 |
5215 |
2.11 |
| NPM–ATES (5%) |
2441 |
5567 |
2.28 |
| NPM–ATES (10%) |
8543 |
21043 |
2.46 |
| NPM–VTMS (5%) |
10723 |
27376 |
2.45 |
| NPM–VTMS (10%) |
6064 |
17747 |
2.92 |
| NPM–VTES (5%) |
5756 |
16607 |
2.88 |
| NPM–VTES (10%) |
8488 |
25982 |
3.06 |
Thermal analysis
In addition to being stable under ambient conditions, these copolymers are also thermally stable. Thermal analyses (TGA, DTA, and DSC) were performed on four copolymers, NPM–VTMS, NPM–VTES, NPM–ATMS, and NPM–ATES. All these copolymers were stable up to 400 °C, as indicated in Fig. 5. A small mass change was observed between 50–100 °C, probably due to the loss of adsorbed water. Initial degradation of the copolymers began at different rates where the vinyl copolymers showed 10% weight loss (T10% = 340–365 °C) at lower temperatures than allyl copolymers (T10% = 427–430 °C). Major degradation began at 400 °C, with the copolymers losing 50% of their mass in the 450–460 °C range. The maximum mass change was observed between 470 °C and 490 °C for all four copolymers. DSC analysis exhibited glass transition temperatures (Tg) for the vinyl and allyl copolymers at 130 and 190 °C respectively (Fig. 5a).
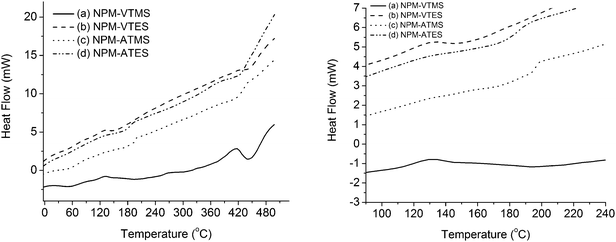 |
| | Fig. 5 (a) DSC thermograms of copolymers: NPM–VTMS (a), NPM–VTES (b), NPM–ATMS (c), and NPM–ATES (d). (b) A blow up of the region of glass transition temperatures of the polymers. | |
For the ethoxysilane copolymers, there was no other significant change in the thermogram until 420 °C, corresponding to the onset of degradation observed in the TGA and DTA curves. The methoxysilane copolymers show a small, broad exothermic peak at 60 °C which is also seen in the DTA curves with the loss of bound solvent. NPM–VTMS has a well defined exothermic peak at 440 °C which is evident in the DTA curves where T50% degradation occurs. Fig. 5b shows a blow up of the regions to clearly show the glass transition temperature.
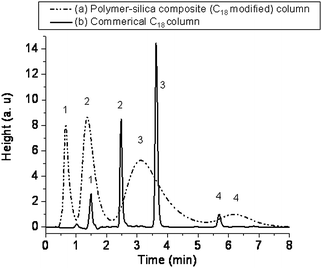
Chromatographic evaluation
Since these silane-based copolymers are thermally stable up to 400 °C a potential application for them could be high temperature and/or highly aqueous separations. Hence, these polymers were also tested as stationery phases in high performance liquid chromatography (HPLC). Chromatograms were obtained (Fig. 6) using NPM–VTMS coated on 10 µm silica particles. A commercial C18 column was used to separate substituted benzenes with a mobile phase containing 80% water and 20% acetonitrile. In the current work, we have used the designed copolymers to perform more effectively under aqueous conditions.
The NPM–VTMS chromatogram showed more symmetric peaks (slightly broader). Preliminary evaluation of these two chromatograms showed that the polymer–silica material was more efficient than C18 under the test conditions. In addition, use of 100% water as the eluent and the ability to narrow the peaks by increasing column temperature (subcritical water ranges) could greatly increase the efficiency of the stationary phase and is a topic of interest for future work.
Experimental
Materials
Ammonium hydroxide, acetonitrile (HPLC grade), and chloroform (HPLC grade) were purchased from Fisher Scientific (Fair Lawn, NJ). Ethanol (200 proof) was purchased from Pharmo Products (Brookfield, CT). Maleic anhydride (≫98%, Fluka, Seelze, Germany), 4-bromoaniline (98%, Alfa Aesar, Ward Hill, MA), aniline (Matheson, Coleman & Bell, Rutherford, NJ), 4-fluoroaniline, 4-chloroaniline (≫97%, Fluka), sodium acetate (99+%, ACROS Organics, Geel, Belgium), and acetic anhydride (99%, ACROS Organics) were used in the synthesis of the maleimides. Alkoxysilanes (vinyltrimethoxysilane, vinyltriethoxysilane, allyltrimethoxysilane, and allyltriethoxysilane, 98%, ACROS Organics) and 2,2′-azobisisobutyronitrile (AIBN, 98%, Aldrich) were used in the synthesis of the copolymers. Water was purified by a Millipore Synergy 185 Ultrapure Water System. Chloroform-d with 1 v/v% of tetramethylsilane (TMS), ACROS Organics, was used as the solvent for NMR analysis. For the separations, LiChrosorb Si60 10 µm silica particles (Merck) and nitrobenzene (99%, ACROS Organics) were purchased while benzoic acid, and benzamide were supplied by chemistry department. A Dionex C18 column (150 mm × 4.6 mm, pore size 300 Å) was compared to the polymer–silica stationary phases.
Monomer preparation
The vinyl and allylalkoxysilanes were acquired commercially. The various maleimides were prepared in-house through a two step process outlined in Scheme 1. Typically a 1 : 1 mole ratio of maleic anhydride (1) and functionalized aniline (2) were dissolved in 40 mL chloroform and allowed to stir at ambient conditions forming the corresponding maleamic acid (3). This was collected and refluxed for 24 h at 70 °C with a 1 : 1 mole ratio of sodium acetate in 30 mL acetic anhydride to close the ring and form the maleimide (4). Spectroscopic analysis confirmed the completion of reaction and formation of the maleimide.
The copolymer was prepared by radical polymerization of a phenylmaleimide (4) with either a vinyl or allylalkoxysilane (5) according to Scheme 2. The monomers were combined in a 1 : 1 mole ratio, and dissolved in 20 mL chloroform to which 2,2′-azobisisobutyronitrile (AIBN), the radical initiator was added and refluxed at 50–60 °C for 24 h. The AIBN content was 1, 5, or 10 mol% in an attempt to control polymer molecular weight. The resulting polymer (6) was recrystallized as a powdery precipitate from hexanes.
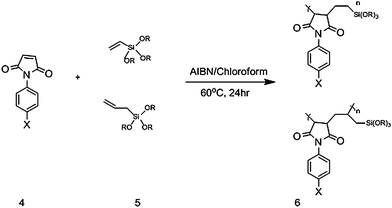 |
| | Scheme 2 General phenylmaleimide-co-alkoxysilane (6) synthesis where various phenylmaleimides (4) and alkoxysilanes (5) were utilized. | |
Instrumentation and measurement
FTIR-ATR analysis was performed on a Thermo Nicolet Nexus 870 FT-IR E.S.P. using OMNIC™ computer software from the Thermo Electron Corporation. Monomer and copolymer products were verified by IR spectra. 1H and 13C NMR analysis was performed using Bruker 300 MHz NMR spectrometer in deuterated chloroform (CDCl3) with 1 v/v% of tetramethylsilane (TMS). Gel permeation chromatography (Agilent 1100) was used to determine polymer molecular weight and polydispersity. Tetrahydrofuran (THF) was used as a solvent, with a flow rate of 0.5 mL min−1 using a refractive index detector. Thermal analysis of the copolymers was performed by using a Q 100 V8 build 261 DSC instrument. A heating rate of 10 °C min−1 was used to analyze the copolymers in the temperature range of 30 to 450 °C. The HPLC system contained a Hitachi L-7455 LaChrom Diode Array detector, Hitachi L-7200 Autosampler, Hitachi D-7000 Interface and D-7000 Multi HSM computer software.
Conclusions
N-Phenylmaleimide monomers and silane-based copolymers have been successfully synthesized and characterized. The phenylmaleimides were synthesized by reacting functionalized aniline and maleic anhydride. IR spectra confirmed the complete conversion of the amine group from aniline derivatives to the maleimide. IR also confirmed the stability of these copolymers under ambient conditions. The copolymers have a measured polydispersity range of 1.87–3.06. Thermal analysis established that these copolymers are thermally stable up to 400 °C. The copolymers prepared would be an improved polymer stationary phase over polystyrene which is stable only up to 150 °C. In conclusion, the copolymers reported in this work have proven to be stable throughout synthesis, ambient and high temperature conditions and have been successfully developed into polymer–silica stationary phases for application in liquid chromatography.
Acknowledgements
The authors would like to thank The Department of Chemistry at The University of Rhode Island for making several analytical equipments used in this work available for use. AMV would also like to thank Prof. Rod Ruoff at UT Austin and K. J. Ganesh for valuable discussions.
References
- K. Kobayashi, S. Kitagawa and H. Ohtani, J. Chromatogr., A, 2006, 1110, 95 CrossRef CAS.
- Y. Zhang, Z. Guo, J. Ye, Q. Xu, X. Liang and A. Lei, J. Chromatogr., A, 2008, 1191, 188 CrossRef CAS.
- M. Xu, D. S. Peterson, T. Rohr, F. Svec and J. M. J. Fréchet, Anal. Chem., 2003, 75, 1011 CrossRef CAS.
- H. Guan-Sajonz, G. Guiochon, E. Davis, K. Gulakowski and D. W. Smith, J. Chromatogr., A, 1997, 773, 33 CrossRef CAS.
- D. J. Miller and S. B. Hawthorne, Anal. Chem., 1997, 69, 623 CrossRef CAS.
- R. M. Smith and R. J. Burgess, J. Chromatogr., A, 1997, 785, 49 CrossRef CAS.
- D. V. McCalley, J. Chromatogr., A, 2000, 902, 311 CrossRef CAS.
- K. Hosoya, M. Teramachi, N. Tanaka, A. Kobayashi, T. Kanda and Y. Ohtsu, Anal. Chem., 2001, 73, 5852 CrossRef CAS.
- K. Hosoya, Y. Watabe, T. Kubo, N. Hoshino, N. Tanaka, T. Sano and K. Kaya, J. Chromatogr., 2004, 1030, 237 CrossRef CAS.
- H. A. Claessens, TrAC, Trends Anal. Chem., 2001, 20, 563 CrossRef CAS.
- H. A. Claessens and M. A. van Straten, J. Chromatogr., A, 2004, 1060, 23 CrossRef CAS.
- M. Liu, Y. Liu, Z. Zeng and T. Peng, J. Chromatogr., A, 2006, 1108, 149 CrossRef CAS.
- M. R. Buchmeiser, J. Chromatogr., A, 2001, 918, 233 CrossRef CAS.
- W. Li, D. P. Fries and A. Malik, J. Chromatogr., A, 2004, 1044, 23 CrossRef CAS.
- Y. S. Li, B. Li, N. Y. Han and B. J. Xu, J. Chromatogr., A, 2003, 1021, 183 CrossRef CAS.
- H. Sertchook, H. Elimelech and D. Avnir, Chem. Mater., 2005, 17, 4711 CrossRef CAS.
- S. Constantin, R. Freitag, D. Solignac, A. Sayah and M. A. M. Gijs, Sens. Actuators, B, 2001, 78, 267 CrossRef.
- X. Li, Z. Zeng, S. Gao and H. Li, J. Chromatogr., A, 2004, 1023, 15 CrossRef CAS.
- K. D. Wyndham, J. E. O'Gara, T. H. Walter, K. H. Glose, N. L. Lawrence, B. A. Alden, G. S. Izzo, C. J. Hudalla and P. C. Iraneta, Anal. Chem., 2003, 75, 6781 CrossRef CAS.
- T.-Y. Kim, K. Alhooshani, A. Kabir, D. P. Fries and A. Malik, J. Chromatogr., A, 2004, 1047, 165 CAS.
- L. He and C.-S. Toh, Anal. Chim. Acta, 2006, 556, 1 CrossRef CAS.
- R. J. Hodgson, Y. Chen, Z. Zhang, D. Tleugabulova, H. Long, X. Zhao, M. Organ, M. A. Brook and J. D. Brennan, Anal. Chem., 2004, 76, 2780 CrossRef CAS.
- E. D. Lipp, Appl. Spectrosc., 1991, 45, 477 CrossRef CAS.
- P. Innocenzi, J. Non-Cryst. Solids, 2003, 316, 309 CrossRef CAS.
- I. Simon and H. O. McMahon, J. Chem. Phys., 1952, 20, 905 CrossRef CAS.
- P. Kohli, A. B. Scranton and G. J. Blanchard, Macromolecules, 1998, 31, 5681 CrossRef CAS.
- S. Yamamoto, F. Sanda and T. Endo, Macromolecules, 1999, 32, 5501 CrossRef CAS.
- M. Spencer, J. S. Parent and R. A. Whitney, Polymer, 2003, 44, 2015 CrossRef CAS.
- C.-S. Tan, C.-C. Juan and T.-W. Kuo, Polymer, 2003, 45, 1805.
- R.-H. Lin, W.-H. Lu and C.-W. Lin, Polymer, 2003, 45, 4423.
- J. S. Major and G. J. Blanchard, Chem. Mater., 2002, 14, 2567 CrossRef CAS.
Footnotes |
| † Present address: Department of Mechanical Engineering and The Texas Materials Institute, 1 University Station, C 2200, The University of Texas at Austin, Austin, TX, 78712. E-mail: avelamakanni@mail.utexas.edu |
| ‡ Present address: USDA-ARS, Center for Grain & Animal Health Research, 515 College Ave., Manhattan, KS 66502. E-mail: deidre.blackwell@ars.udsa.gov |
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.