Quantitative structure–reactivity modeling of copper-catalyzed atom transfer radical polymerization†
Fabio
di Lena
* and
Christina L. L.
Chai
Institute of Chemical and Engineering Sciences, Agency of Science, Technology and Research, 1 Pesek Road, Jurong Island, 627833, Singapore. E-mail: fabio_di_lena@ices.a-star.edu.sg
First published on 13th April 2010
Abstract
In this work, we present the first successful application of in silico modeling to the construction of quantitative and predictive relationships between the set of constants kact, kdeact and KATRP and the structures and properties of various ATRP catalysts and initiators. The results are consistent not only with the generally accepted ATRP mechanistic picture but also provide valuable insights into this complex polymerization reaction. The models, built using the genetic function approximation algorithm, highlight and quantify the pivotal roles played in the ATRP process by energetic and steric factors of both catalysts and initiators as well as by the reaction medium. Moreover, the models suggest the existence of long-range interactions in catalyst–initiator recognition and subsequent binding. We believe that the approach will prove to be a powerful tool for the discovery of improved catalysts for ATRP.
Introduction
In order to lower the cost and shorten the time to market of new compounds, automated high-throughput synthesis, screening and characterization techniques have become increasingly common in chemical laboratories.1–11 Notwithstanding the capability of these methodologies to carry out thousands of experiments per day, it has become apparent that a thorough exploration of the chemical space,12 which is believed to exceed 1060 compounds, is materially impossible. This has led to a greater use of computer-aided molecular design (CAMD) with the objective of finding a more reasonable number of molecules covering the chemical space as efficiently as possible.13–22 The first step in a typical CAMD approach usually consists of the creation, on the basis of literature data, chemical knowledge and intuition, of virtual libraries of compounds (reaction space) whose structural and chemical properties are translated into numerical terms by calculating, for each of them, hundreds of molecular descriptors. The descriptors are mathematically treated, for instance with principal component analysis (PCA),23–25 in order to select subsets of test items that cover the largest spread of molecular properties (diversity search). Once the actual experiments have been carried out and the reaction responses recorded, the reaction space is mathematically correlated with the response space by means of regression techniques. The resulting models are then used to design, in silico, the subsequent libraries of compounds to be tested in vitro. In general, when an initial hit is identified, new sets of compounds that sample smaller regions of the space around it are created and evaluated (similarity search). Several modeling/experiment cycles may be necessary before finding a compound with the specific combination of properties. While extensively employed in pharmaceutical17,19,20 and agrochemical16,18,21 research, virtual screening approaches have only started to be applied to the development of catalysts26–41 and materials42–47 recently.Atom transfer radical polymerization (ATRP)48–51 is one of the methods for controlled radical polymerization that have revolutionized the field of polymer chemistry in the last decade, the others being nitroxide mediated (NMP)52,53 and reversible addition–fragmentation chain transfer (RAFT)53,54 radical polymerizations. Macromolecules with precisely controlled architecture and functionality have been synthesized from a wide range of monomers under conditions that are much less stringent than those previously required for ionic living polymerizations.50–52,54 Mechanism wise, ATRP is a redox-based process in which a transition-metal complex acts as a mediator for the intermittent generation of propagating radicals from alkyl halides (Scheme 1).48,49 The polymerization control depends largely on the appropriate equilibrium between the activation (generation of radicals, kact) and the deactivation (formation of alkyl halides, kdeact) processes, which determines the concentration of radicals and thus the rates of polymerization and termination as well as polydispersities. In particular, KATRP = kact/kdeact should be sufficiently small in order to maintain a low concentration of radicals and to minimize termination reactions, whereas both kact and kdeact should be large enough (though kact ≪ kdeact) to provide good control over the polymerization while providing a reasonable polymerization rate. The values of KATRP, kact and kdeact depend on the catalyst, initiator and monomer structure, as well as on the type of solvent and the reaction conditions. Understanding how these parameters affect the three reaction constants is crucial for the rational development of more efficient ATRP catalysts.
 | ||
| Scheme 1 Atom transfer radical polymerization equilibrium. | ||
Recently, large datasets of KATRP, kact and kdeact, either experimentally determined55,56 or extrapolated by using the constant selectivity principle, were reported for a number of copper catalysts and alkyl halide initiators.57 Semi-quantitative correlations between the structures of these compounds and KATRP57 or kact57–59 were also described. In contrast, due to the difficulty of measuring the relative fast process, no such correlations are presently available for kdeact.
In this work, we present the first, successful application of in silico modeling to the construction of fully quantitative relationships between the set of constants kact, kdeact and KATRP and the structures and properties of ATRP catalysts and initiators.
Computational details
All calculations were performed with the software package Materials Studio® 4.460 on a Windows Server 2008 SP1 64-bit equipped with 16 Intel Xeon 2.6GHz CPUs and 20GB RAM.Geometry optimization
The geometry of catalysts and initiators was first pre-optimized with Forcite, an advanced classical molecular mechanics tool, using the Universal force field61 together with the SMART minimizer at the ultrafine quality level. The resulting conformers were taken as input structures in Dmol3,62,63 a software for DFT calculations, using the following settings. Functional: generalized gradient approximation (GGA)64,65 in combination with Becke exchange66,67 plus Lee–Yang–Parr correlation68 (BLYP); Basis set: double numerical plus d-function (DND); Basis file: 3.5; Core treatment: all electrons; Quality: fine; Integration accuracy: fine; SCF tolerance: fine; Max. SCF cycles: 50; Multipolar expansion: octapolar; DIIS: yes; DIIS size: 6; Orbital cutoff quality: fine; Orbital cutoff scheme: global; Solvation model: COSMO; Solvent: acetonitrile; Symmetry: yes; Energy convergence tolerance: 10−5 Ha; Max. force convergence tolerance: 2 × 10−3 Ha Å−1; Max. displacement convergence tolerance: 5 × 10−3 Å; Max. iterations: 50; Max. step size: 0.3 Å.Molecular descriptors
A large variety of molecular descriptors were calculated for both catalysts and initiators, including: quantum mechanical descriptors (total energy, HOMO, LUMO, HOMO–LUMO gap, binding energy, dielectric energy, solvation energy, surface area, cavity volume and dipoles, via Dmol3); molecular mechanical descriptors (bond and non-bond energies, van der Waals energy, electrostatic energy, valence energy (diagonal terms), angle energy, torsion energy and inversion energy, via Forcite); topological descriptors (Wiener index, Zagreb index, Balaban indices, mχ connectivity indices, mχv valence connectivity indices, mκ shape indices, α-modified mκ shape indices, molecular flexibility index, subgraph counts, edge adjacency and distance (magnitude), vertex adjacency and distance (magnitude and equality), bond, complementary and structural information content); electrotopological descriptors (E-state indices); spatial descriptors (molecular area, molecular volume, molecular density, principal moments of inertia, moments of inertia, radius of gyration, ellipsoidal volume, shadow areas, shadow area fractions, shadow lengths, shadow ratio, molecular shadow areas, molecular shadow area fractions, molecular shadow lengths, molecular shadow ratio, Connolly surface area, Connolly surface occupied volume, van der Waals surface area, solvent surface area, solvent-accessible surface area, van der Waals surface occupied volume, solvent surface occupied volume, and solvent-accessible occupied volume); thermodynamical descriptors (molecular refractivity, AlogP, and AlogP98); and constitutional descriptors (total molecular mass, atom count, element count, average N–Cu bond length, Cu–X bond length, C–X bond length, and count of various molecular fragments).69Principal components analysis (PCA)
PCA was carried out by standardizing the descriptor data by using the mean and scaling the results using unit standard deviation. Among all the resulting principal components, only those with an eigenvalue ≥ 1 were used subsequently.Hierarchical cluster analysis (HCA)
Hierarchical cluster analysis was performed by applying the Ward's clustering method70 along with Murtagh's reciprocal nearest neighbors (RNN) algorithm71 to the first n principal components for which the total variance explained was ≥0.9. The data were centered on the mean.Regression analysis
Quantitative structure–reactivity relationships (QSRR) were built using the genetic function approximation (GFA) algorithm.72 Unless otherwise specified, the literature values of kact and kdeact for copper complexes used in this study were those reported at 22 °C in MeCN with EtBriB as the initiator, whereas those of kact and KATRP for the initiators refer to those reported at 22 °C in MeCN with PMDETA/CuBr as the catalyst.57 Basis functions: linear polynomial and spline; Additional terms: none; Initial equation length: 5 terms; Maximum equation length: 9 terms; Population: up to 2000; Maximum generations: up to 5000; Number of top equations returned: 10; Mutation probability: 0.4; Total spline mutation probability: 0.1; Knot shift mutation probability: 0.5; Maximum knot shift fraction: 0.1; Random number seed: 9999; Minimum prediction fraction for term inclusion: 0.0001; Scoring function: Friedman's LOF;73 Scaled LOF smoothness parameter: 0.5.Results and discussion
Catalysts
Quantitative structure–reactivity relationship (QSRR) models were constructed utilizing 20 complexes that contain copper and multidentate nitrogen-based ligands (Chart 1) for which the values for the set of constants kact, kdeact and KATRP had been reported in the literature.57 In particular, the rate constants kact and kdeact were quantitatively correlated with what are believed to be the structures of the activators and the deactivators in solution, respectively. | ||
| Chart 1 Ligands for copper-catalyzed atom transfer radical polymerization used in this study. | ||
The complexes with CuIX and either bidentate (bpy, dNbpy, NPPMI, NOPMI) or tetradentate (HMTETA, [2.2.3], [2.3.2], [3.2.3], BPED, Me6TREN, Et6TREN, MA6TREN, BA6TREN, AN6TREN, TPMA, Me4Cyclam, DMCBCy) ligands have been reported to exist in solution preferentially as [CuI(κ2-Lig)2]+ and [CuI(κ4-Lig)]+ respectively, whereas those with CuIIX2 as [CuII(κ2-Lig)2X]+ and [CuII(κ4-Lig)X]+.74,75 On the other hand, in the presence of tridentate ligands such as PMDETA and BPMPA, CuIX and CuIIX are likely to form neutral complexes of the type [CuI(κ3-Lig)X] and [CuII(κ3-Lig)X2] though the displacement of a halogen atom by the solvent or monomer is feasible in both species.74,75 Finally, the complex with CuIX and TPEN may exist both as a binuclear tricoordinated complex, [CuI(κ3-TPEN)X]2, and a mononuclear pentacoordinated complex, [CuI(κ5-TPEN)]+, the latter likely being the active species, while CuIIX2 with TPEN can only exist as a mononuclear and hexacoordinated compound [CuII(κ5-TPEN)X]+.76
After optimizing the geometries, molecular descriptors were calculated for each of the copper complexes above, and PCA was performed on the Cu(I) and Cu(II) species separately. The first 6 principal components (PCs) of each of the two groups of complexes were subjected to hierarchical cluster analysis (Fig. S1 and S2, ESI†). Test sets of diverse molecules were obtained by selecting one complex from each of the 4 most distant clusters such that the corresponding points in the figure-of-merit space were homogeneously spaced out. Accordingly, [CuI(NPPMI)Br], [CuI(Me6TREN)]+, [CuI(MA6TREN)]+ and [CuI(TPEN)]+ were chosen as a test set for the modeling of kact, whereas [CuII(BPMPA)Br2], [CuII(MA6TREN)Br]+, [CuII(DMCBCy)Br]+ and [CuII(TPEN)Br]+ as a test set for kdeact. Furthermore, in order to ensure the inclusion of the most relevant molecular information, the initial group of descriptors used for modeling the remaining 16 activators and 16 deactivators (training sets) was chosen among the peripheral points in the loading plots PC1 vs. PC2 vs. PC3 (Fig. S3 and S4, ESI†). Generally accepted mechanistic considerations of the ATRP process were also taken into consideration. The number of descriptors was progressively reduced by iteratively subjecting them to GFA algorithm and discarding, after each regression cycle, those with the lowest occurrences in the populations.
Of all the molecular descriptors calculated for Cu(I) complexes, HOMO energy (HOMO), cavity volume (CV), molecular area (MA), moment of inertia Z (IZ), van der Waal's surface area (vSA), solvent surface area (SSA), 5χpath, 5χchain, 5χvchain, 6χpath, and 6χvpath survived the selection process for kact. These were used to build quantitative structure–reactivity relationships and the GFA algorithm with linear polynomials returned the top 10 models in Table 1. The values of Friedman's LOF, R2, adjusted R2, cross-validated R2, significance-of-regression (SOR) F-value, and critical SOR F-value (95%) indicate that all the regressions are statistically significant. Interestingly, eqn (10) rather than the one with the lowest Friedman's LOF value yields the best prediction for the test set (Fig. 1). A close look at the models reveals a direct proportionality between kact and HOMO, which mirrors the experimental observation that the higher the reducing power of a Cu(I) complex, the higher its ATRP activity.57 The regressions highlight also the pivotal role played in the reactivity of Cu(I) compounds by topological descriptors, which differentiate the molecules according to their size, degree of branching, flexibility and overall shape. It is worth noting that only few of the tens of topological indices computed in the present study ended up in the models. This should minimize the risk of “fortuitous explanation” of the figure-of-merit that is sometimes observed with relationships containing a large number of such parameters.77 Surface area-based descriptors, which measure the extent to which molecules expose themselves to the external environment, have been generally related to molecular factors such as transport and solubility. It is therefore interesting to find out that MA and vSA, although positively correlated, not only have opposite effects on kact but also, as pointed out by control GFA analysis, both parameters need to be present in the model in order to obtain a reliable regression. According to the role of CV in the equations, metal accessibility is another parameter that should be taken into consideration when designing an ATRP catalyst (vide infra). The moments of inertia depend on both the mass of each atom in the molecule and the overall molecular geometry. The presence of Iz in five out of ten models can be therefore taken as a further evidence of the importance of the geometry of the activator on the activation process. In contrast, the absence of the average N–Cu bond length in all the models may be symptomatic of a secondary role, with respect to the other parameters, played by the stability of Cu(I) complexes in the activation process. The relatively small preference of Cu(I) for specific donor atoms as well as the much smaller variation, compared to Cu(II) compounds, of the stabilities of its complexes as the ligand is altered have been recently pointed out.78
| Model | Friedman's LOF | R 2 | Adjusted R2 | Cross-validated R2 | SOR F-value | Critical SOR F-value (95%) | |
|---|---|---|---|---|---|---|---|
| 1 | log(kact) = 47(HOMO) + 6.7 × 10−3(CV) − 3.1(5χpath) + 5.1(6χpath) + 42(5χvchain) − 4.3(6χvpath) + 0.46(MA) + 1.6 × 10−3(IZ) − 0.54(vSA) + 11 | 0.450 | 0.990 | 0.974 | 0.937 | 63.6 | 4.15 |
| 2 | log(kact) = 58(HOMO) + 8.1 × 10−3(CV) − 2.5(5χpath) + 30(5χchain) + 4.0(6χpath) − 3.2(6χvpath) + 0.37(MA) + 1.8 × 10−3(IZ) − 0.47(vSA) + 12 | 0.497 | 0.988 | 0.971 | 0.807 | 57.6 | 4.15 |
| 3 | log(kact) = 59(HOMO) + 7.9 × 10−3(CV) − 3.5(5χpath) + 5.9(6χpath) + 45(5χvchain) − 5.3(6χvpath) + 0.54(MA) − 0.64(vSA) + 12 | 0.510 | 0.978 | 0.952 | 0.874 | 38.3 | 3.75 |
| 4 | log(kact) = 58(HOMO) + 11 × 10−3(CV) − 3.4(5χpath) + 5.6(6χpath) + 44(5χvchain) − 5.2(6χvpath) + 0.55(MA) − 0.67(vSA) + 7.3 × 10−3(SSA) + 11 | 0.588 | 0.986 | 0.966 | 0.780 | 48.5 | 4.15 |
| 5 | log(kact) = 71(HOMO) + 13 × 10−3(CV) − 2.8(5χpath) + 32(5χchain) + 4.5(6χpath) − 4.2(6χvpath) + 0.47(MA) − 0.61(vSA) + 8.7 × 10−3(SSA) + 11 | 0.611 | 0.986 | 0.964 | 0.742 | 46.7 | 4.15 |
| 6 | log(kact) = 73(HOMO) + 9.5 × 10−3(CV) − 2.9(5χpath) + 32(5χchain) + 4.8(6χpath) − 4.3(6χvpath) + 0.47(MA) − 0.56(vSA) + 12 | 0.612 | 0.973 | 0.942 | 0.553 | 31.7 | 3.75 |
| 7 | log(kact) = 36(HOMO) − 2.3 × 10−3(CV) − 1.6(5χpath) + 26(5χchain) + 2.2(6χpath) + 5.4 × 10−3(IZ) − 14 × 10−3(SSA) + 12 | 0.906 | 0.935 | 0.879 | 0.735 | 16.6 | 3.51 |
| 8 | log(kact) = 63(HOMO) + 8.3 × 10−3(CV) − 3.3(5χpath) + 9.9(5χchain) + 5.6(6χpath) + 31(5χvchain) − 5.0(6χvpath) + 0.52(MA) − 0.61(vSA) + 12 | 0.927 | 0.979 | 0.946 | 0.554 | 30.6 | 4.15 |
| 9 | log(kact) = 38(HOMO) − 1.6(5χpath) + 26(5χchain) + 2.1(6χpath) + 5.1 × 10−3(IZ) − 20 × 10−2(vSA) − 8.9 × 10−3(SSA) + 12 | 0.967 | 0.931 | 0.871 | 0.518 | 15.4 | 3.51 |
| 10 | log(k act ) = 35(HOMO) − 2.8( 5 χ path ) + 4.6( 6 χ path ) + 41( 5 χ v chain ) − 3.0( 6 χ v path ) + 0.32(MA) + 3.5 × 10 −3 (I Z ) − 0.34(vSA) + 11 × 10 −3 (SSA) + 12 | 0.986 | 0.977 | 0.943 | 0.762 | 28.7 | 4.15 |
 | ||
Fig. 1 Calculated vs. experimental log(kact) values for Cu(I) activators obtained by genetic function approximation analysis with linear polynomials. Training and test sets are represented by the symbols “ ” and “ ” and “ ”, respectively. ”, respectively. | ||
As to kdeact, which is responsible for the control in ATRP reactions, the following descriptors for Cu(II) complexes outlasted the selection process: 4χvpath, 4χvpath/cluster, 5χvpath/cluster, electrostatic energy (EE), valence energy (VE), binding energy (BE), LUMO–HOMO energy (L–H), dipole Y (δY), surface area (SA), average N–Cu bond length (N–Cu), and Cu–Br bond length (Cu–Br). The comparison of these parameters with those obtained for the activation reaction highlights a higher incidence of energetic terms on the deactivation reaction. The GFA algorithm with linear polynomials returned the statistically significant top 10 models in Table 2. To the best of our knowledge, this is the first time that correlations between kdeact and the structure of ATRP catalysts are reported. Similar to what was found for kact, model 14 rather than the one with the lowest Friedman's LOF value exhibits the highest predictive power (Fig. 2), despite the negative value of cross-validated R2. Likewise topological descriptors, whose importance has already been discussed above, L–H and SA appear in all the 10 equations. In particular, the fact that the rate of deactivation is roughly proportional to the width of the L–H gap is consistent with the known, high kinetic reactivity of compounds having a narrow band gap.79 On the other hand, the direct proportionality between SA and kdeact can be rationalized on the basis of the different affinities of Cu(II) complexes for the reaction medium. The Cu–Br bond length, which turns out to be inversely proportional to the deactivation rate constant, is another important parameter that emerges from the modeling. Intuitively it is expected that the shorter the length of the Cu–Br bond, the stronger is the bond and hence the more favored is the deactivation process. This relates also to the concept of metal “halidophilicity”. It is experimentally observed that the degree of control in ATRP is lower in the presence of metal complexes with low halide affinity and/or of solvents that, like water, favor the dissociation of halides by effectively solvating them, thus reducing the amount of deactivator in solution.80,81 Interestingly, albeit not included in model 14, N–Cu is present in four out of the ten regressions suggesting that, unlike Cu(I), the stabilities of Cu(II) complexes should be taken into consideration when designing an ATRP catalyst.82,83
| Model | Friedman's LOF | R 2 | Adjusted R2 | Cross-validated R2 | SOR F-value | Critical SOR F-value (95%) | |
|---|---|---|---|---|---|---|---|
| 11 | log(kdeact) = −2.3(4χvpath) − 1.8(4χvpath/cluster) + 1.4(5χvpath/cluster) − 9.1 × 10−3(VE) + 1.3(BE) + 4.1 × 102(L–H) − 0.25(δY) + 19 × 10−3(SA) − 0.25 | 1.30 | 0.906 | 0.800 | −1.37 | 8.48 | 3.75 |
| 12 | log(kdeact) = −2.0(4χvpath) − 1.0(4χvpath/cluster) + 0.90(5χvpath/cluster) − 20 × 10−3(VE) + 14(N–Cu) − 6.1(Cu–Br) + 4.3 × 102(L–H) + 8.2 × 10−3(SA) − 10 | 1.40 | 0.899 | 0.784 | 0.0716 | 7.81 | 3.75 |
| 13 | log(kdeact) = −2.1(4χvpath) − 1.2(4χvpath/cluster) + 1.1(5χvpath/cluster) − 9.6 × 10−3(VE) − 5.0(Cu–Br) + 0.82(BE) + 4.3 × 102(L–H) + 14 × 10−3(SA) + 13 | 1.54 | 0.889 | 0.762 | 0.0711 | 7.02 | 3.75 |
| 14 | log(k deact ) = −2.0( 4 χ v path ) − 1.0( 4 χ v path/cluster ) + 0.79( 5 χ v path/cluster ) + 4.3 × 10 −3 (EE) − 5.9(Cu–Br) + 4.0 × 10 2 (L–H) − 0.18(δ Y ) + 8.0 × 10 −3 (SA) + 17 | 1.78 | 0.872 | 0.725 | −3.40 | 5.94 | 3.75 |
| 15 | log(kdeact) = −2.0(4χvpath) − 1.4(4χvpath/cluster) + 1.0(5χvpath/cluster) + 2.5 × 10−3(EE) − 0.87(BE) + 3.6 × 102(L–H) − 0.25(δY) + 14 × 10−3(SA) + 0.59 | 1.81 | 0.869 | 0.720 | −4.52 | 5.82 | 3.75 |
| 16 | log(kdeact) = −1.9(4χvpath) − 1.1(4χvpath/cluster) + 0.97(5χvpath/cluster) − 12 × 10−3(VE) + 9.3(N–Cu) + 0.76(BE) + 4.2 × 102(L–H) + 13 × 10−3(SA) − 18 | 1.95 | 0.859 | 0.699 | −3.68 | 5.35 | 3.75 |
| 17 | log(kdeact) = −2.0(4χvpath) − 0.91(4χvpath/cluster) + 0.81(5χvpath/cluster) + 2.6 × 10−3(EE) − 5.1 × 10−3(VE) − 7.6(Cu–Br) + 4.4 × 102(L–H) + 7.7 × 10−3(SA) + 21 | 1.98 | 0.857 | 0.693 | −3.88 | 5.24 | 3.75 |
| 18 | log(kdeact) = −1.8(4χvpath) − 0.78(4χvpath/cluster) + 0.69(5χvpath/cluster) + 2.7 × 10−3(EE) − 5.1(Cu–Br) + 0.40(BE) + 3.8 × 102(L–H) + 9.8 × 10−3(SA) + 14 | 2.06 | 0.852 | 0.682 | −6.90 | 5.03 | 3.75 |
| 19 | log(kdeact) = −1.8(4χvpath) − 0.73(4χvpath/cluster) + 0.65(5χvpath/cluster) + 3.5 × 10−3(EE) − 0.99(N–Cu) − 7.0(Cu–Br) + 4.1 × 102(L–H) + 7.0 × 10−3(SA) + 22 | 2.23 | 0.839 | 0.655 | −3.89 | 4.56 | 3.75 |
| 20 | log(kdeact) = −2.0(4χvpath) − 1.2(4χvpath/cluster) + 0.94(5χvpath/cluster) − 19 × 10−3(VE) + 18(N–Cu) + 3.9 × 102(L–H) − 0.17(δY) + 8.2 × 10−3(SA) − 32 | 2.32 | 0.833 | 0.643 | −2.52 | 4.37 | 3.75 |
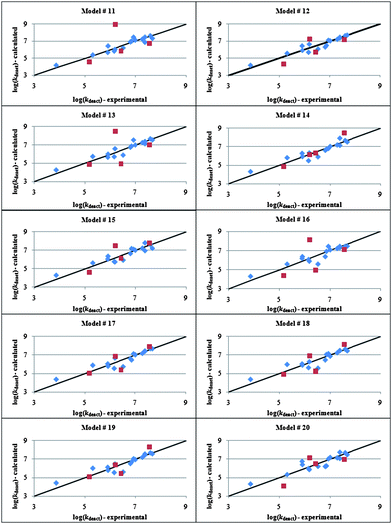 | ||
| Fig. 2 Calculated vs. experimental log(kdeact) values for Cu(II) deactivators obtained by genetic function approximation analysis with linear polynomials. Training and test sets are represented by the symbols “” and “”, respectively. | ||
When linear splines were employed as basis functions in the GFA algorithm, the resulting models not only contained a smaller number of molecular descriptors (Tables S1 and S2, ESI†) but also provided better predictions for the training set (Fig. S5 and S6, ESI†). None of them, however, was able to produce reliable predictions for the test set.
Initiators
The procedure used to build QSRR models for the ATRP initiators was identical to that described above for the catalysts. In particular, the constants kact and KATRP were quantitatively correlated to the structures of the 21 alkyl halides depicted in Chart 2. In contrast, since the function kdeact = f(structure of active radicals) is not bijective as different values of kdeact (e.g., for CH3OCOCH2Cl and CH3OCOCH2Br) may correspond to the structure of a single radical (i.e., CH3OCOCH2˙), it was not possible to model the deactivation rate constants directly. With the help of HCA carried out on the first 6 principal components (Fig. S7, ESI†), MClAc, tBBrP, BrAN and ECPA were chosen as the test set for kact, whereas MClAc, tBBrP, BrAN and AllBr as the one for KATRP. | ||
| Chart 2 Alkyl halide initiators for atom transfer radical polymerization used in this study. | ||
The descriptors selected for kact, namely nitrile fragment counts (CN), total molecular mass (Mw), molecular volume (MV), molecular refractivity (MR), Wiener index (W), 1κ, 0χv, 3χpath, 3χcluster, principal moment of inertia X (IX), radius of gyration (RoG), ellipsoidal volume (EV), solvation energy (SE) and LUMO energy (LUMO) were used in the GFA algorithm, which returned the top 10 linear polynomials in Table 3. Interestingly, both regression 22 and 30, which have the second lowest and the highest Friedman's LOF value, respectively, show the highest predictive power for the test set (Fig. 3). The models suggest that shape- and size-based descriptors play a crucial role also in the reactivity of the initiators. This can be rationalized as follows. Since ATRP is believed to occur through an inner sphere electron transfer (ISET) mechanism,84 the halogen of the alkyl halide needs to be in the first coordination sphere of the metal center in order for the atom transfer to take place. It is therefore reasonable to imagine that the more compatible the shape of the initiator is with the steric neighborhood of the metal center in the activator, the more effective is the activation reaction (vide supra). The fact that the solvation energy appears in a number of regressions underlines further the importance of the solvent in the ATRP equilibrium.81 Another remarkable finding is the inverse proportionality between kact and LUMO energy. This is consistent with the fact that the alkyl halide is the electron acceptor in the activation reaction and hence the lower the energy of the LUMO, the faster is the reduction. Moreover, the beneficial effect on the activation process resulting from the presence of a nitrile on the activator is due to the ability of this group to stabilize radicals by resonance, a property exploited in azo initiators for conventional free radical polymerization for example the use of 2,2′-azobisisobutyronitrile (AIBN).
| Model | Friedman's LOF | R 2 | Adjusted R2 | Cross-validated R2 | SOR F-value | Critical SOR F-value (95%) | |
|---|---|---|---|---|---|---|---|
| 21 | −log(kact) = −1.9(CN) − 22 × 10−3(W) − 1.6(0χv) + 0.16(MV) − 5.0(RoG) + 41(LUMO) + 9.1 | 0.312 | 0.972 | 0.955 | 0.0595 | 57.5 | 3.23 |
| 22 | −log(k act ) = −2.8(CN) − 25 × 10 −3 (W) − 1.8( 0 χ v ) + 0.19(MV) − 7.4(RoG) − 1.9 × 10 2 (SE) + 45(LUMO) + 9.9 | 0.379 | 0.978 | 0.961 | 0.353 | 57.5 | 3.30 |
| 23 | −log(kact) = −33 × 10−3(W) − 1.8(0χv) + 0.13(MV) + 43(LUMO) + 2.2 | 0.455 | 0.919 | 0.893 | 0.675 | 34.3 | 3.31 |
| 24 | −log(kact) = −25 × 10−3(W) − 1.4(0χv) + 0.10(MV) + 1.9 × 102(SE) + 39(LUMO) + 4.7 | 0.477 | 0.938 | 0.910 | 0.629 | 33.3 | 3.22 |
| 25 | −log(kact) = −1.9(CN) − 22 × 10−3(W) + 12 × 10−3(3χpath) − 1.5(0χv) + 0.16(MV) − 5.1(RoG) + 42(LUMO) + 9.1 | 0.488 | 0.972 | 0.950 | −0.0681 | 44.3 | 3.30 |
| 26 | −log(kact) = −0.85(CN) − 30 × 10−3(W) − 1.8(0χv) + 0.12(MV) + 34(LUMO) + 2.7 | 0.498 | 0.935 | 0.906 | −1.08 | 31.8 | 3.22 |
| 27 | −log(kact) = −6.8 × 10−3(W) + 3.9 × 102(SE) + 46(LUMO) + 9.8 | 0.546 | 0.874 | 0.845 | 0.684 | 30.0 | 3.57 |
| 28 | −log(kact) = −15 × 10−3(W) + 0.60(3χpath) + 3.8 × 102(SE) + 45(LUMO) + 9.1 | 0.580 | 0.897 | 0.863 | 0.752 | 26.2 | 3.31 |
| 29 | −log(kact) = −31 × 10−3(W) − 1.7(0χv) + 0.14(MV) − 1.4(RoG) + 49(LUMO) + 3.8 | 0.582 | 0.924 | 0.890 | 0.696 | 26.9 | 3.22 |
| 30 | −log(k act ) = −3.3(CN) − 26 × 10 −3 (W) − 0.48( 3 χ path ) − 2.2( 0 χ v ) + 0.22(MV) − 7.6(RoG) − 2.8 × 10 2 (SE) + 40(LUMO) + 8.5 | 0.582 | 0.981 | 0.962 | 0.386 | 52.0 | 3.45 |
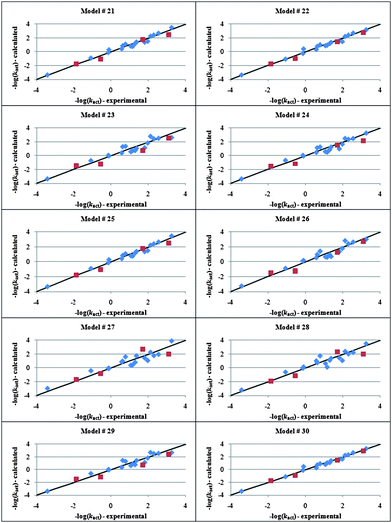 | ||
| Fig. 3 Calculated vs. experimental log(kact) values for alkyl halide initiators obtained by genetic function approximation analysis with linear polynomials. Training and test sets are represented by the symbols “” and “”, respectively. | ||
As far as KATRP is concerned, the following descriptors were found to be important for the modeling: nitrile fragment counts (CN), element count (EC), molecular density (MD), molecular refractivity (MR), Balaban index X (JX), Balaban index Y (JY), 2χ, principal moment of inertia X (IX), dipole Z (δZ), shadow area ZX plane (ShAZX), solvent surface occupied volume (SSOV), solvation energy (SE), total energy (TE), binding energy (BE), HOMO energy (HOMO), LUMO–HOMO energy (L–H) and C–X bond distance (C–X). When these parameters were used with the GFA algorithm, the linear polynomials in Table 4 were obtained, among which models 38 and 40 distinguished themselves for the reliability of the predictions (Fig. 4). The presence in the models of the highly discriminating Balaban index, whose value is substantially independent of the size of the molecule, as well as of the solvent surface occupied volume and solvation energy adds to what has already been said about the relevance of, respectively, topological descriptors and reaction medium in ATRP. By the same token, the fact that most equations contain the moment of inertia Ix goes back to the previously discussed importance of the geometry of the species involved in the ATRP equilibrium. Notably, since dipolar properties have been correlated to long-range electrostatic interactions,69 which are believed to strongly influence the mutual orientation of ligands and receptors prior to Brownian collision and subsequent binding,85 the inclusion of δZ in the regressions may indicate the existence of analogous long-range recognition processes between ATRP catalysts and initiators. Last but not least, a thorough analysis of the models points out the marked incidence of the total energy of the initiator on KATRP. TE is a measure of the overall reactivity of a molecule and can be related, in the present case, to both the reactivity of the carbon–halogen bond and the presence of radical stabilizing groups in the initiators.
| Model | Friedman's LOF | R 2 | Adjusted R2 | Cross-validated R2 | SOR F-value | Critical SOR F-value (95%) | |
|---|---|---|---|---|---|---|---|
| 31 | −log(KATRP) = 5.9(EC) − 2.5(MR) + 2.7(JY) − 4.8(2χ) − 6.6 × 10−3(IX) − 1.5 × 102(SE) − 3.5 × 10−3(TE) − 15(BE) + 13 | 86.7 × 10−3 | 0.996 | 0.992 | 0.956 | 259 | 3.45 |
| 32 | −log(KATRP) = −6.1(JX) + 7.9(JY) − 3.4(0χv) − 8.8 × 10−3(IX) + 49 × 10−3(SSOV) + 45(HOMO) − 1.2(δZ) + 9.9(C–X) − 8.0 | 88.5 × 10−3 | 0.996 | 0.992 | 0.977 | 254 | 3.45 |
| 33 | −log(KATRP) = −19(JX) + 21(JY) − 1.5(0χv) + 3.1(MD) − 9.7 × 10−3(IX) − 39 × 10−3(SSOV) + 1.1 × 103(SE) − 69(LUMO) − 3.3(δZ) − 1.1 | 92.5 × 10−3 | 0.998 | 0.995 | 0.983 | 342 | 3.70 |
| 34 | −log(KATRP) = 4.1(EC) − 1.9(MR) + 2.4(JY) − 4.0(2χ) − 7.2 × 10−3(IX) − 2.7 × 10−3(TE) − 13(BE) − 0.58(δZ) + 12 | 93.5 × 10−3 | 0.996 | 0.992 | 0.984 | 240 | 3.45 |
| 35 | −log(KATRP) = 4.6(EC) − 2.3(MR) − 2.2(2χ) − 7.6 × 10−3(IX) − 0.14(ShAZX) − 3.3 × 10−3(TE) − 16(BE) + 22 | 98.1 × 10−3 | 0.993 | 0.988 | 0.966 | 186 | 3.30 |
| 36 | −log(KATRP) = −11(JX) + 12(JY) − 1.0(0χv) − 9.3 × 10−3(IX) + 21 × 10−3(SSOV) + 4.7 × 102(SE) − 2.6 × 10−4 (TE) − 31(L–H) − 2.0(δZ) + 11 | 99.7 × 10−3 | 0.998 | 0.994 | 0.970 | 320 | 3.70 |
| 37 | −log(KATRP) = −2.1(CN) − 0.59(EC) + 2.5(JY) − 3.5(0χv) − 9.8 × 10−3(IX) + 66 × 10−3(SSOV) − 1.8(δZ) + 9.2(C–X) − 21 | 0.107 | 0.995 | 0.990 | 0.965 | 207 | 3.45 |
| 38 | −log(K ATRP ) = −9.5(J X ) + 10(J Y ) − 9.8 × 10 −3 (I X ) + 4.8 × 10 2 (SE) − 3.4 × 10 −4 (TE) − 0.89(BE) − 1.7(δ Z ) + 9.1 | 0.109 | 0.992 | 0.986 | 0.974 | 167 | 3.30 |
| 39 | −log(KATRP) = 6.1(EC) − 2.7(MR) + 2.9(JX) − 5.5(2χ) − 6.4 × 10−3(IX) − 2.1 × 10−3(SE) − 3.8 × 10−3 (TE) − 18(BE) + 14 | 0.111 | 0.995 | 0.990 | 0.960 | 202 | 3.45 |
| 40 | −log(K ATRP ) = −9.1(J X ) + 10(J Y ) − 10 × 10 −3 (I X ) + 10 × 10 −3 (SSOV) + 51 × 10 2 (SE) − 2.8 × 10 −4 (TE) − 1.8(δ Z ) + 6.6 | 0.111 | 0.992 | 0.986 | 0.961 | 163 | 3.30 |
 | ||
| Fig. 4 Calculated vs. experimental log(KATRP) values for alkyl halide initiators obtained by genetic function approximation analysis with linear polynomials. Training and test sets are represented by the symbols “” and “”, respectively. | ||
As seen with the catalysts, when linear splines were used as basis functions in the GFA algorithm, the resulting models, while containing less molecular descriptors (Tables S3 and S4, ESI†) and providing better predictions for the training set, failed to return reliable values for the test set (Fig. S8 and S9, ESI†).
Conclusions
In this work, in silico modeling has been applied for the first time to the construction of quantitative and predictive relationships between the set of constants kact, kdeact and KATRP and the structures and properties of various ATRP catalysts and initiators. The results are consistent not only with the generally accepted mechanistic picture for ATRP but also provide unprecedented insight into the factors governing this complex reaction. The models highlight and quantify the crucial roles played in ATRP by (i) the absolute and relative HOMO and LUMO energies and (ii) the topology (i.e., size, flexibility, geometry, overall shape etc.) of both catalysts and initiators; (iii) the compatibility between the shape of the initiator and the steric neighborhood of the metal center in the activator; (iv) the presence of radical stabilizing groups in the initiator; (v) the Cu–Br and the average N–Cu bond lengths in the deactivator; and (vi) the affinity of both catalysts and initiators for the reaction medium. Moreover, the regressions suggest the existence of long-range interactions in catalyst–initiator recognition and subsequent reaction. The approach is expected to greatly facilitate the design of new generations of atom transfer radical polymerization catalysts of wider academic and industrial interest.Acknowledgements
The Agency for Science, Technology and Research, Singapore, is gratefully acknowledged for financial support.Notes and references
- B. Jandeleit, D. J. Schaefer, T. S. Powers, H. W. Turner and W. H. Weinberg, Angew. Chem., Int. Ed., 1999, 38, 2494–2532 CrossRef CAS.
- J. P. Stambuli and J. F. Hartwig, Curr. Opin. Chem. Biol., 2003, 7, 420–426 CrossRef CAS.
- J. G. de Vries and A. H. M. de Vries, Eur. J. Org. Chem., 2003, 799–811 CrossRef CAS.
- C. Gennari and U. Piarulli, Chem. Rev., 2003, 103, 3071–3100 CrossRef CAS.
- I. Takeuchi, J. Lauterbach and M. J. Fasolka, Mater. Today, 2005, 8, 18–26 CrossRef CAS.
- W. F. Maier, K. Stoewe and S. Sieg, Angew. Chem., Int. Ed., 2007, 46, 6016–6067 CrossRef CAS.
- D. L. Kale, S. C. Chaturvedi, D. Patil and R. B. Kakde, Pharm. Rev., 2008, 6, 46–51 Search PubMed.
- L. M. Mayr and D. Bojanic, Curr. Opin. Pharmacol., 2009, 9, 580–588 CrossRef CAS.
- J. Inglese and D. S. Auld, in Wiley Encyclopedia of Chemical Biology, ed. T. P. Begley, John Wiley & Sons, Inc., Hoboken, NJ, 2009, pp. 260–274 Search PubMed.
- H. W. Turner, A. F. Volpe and W. H. Weinberg, Surf. Sci., 2009, 603, 1763–1769 CrossRef CAS.
- V. Busico, R. Pellecchia, F. Cutillo and R. Cipullo, Macromol. Rapid Commun., 2009, 30, 1697–1708 CrossRef CAS.
- P. Kirkpatrick and C. Ellis, Nature, 2004, 432, 823 CrossRef CAS and references therein.
- W. P. Walters, M. T. Stahl and M. A. Murcko, Drug Discovery Today, 1998, 3, 160–178 CrossRef CAS.
- R. Carlson, J. Carlson and A. Grennberg, J. Chemom., 2001, 15, 455–474 CrossRef CAS.
- B. K. Shoichet, Nature, 2004, 432, 862–865 CrossRef CAS.
- S. D. Lindell and J. Scherkenbeck, Comb. Chem. High Throughput Screening, 2005, 8, 555–562 CrossRef CAS.
- C. McInnes, Curr. Opin. Chem. Biol., 2007, 11, 494–502 CrossRef CAS.
- K.-J. Schleifer, Virtual Screening in Crop Protection Research, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2007 Search PubMed.
- U. Rester, Curr. Opin. Drug Discovery Dev., 2008, 11, 559–568 CAS.
- J. M. Rollinger, H. Stuppner and T. Langer, Prog. Drug Res., 2008, 65, 211–249 Search PubMed.
- S. D. Lindell, L. C. Pattenden and J. Shannon, Bioorg. Med. Chem., 2009, 17, 4035–4046 CrossRef CAS.
- A. Varnek and A. Tropsha, Chemoinformatics Approaches to Virtual Screening, Royal Society of Chemistry, Cambridge, UK, 2009 Search PubMed.
- K. Pearson, Philos. Mag., 1901, 2, 559–572.
- H. Hotelling, J. Educ. Psychol., 1933, 24, 417–441.
- H. Hotelling, J. Educ. Psychol., 1933, 24, 498–520 CrossRef.
- R. Scordamaglia and L. Barino, Macromol. Theory Simul., 1998, 7, 399–405 CrossRef CAS.
- R. Scordamaglia and L. Barino, Macromol. Theory Simul., 1998, 7, 407–419 CrossRef.
- S. Yao and Y. Tanaka, Macromol. Theory Simul., 2001, 10, 850–854 CrossRef CAS.
- K. B. Lipkowitz and M. C. Kozlowski, Synlett, 2003, 1547–1565 CrossRef CAS.
- E. Burello, D. Farrusseng and G. Rothenberg, Adv. Synth. Catal., 2004, 346, 1844–1853 CrossRef CAS.
- E. Burello, P. Marion, J.-C. Galland, A. Chamard and G. Rothenberg, Adv. Synth. Catal., 2005, 347, 803–810 CrossRef CAS.
- V. L. Cruz, J. Ramos, S. Martinez, A. Munoz-Escalona and J. Martinez-Salazar, Organometallics, 2005, 24, 5095–5102 CrossRef CAS.
- J. B. van der Linden, E.-J. Ras, S. M. Hooijschuur, G. M. Klaus, N. T. Luchters, P. Dani, G. Verspui, A. A. Smith, E. W. P. Damen, B. McKay and M. Hoogenraad, QSAR Comb. Sci., 2005, 24, 94–98 Search PubMed.
- E. Burello and G. Rothenberg, Int. J. Mol. Sci., 2006, 7, 375–404 Search PubMed.
- G. Occhipinti, H.-R. Bjørsvik and V. R. Jensen, J. Am. Chem. Soc., 2006, 128, 6952–6964 CrossRef CAS.
- J. A. Hageman, J. A. Westerhuis, H.-W. Fruhauf and G. Rothenberg, Adv. Synth. Catal., 2006, 348, 361–369 CrossRef CAS.
- V. L. Cruz, J. Martinez, J. Martinez-Salazar, J. Ramos, M. L. Reyes, A. Toro-Labbe and S. Gutierrez-Oliva, Polymer, 2007, 48, 7672–7678 CrossRef CAS.
- V. L. Cruz, S. Martinez, J. Martinez-Salazar, D. Polo-Cero, S. Gomez-Ruiz, M. Fajardo and S. Prashar, Polymer, 2007, 48, 4663–4674 CrossRef CAS.
- M. L. Drummond and B. G. Sumpter, Inorg. Chem., 2007, 46, 8613–8624 CrossRef CAS.
- A. G. Maldonado, J. A. Hageman, S. Mastroianni and G. Rothenberg, Adv. Synth. Catal., 2009, 351, 387–396 CrossRef CAS.
- A. G. Maldonado and G. Rothenberg, Chem. Soc. Rev., 2010 10.1039/b921393g.
- C. H. Reynolds, J. Comb. Chem., 1999, 1, 297–306 CrossRef CAS.
- J. S. Duca, Y.-F. Tseng and A. J. Hopfinger, Adv. Mater., 2001, 13, 1713–1717 CrossRef CAS.
- J. R. Smith, A. Seyda, N. Weber, D. Knight, S. Abramson and J. Kohn, Macromol. Rapid Commun., 2004, 25, 127–140 CrossRef CAS.
- N. Adams and P. Murray-Rust, Macromol. Rapid Commun., 2008, 29, 615–632 CrossRef CAS.
- T. Puzyn, D. Leszczynska and J. Leszczynski, Small, 2009, 5, 2494–2509 CrossRef CAS.
- D. Farrusseng, F. Clerc, C. Mirodatos and R. Rakotomalala, Comput. Mater. Sci., 2009, 45, 52–59 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689–3745 CrossRef CAS.
- K. Matyjaszewski and N. V. Tsarevsky, Nat. Chem., 2009, 1, 276–288 Search PubMed.
- M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963–5050 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Acc. Chem. Res., 2008, 41, 1133–1142 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- T. Pintauer, P. Zhou and K. Matyjaszewski, J. Am. Chem. Soc., 2002, 124, 8196–8197 CrossRef CAS.
- W. Tang, N. V. Tsarevsky and K. Matyjaszewski, J. Am. Chem. Soc., 2006, 128, 1598–1604 CrossRef CAS.
- W. Tang, Y. Kwak, W. Braunecker, N. V. Tsarevsky, M. L. Coote and K. Matyjaszewski, J. Am. Chem. Soc., 2008, 130, 10702–10713 CrossRef CAS and references therein.
- W. Tang and K. Matyjaszewski, Macromolecules, 2006, 39, 4953–4959 CrossRef CAS.
- W. Tang and K. Matyjaszewski, Macromolecules, 2007, 40, 1858–1863 CrossRef CAS.
- Accelrys Software Inc., San Diego, CA, USA, http://accelrys.com/products/materials-studio/.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. I. Goddard and W. M. Skid, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef CAS.
- B. Delley, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS.
- B. Delley, Theor. Comput. Chem., 1995, 2, 221–254 Search PubMed.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter, 1992, 46, 6671–6687 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
- R. Todeschini and V. Consonni, Handbook of Molecular Descriptors, Wiley-VCH, Weinheim, 2002 Search PubMed.
- J. H. Ward, J. Am. Stat. Assoc., 1963, 58, 236–244.
- F. Murtagh, Comput. J., 1983, 26, 354–359 Search PubMed.
- D. Rogers and A. J. Hopfinger, J. Chem. Inf. Comput. Sci., 1994, 34, 854–866 CrossRef CAS.
- J. H. Friedman, Ann. Stat., 1991, 19, 1–67 CrossRef.
- T. Pintauer and K. Matyjaszewski, Coord. Chem. Rev., 2005, 249, 1155–1184 CrossRef CAS.
- T. Pintauer and K. Matyjaszewski, Top. Organomet. Chem., 2009, 26, 221–251 CAS.
- H. Tang, N. Arulsamy, J. Sun, M. Radosz, Y. Shen, N. V. Tsarevsky, W. A. Braunecker, W. Tang and K. Matyjaszewski, J. Am. Chem. Soc., 2006, 128, 16277–16285 CrossRef CAS.
- J. Devillers, Topological Indices and Related Descriptors in QSAR and QSPR, Gordon and Breach Science Publishers, Amsterdam, 1999, ch. 1, and references therein Search PubMed.
- D. B. Rorabacher, Chem. Rev., 2004, 104, 651–697 CrossRef CAS.
- A. Rauk, Orbital Interaction Theory of Organic Chemistry, Wiley-Interscience, New York, 2000, ch. 13, and references therein Search PubMed.
- W. A. Braunecker, W. C. Brown, B. C. Morelli, W. Tang, R. Poli and K. Matyjaszewski, Macromolecules, 2007, 40, 8576–8585 CrossRef CAS.
- W. A. Braunecker, N. V. Tsarevsky, A. Gennaro and K. Matyjaszewski, Macromolecules, 2009, 42, 6348–6360 CrossRef CAS.
- N. V. Tsarevsky, W. A. Braunecker, W. Tang, S. J. Brooks, K. Matyjaszewski, G. R. Weisman and E. H. Wong, J. Mol. Catal. A: Chem., 2006, 257, 132–140 CrossRef CAS.
- N. V. Tsarevsky and K. Matyjaszewski, Chem. Rev., 2007, 107, 2270 CrossRef CAS.
- K. Matyjaszewski, Macromol. Symp., 1998, 134, 105–118 CAS.
- L. N. Drozdov-Tikhomirov, D. M. Linde, V. V. Poroikov, A. A. Alexandrov, G. I. Skurida, P. V. Kovalev and V. Y. Potapov, Mol. Biol., 2003, 37, 148–155 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: cluster analysis dendrograms, loading plots, linear spline models and best fits. See DOI: 10.1039/c0py00058b |
| This journal is © The Royal Society of Chemistry 2010 |