Solution, thermal and optical properties of new poly(pyridinium salt)s derived from bisquinoline diamines†‡
Alexi K.
Nedeltchev
,
Haesook
Han
and
Pradip K.
Bhowmik
*
Department of Chemistry, University of Nevada Las Vegas, 4505 Maryland Parkway, Las Vegas, NV 89154-4003, USA. E-mail: pradip.bhowmik@unlv.edu; Fax: +1-702-895-4072; Tel: +1-702-895-0885
First published on 6th May 2010
Abstract
Several novel poly(pyridinium salt)s with heterocyclic bisquinoline moieties having both tosylate and triflimide counterions were prepared by either ring-transmutation or metathesis reaction. Their chemical structures were established by using 1H and 13C NMR, FTIR and elemental analysis. Their number-average molecular weights (Mn) ranged from 53,000 to 79,000 and their polydispersities ranged from 1.30 to 3.24 as determined by gel permeation chromatography. They had excellent thermal stabilities in the temperature range of 336–447 °C and glass transition temperatures in the range of 119–174 °C. They had good solubilities in common organic solvents and in several cases their critical concentrations (*C) were exceeded to form lyotropic liquid-crystalline phases. Additionally, they emitted light in the blue and green region in both solution and solid state. Their quantum yields were also determined by using a spectrofluorometer.
Introduction
Development of novel smart materials that combine unique properties are of interest in both industry and academia, since they are useful for the fabrication of opto-eletronic devices such as organic light-emitting diodes (OLEDs), organic field-effect transistors (OFETs), organic photovoltaic cells and organic lasers.1 Generally, they have π-conjugated structures to serve as organic semiconductors by transporting charge, either hole or electron, from the anode and the cathode, respectively.1 Many electron rich materials have been designed and introduced as hole transporting/electron blocking layers (HTLs).1 However, fewer electron poor materials have been made as efficient electron transporting/hole blocking layers (ETLs). Various electron deficient moieties have been introduced in both molecular and polymeric materials to serve as ETLs, which have been reviewed by Jenekhe and co-workers.2 Additionally, heterocyclic moieties are beneficial since they improve the thermal stability of both molecular3 and polymeric4 materials. However, their solubility in common organic solvents, in many cases, is severely compromised. To overcome this problem, several approaches have been undertaken that included the attachment of flexible alkyl chains as pendant groups,5a or incorporation of kinks and twists in the π-conjugated moieties of both molecular materials and polymers.5b An additional approach is to introduce electrostatic charges in the main chain of polymers. Such are the cases with viologen polymers6 and poly(pyridinium salt)s.7–9 Having ionic charges in the polymer chains have other benefits. It has been largely demonstrated the formation of layer-by-layer assemblies to produce nanocomposites both on substrates10a,d or freestanding films10b,c with appropriate polyanions and polycations via dipping10a,b or spin-coating10c,d methods. Moreover, the ionic conductivity of electrostatic charges have found to be useful for the preparation of light-emitting electrochemical cells (LECs).11 In addition to electroluminescent properties, a photovoltaic response has also been identified in ionic junctions of sandwiched electrostatic materials.11c Poly(pyridinium salt)s are a class of polyelectrolytes that combine many of these benefits.Since the conception of poly(pyridinium salt)s, they have undergone a great deal of improvements with regard to synthesis and physical properties. Initially, they had very low molecular weights, limited solubility and low thermal stability.7 Harris et al. were able to improve the methodology of the ring-transmutation polymerization reaction thereby significantly improving their molecular weights.8a Their solubility and thermal stability were significantly improved with the introduction of organic counterions for the main-chain ionic polymers.8b,9a,b Since then we have continuously explored various poly(pyridinium salt)s containing both aromatic and aliphatic moieties for their various properties.9 They exhibited photoluminescence in various organic solvents as well as film state ranging from UV to green light.9d Moreover, they are amorphous having glass transition temperatures Tg > 150 °C even for polymers with oxyalkylene units in their main chains.9f,g These ionenes are thermally robust with decomposition temperatures (Td) > 400 °C9a–d for fully aromatic polymers and Td > 200 °C9e for alkyl ones. They could be easily processed into thin films and multilayer assemblies since they are polycations having excellent solubility in common organic solvents. Furthermore, they exhibit both thermotropic (in melt) and lyotropic (in solution) liquid-crystalline properties.9 This compilation of properties makes these polymers potential candidates for various opto-electronic applications such as charge-transporting materials in OLEDs or materials for LECs. In this article, we describe a new series of wholly aromatic poly(pyridinium salts), with tosylate or triflimide counterions, having three different bisquinoline moieties in their main chains, prepared through ring-transmutation and metathesis reactions. Their chemical structures were established by using NMR, FTIR and elemental analysis. Their solution, thermal, and photoluminescence properties in both solutions and film states were also examined. The general structures and designation of this class of poly(pyridinium salt)s, which were used in this work, are shown in Scheme 1.
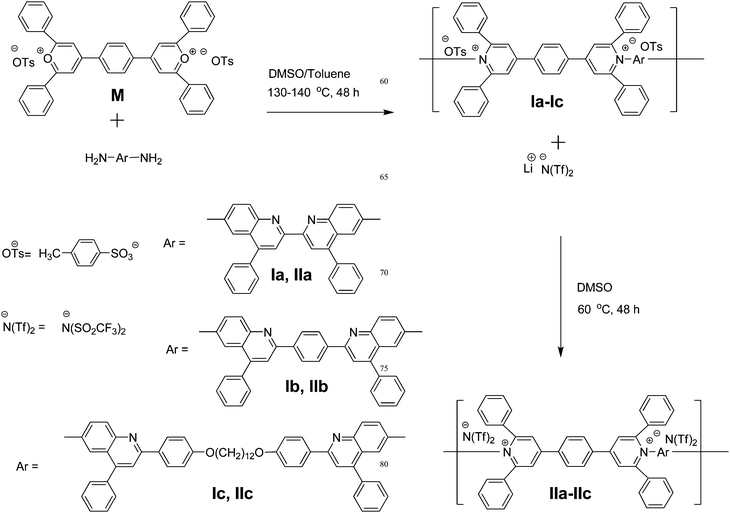 | ||
| Scheme 1 Synthesis of poly(pyridinium salt)s. | ||
Results and discussions
Chemical structures
The chemical structures of poly(pyridinium salt)s described herein are consistent with their spectroscopy analysis via1H and 13C NMR, FTIR as well as elemental analysis. Disappearance of proton and carbon signals of the vinylogous groups in polymers Ia–Ic spectra suggested that the ring-transmutation reaction proceeded to completion.12 The metathesis reaction was followed by the disappearance of the tosylate counterion characteristic peaks in polymers IIa–IIc. The 1H NMR peaks for all polymers are broad due to their high viscosities and high molecular weights, which were also confirmed by GPC measurements (vide infra). The FTIR spectrum showed the following representative, characteristic peaks for polymer Ia: 3058 (=C–H aromatic stretching), 1618–1449 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CN aromatic ring stretching), 1197 (C–N+), 1121 (SO asymmetric stretching), 1034–1012 (SO symmetric stretching) and 897–680 cm−1 (C–H out-of-plane bending); and those for polymer IIa: 3063 (C–H aromatic stretching), 1617–1452 (CC and CN aromatic ring stretching), 1349 (C–F stretching), 1193 (C–N+), 1136 (SO asymmetric stretching), 1058 (SO symmetric stretching) and 899–699 cm−1 (=C–H out-of-plane bending). For detailed information all the NMR and FTIR spectra are provided in the ESI.†
C and CN aromatic ring stretching), 1197 (C–N+), 1121 (SO asymmetric stretching), 1034–1012 (SO symmetric stretching) and 897–680 cm−1 (C–H out-of-plane bending); and those for polymer IIa: 3063 (C–H aromatic stretching), 1617–1452 (CC and CN aromatic ring stretching), 1349 (C–F stretching), 1193 (C–N+), 1136 (SO asymmetric stretching), 1058 (SO symmetric stretching) and 899–699 cm−1 (=C–H out-of-plane bending). For detailed information all the NMR and FTIR spectra are provided in the ESI.†
Molecular weight determination
The molecular weight data for the the poly(pyridinium salt)s are summarized in Table 1, which also shows Mark-Houwink constants α and K as well as dn/dc values. Their intrinsic viscosities were in the range 0.65–1.29, which are similar to other poly(pyridinium salt)s9f,g but lower than other neutral polyquinolines.4a,13 The exchange of counterion from tosylate to triflimide resulted in an increase in the intrinsic viscosity for each of the polymers. Surprisingly, the polymers Ic and IIc had higher viscosities despite the presence of flexible alkyl chains in each of them when compared with the other completely aromatic polymers in this series. Number-average molecular weights (Mn) for this series of polymers were as low as 53,000 and as high as 79,000. Their weight-average molecular weights (Mw) ranged from 78,000 to 194,000. Generally, both the Mn and Mw increased when exchanging the counterion from tosylate to triflimide, which is expected since the triflimide counterion had a higher molecular weight than the tosylate counterion (Fig. 1). The polydispersity indices (Mw/Mn) for polymers Ia and IIb were relatively broad and 2.89 and 3.24, respectively. Other polymers had quite narrow polydispersities (1.30–1.35), which are in excellent agreement with those reported in the literature.9f,g Overall, these polymers had substantially high molecular weights to properly assess their physical propeties without concerns with regard to the effect of molecular weights on these properties.| Polymer | IV dL g−1 | M n | M w | M w/Mn | dn/dc (ml g−1) | α | K |
|---|---|---|---|---|---|---|---|
| Ia | 0.65 | 53,193 | 153,517 | 2.89 | 0.1200 | 0.708 | 1.96 × 10−4 |
| IIa | 1.10 | 59,950 | 194,346 | 3.24 | 0.1500 | 0.708 | 2.83 × 10−4 |
| Ib | 0.28 | 60,188 | 78,174 | 1.30 | 0.0700 | 1.415 | 3.28 × 10−8 |
| IIb | 0.74 | 71,921 | 125,614 | 1.33 | 0.1500 | 1.208 | 7.08 × 10−7 |
| Ic | 1.22 | 77,628 | 101,248 | 1.30 | 0.1400 | 0.927 | 3.05 × 10−5 |
| IIc | 1.29 | 79,266 | 107,266 | 1.35 | 0.1400 | 0.893 | 4.59 × 10−5 |
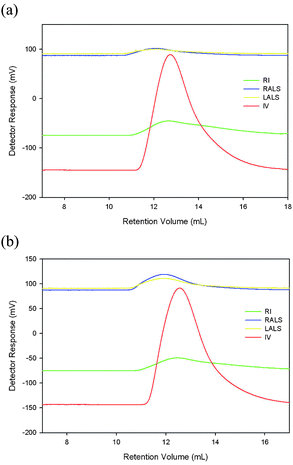 | ||
| Fig. 1 Gel permeation chromatography plots of (a) polymer Ib and (b) polymer IIb. | ||
Solution properties
Solubility in common organic solvents is a desirable property in order to process polymers into thin films. Highly rigid polymers like Kevlar, which is a well-known para-linked aramid,14 have generally limited solubility in strong acids and therefore are difficult and expensive to make into functional materials. Poly(pyridinum salt)s have good solubility in common organic solvents since they have formal positive charges in their backbones (vide supra). All of the ionic polymers described herein are soluble in solvents such as methanol, acetonitrile, and DMSO, among others, despite the presence of bisquinoline moieties in their main chains. Neutral polyquinolines are soluble only in high boiling solvents or completely insoluble, thereby limiting their potential use.4a,15 In contrast, the ionic polymers could not only be easily processed into thin functional films but also they could be utilized for preparation of nanocomposites with proper polyanions.10 Furthermore, we were motivated to study their lyotropic liquid-crystalline (LC) properties because poly(pyridinium salt)s have been demonstrated to form mosaic textures or Maltese crosses when critical concentraions (C*) are exceeded.9 The solution properties of these ionic polymers in this study are summarized in Table 2. Polymer Ia, which had a tosylate counterion had low solubility in methanol and acetonitrile, but developed only biphasic solutions of anisotropic and isotropic phase up to 20 wt% in both cases. Its solubility in DMSO was excellent. It retained isotropic solutions at relatively high concentrations, but at 41 wt% in this solvent it exhibited fully developed lyotropic LC phase as detemined by polarizing optical microscope (POM) studies. Triflimide polymer IIa was not soluble in metanol but formed lyotropic LC phase in both actetonitrile and DMSO at relatively low concentrations (Table 2). Next polymer Ib showed a lyotropic LC phase, biphasic solution and isotropic solution in methanol, acetonitrile and DMSO, respectively. Polymer IIb was not soluble in methanol similar to IIa but formed a lyotropic LC phase in acetonitrile at relatively low concentration (Fig. 2), but formed a biphasic solution in DMSO at relatively high concentraion of 41 wt%.| Polymer | Ia | IIa | Ib | IIb | Ic | IIc |
|---|---|---|---|---|---|---|
| CH3OH (ε = 32.6) | 1%–20% biphasic | — | 10% lyotropic | — | Up to 20% isotropic | 20% biphasic |
| CH3CN (ε = 37.5) | 1%–20% biphasic | 10% lyotropic | 19% biphasic | 29% lyotropic | Up to 21% isotropic | 30% biphasic |
| DMSO (ε = 48.9) | 41% lyotropic | 19%–39% lyotropic | Up to 41% isotropic | 41% biphasic | Up to 41% isotropic | Up to 41% isotropic |
 | ||
| Fig. 2 Photomicrograph of polymer IIb at 29 wt% CH3CN under cross polarizers exhibiting lyotropic LC phase (magnification 400×). | ||
Tosylate polymer Ic had very good solubility in the chosen solvents (Table 2) but no LC was observed in any of these solvents. On the other hand, its triflimide counterpart, polymer IIc, formed biphasic solutions in methanol and acetonitrile, but it did not form a fully-grown lyotropic phase in any of these solvents (Table 2). Like polymer Ic, it also formed an isotropic phase at relatively high concentration of 41 wt%. Overall, two trends were clearly observed. First, the completely conjugated polymers were able to form lyotropic LC phase more efficiently than the polymers with flexible alkyl chains. Second, the exchange of the counterion from tosylate to triflimide increased the polymer solubility in acetonitrile but decreased its solublity in methanol, which also affected the formation of the lyotropic LC phase.
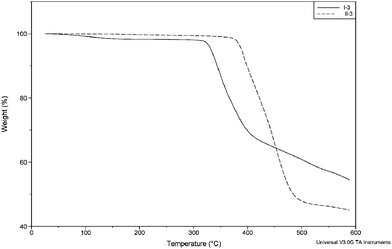
Thermal properties
| Polymer | Ia | IIa | Ib | IIb | Ic | IIc |
|---|---|---|---|---|---|---|
| a Glass transition temperatures recorded from the second heating cycles of DSC thermograms. b Thermal decomposition temperatures recorded at 5% weight loss by TGA in nitrogen. | ||||||
| T g | — | — | — | — | 174 °C | 119 °C |
| T d | 367 °C | 445 °C | 367 °C | 447 °C | 336 °C | 389 °C |
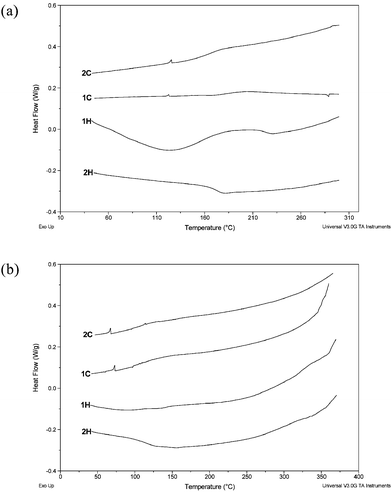 | ||
| Fig. 4 DSC thermograms of (a) polymer Ic and (b) polymer IIc obtained at heating and cooling rates of 10 °C min−1 in nitrogen. | ||
Optical properties
Light-emitting polymers are of particular interest for the development of optoelectronic devices.1 Introducing heterocyclic moieties in the polymer chains to develop this class of polymers is also gaining considerable attention since their properties can be tuned via protonation of the nitrogen heterocycles.5a,18 Many neutral, photoresponsive molecular and polymeric quinoline materials have been developed in recent years.13,15 In the series of poly(pyridinium salt)s described herein we explore the effect of bisquinoline moieties on their light-emitting properties both in solutions and solid states. Their optical properties including UV-Vis absorption spectra, photoluminescence, and fluorescence quantum yields are summarized in Table 4. The photoluminescent spectra of tosylate polymers Ia–Ic in methanol solutions are displayed in Fig. 5a. Polymer Ia exhibited an emission peak (λmax) at 502 nm when excited with 340 nm light. Polymer Ib showed a large bathochromically shifted λmax at 540 nm when excited with 345 nm light. This shift is expected since polymer Ib has a long conjugation length. On the other hand, polymer Ic exhibited a hypsochromic shift at 461 nm, when excited with 275 nm light, compared with those of polymers Ia and Ib because its conjugation was broken by the presence of twelve methylene units in its main chain. Similar behavior was observed for photoluminescence of polymers IIa–IIc in acetonitrile (Fig. 5b). They showed λem at 505, 547, and 448 nm when excited with 340, 340 and 385 nm wavelength of light, respectively. In some cases, exchange of the counterion from tosylate to triflimide resulted in a large shift in the wavelength of light emission in solution (polymers Ib and IIb). In other cases, the shift was negligible or non-existing (polymers Ic and IIc). Their full-width half-maximum (fwhm) values were generally quite large (>100 nm), which suggested that their fluorescence properties stem from multiple chromophores present in these ionic polymers, which are in good agreement with those in other poly(pyridinium salt)s reported previously.9 Mostly, their quantum yields were moderate (0.85%–1.2%) except for polymer Ib, which had fluorescence quantum efficiency of 15%. Additionally, the optical band gaps (Eg) were assessed from the onset of the UV-Vis spectra for these polymers. Their values ranged from 2.88–3.20 eV, which is typical for poly(pyridinium salt)s9 but generally high for neutral light-emitting polymers.1| Polymer | Ia | IIa | Ib | IIb | Ic | IIc |
|---|---|---|---|---|---|---|
| a Quantum yields were calculated with diphenyl anthracene as standard (ΦF = 0.9). b sh = shoulder. | ||||||
| UV abs CH3CN/nm | 350 | 355 | 305, 345 | 300 sh,b 360 | 345 | 295 sh,b 345 |
| Band Gap/eV | 3.08 | 3.20 | 2.88 | 3.12 | 3.14 | 3.20 |
| PL λem CH3OH/nm | 502 | 500 | 540 | 471 | 461 | 461 |
| PL λem CH3CN/nm | 451, 481 | 505 | 454 | 547 | 445 | 448 |
| PL λem film/nm | 472 | 495 | — | 513 | 530 | 468 |
| ΦF (%)a | 1.2 | 1.1 | 15.0 | 0.9 | 1.1 | 0.85 |
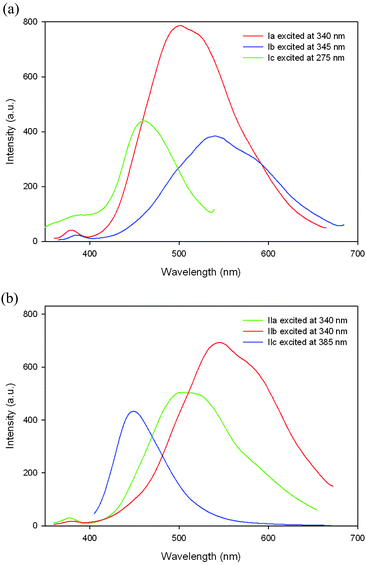 | ||
| Fig. 5 Emission spectra of (a) polymers Ia–Ic in methanol and (b) polymers IIa–IIc in acetonitrile at various excitation wavelengths. | ||
To determine effectively the potential for optoelectronic applications of these ionic polymers, we studied their optical properties in the solid state. Fig. 6a shows thin film emissions of the tosylate polymers Ia and Ic cast from acetonitrile. Polymer Ia exhibited λem at 472 nm when excited with 335 nm light. It showed a hypsochromic shift when compared to that of its solution spectrum. This result is consistent with the results of other poly(pyridinium salt)s reported previously.9 Interestingly, polymer Ic showed a light emission peak at 530 nm, when excited with 345 nm light in thin film, which was bathochromically shifted compared to that in solution spectrum. This result suggested that in the solid state it was more ordered than that in solution, which is common for many neutral light-emitting polymers.1 This unusual behavior is attributed to the long flexible carbon chain present in polymer Ic, which allowed an efficient polymer packing. Fluorescence of polymer Ib was too weak to measure in the thin film state. Fig. 6b displayed the photoluminescence spectra of polymers IIa–IIb in thin films cast from acetonitrile. They showed λem at 495, 513 and 468 nm when monitored with 340, 350 and 385 nm wavelength of light, respectively. Polymer IIa and IIb showed slightly hypsochromically shifted emissions when compared to those of their solution spectra in acetonitrile. On the other hand, polymer IIc showed slightly bathochromically shifted emission to that of its solution spectrum similar to polymer Ic. Exchange of the counterion had somewhat variable effect on the fluorescence properties in the thin film state. Polymers Ia and IIa exhibited a bathochromic shift of 22 nm, while polymers Ic and IIc showed a hypsochromic shift of 62 nm. This difference in light emission implied that the counterion in conjuction with the variation of chemical structures of polymer chains were responsible for the emission of light of variable wavelengths.
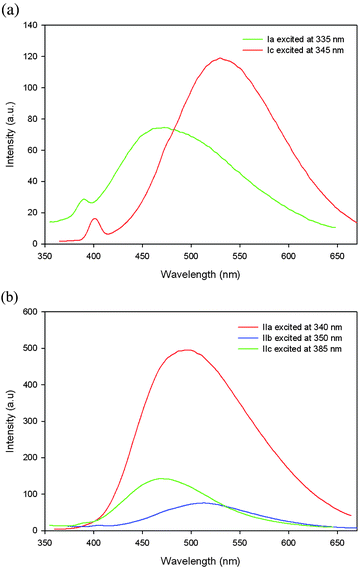 | ||
| Fig. 6 Emission spectra of thin films of (a) polymers Ia,Ic and (b) polymers IIa–IIc cast from acetonitrile at various excitation wavelengths. | ||
Conclusions
Six novel poly(pyridinium salt)s with tosylate and triflimde counterions were prepared via either the ring-transmutation or the metathesis reaction. Their chemical structures were confirmed by using NMR, FTIR and elemental analysis. Their inherent visicosities ranged from 0.65–1.29 dL/g and their weight-average molecular weights (Mw) ranged from 78,000 to 194,000 as determined by GPC measurements. The thermal stabilities of these polymers depended strongly on their counterions and were as high as 447 °C. The conjugated ionic polymers in this series did not have detectable glass transitions before their decomposition temperatures, but the polymers containing flexible twelve methylene units in their main chain had also high Tgs. They had good solubilities in common organic solvents such as methanol, acetonitrile, and DMSO, which are essential for their processability in preparation of thin films. Moreover, they exhibited full-grown lyotropic LC textures in several organic solvents.All of these polymers emitted blue and green light in both solutions and solid states. Exchange of the counterion from tosylate to triflimide affected the light emission in both solutions and films. Their fwhm values were over 100 nm suggesting that their photoluminescent properties stem from the multiple chromophores present in each of these polymers. Their quantum yields were comparable to those of π-conjugated light-emitting polymers.1
The combination of high molecular weights, good thermal stability, high Tgs, good solubility in common organic solvents, lyotropic LC properties and light emission in both solutions and solid states makes these polymers as attractive materials for various electronic and opto-electronic applications such as OLEDs, OFETs, organic photovoltaic cells.1 Since they have ionic charges they could also be used for light-emitting electrochemical cells.11 Moreover, their thermal and optical properties could be fine tuned by exchange of the counterion as required. Finally, these polymers could serve as polycations with appropriate polyanions to produce multilayered nanoassemblies by either dipping or spin-assisted methods.10
Experimental
Monomer synthesis
The 4,4′-(1,4-phenylene)bis(2,6-diphenylpyrylium)ditosylate, M, was synthesized according to the known procedure.9a The synthesis of the three bisquinoline monomers will be reported elsewhere, but a synthetic scheme is provided for their preparation in the ESI.†Polymer synthesis
Polymer Ia. Found: C, 76.00; H, 5.49; N, 4.27; S, 4.71. Calc. for C84H60N4O6S2 (1285.56): C, 78.48; H, 4.70; N, 4.36; S, 4.99%.
Polymer Ib. Found: C, 75.84; H, 4.88; N, 4.19; S, 5.17. Calc. for C90H64N4O6S2 (1361.66): C, 79.84; H, 4.74; N, 4.11; S, 4.71%.
Polymer Ic. Found: C, 76.80; H, 6.36; N, 3.47; S, 4.48. Calc. for C108H92N4O8S2 (1669.06): C, 77.72; H, 5.56; N, 3.36; S, 3.84%.
Polymer IIa. Found: C, 60.66; H, 3.61; N, 5.68; S, 7.77. Calc. for C74H46N6O8S4F12 (1534.42): C, 57.93; H, 3.02; N, 5.48; S, 8.36%.
Polymer IIb. Found: C, 61.28; H, 3.56; N, 5.50; S, 8.48. Calc. for C80H60N6O8S4F12 (1579.54): C, 60.83; H, 3.19; N, 5.32; S, 8.12%.
Polymer IIc. Found: C, 63.80; H, 4.13; N, 4.61; S, 7.40. Calc. for C98H78N6O10S4F12 (1855.97): C, 63.42; H, 4.24; N, 4.53; S, 6.91%.
Polymer characterization
The 1H and 13C NMR spectra were recorded with Varian NMR 400 spectrometer equipped with three RF channels operating at 400 MHz and 100 MHz, respectively. Polymer solutions were prepared by dissolving 35–40 mg of polymers per millilitre of d6-DMSO with TMS as an internal standard. FTIR spectra were recorded with a Shimadzu spectrometer. Polymer samples were prepared by coating NaCl plates with polymers, which were subsequently vacuum dried at 70 °C overnight. Polymers phase transitions were measured using TA 2100 differential scanning calorimetry (DSC) under nitrogen at a rate of 10 °C min−1. The temperature axis of the DSC thermogram was calibrated with reference standards of high purity indium and tin before use. Polymer samples of 8–10 mg were used in these measurements. Thermal stability of each of these polymers was analyzed through thermogravimetric analysis (TGA) using TA 2100 instrument at a rate of 10 °C min−1 under nitrogen using samples no less than 10 mg. Liquid-crystalline properties both lyotropic and thermotropic were assessed using a polarized optical microscope (POM) Nikon Labophot 2 equipped with crossed polarizers and hot stage. The UV-Vis absorption spectra of polymer solutions in organic solvents were recorded using Varian Cary 50 Bio UV-Visible spectrophotometer in quartz cuvettes. Their photoluminescent properties in both solutions and film states were analyzed using a Perkin Elmer LS-55 luminescence spectrophotometer. Quantum yields were analyzed by adjusting the solution absorption using the UV-Vis to ca. 0.05 at 350 nm wavelength, the outputs were measured using the luminescence spectrophotometer at the same wavelength and compare it to known 9,10-diphenylanthracene standard using eqn (1):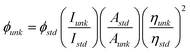 | (1) |
Acknowledgements
P.K.B. acknowledges the University of Nevada Las Vegas (UNLV) for New Investigation Award (NIA), Planning Initiative Award (PIA), and Applied Research Initiative (ARI) grants, and the donors of the Petroleum Research Fund, administered by the American Chemical Society, and Research Corporation for an award (CCSA# CC5589) for the support of this research. A.K.N. acknowledges the Graduate College (UNLV) for providing him a Nevada Stars Graduate Assistantship for the period of 2006–2008.Notes and references
- (a) J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burns and A. B. Holmes, Nature, 1990, 347, 539 CrossRef CAS; (b) A. Kraft, A. C. Grimsdale and A. B. Holmes, Angew. Chem., Int. Ed., 1998, 37, 402 CrossRef; (c) R. H. Friend, R. W. Gymer, A. B. Holmes, J. H. Burroughes, R. N. Marks, C. Taliani, D. D. C. Bradley, D. A. D. Santos, J. L. Bredas, M. Logdlund and W. R. Salaneck, Nature, 1999, 397, 121 CrossRef CAS; (d) U. Mitschke and P. Bauerle, J. Mater. Chem., 2000, 10, 1471 RSC; (e) L. Akcelrud, Prog. Polym. Sci., 2003, 28, 875 CrossRef CAS; (f) M. D. McGehee and A. J. Heeger, Adv. Mater., 2000, 12, 1655 CrossRef CAS.
- A. P. Kulkarni, C. J. Tonzola, A. Babel and S. A. Jenekhe, Chem. Mater., 2004, 16, 4556 CrossRef CAS.
- (a) Y. Zhu, A. P. Kulkarni, P.-T. Wu and S. A. Jenekhe, Chem. Mater., 2008, 20, 4200 CrossRef CAS; (b) C. J. Tonzola, A. P. Kulkarni, A. P. Gifford, W. Kaminsky and S. A. Jenekhe, Adv. Funct. Mater., 2007, 17, 863 CrossRef CAS.
- (a) J. K. Stille, Macromolecules, 1981, 14, 870 CrossRef CAS; (b) D.-J. Liaw, K.-L. Wang and F.-C. Chang, Macromolecules, 2007, 40, 3568 CrossRef CAS; (c) A. Abdolmaleki, Polym. Degrad. Stab., 2007, 92, 292 CrossRef CAS.
- (a) A. P. Monkman, L.-O. Palsson, R. W. T. Higgins, C. Wang, M. R. Bryce, A. S. Batsanov and J. A. K. Howard, J. Am. Chem. Soc., 2002, 124, 6049 CrossRef CAS; (b) G.-S. Liou, Y.-L. Yang and Y. O. Su, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2587 CrossRef CAS.
- H. Han, P. R. Vantine, A. K. Nedeltchev and P. K. Bhowmik, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1541 CrossRef CAS.
- A. R. Katritzky, R. D. Tarr, S. M. Heilmann, J. K. Rasmussen and L. R. Krepski, J. Polym. Sci., Part A: Polym. Chem., 1988, 26, 3323 CrossRef CAS.
- (a) F. W. Harris, K. C. Chuang, S. A. X. Huang, J. J. Janimak and S. Z. D. Cheng, Polymer, 1994, 35, 4940 CrossRef CAS; (b) S. A. X. Huang, K. C. Chuang, S. Z. D. Cheng and F. W. Harris, Polymer, 2000, 41, 5001 CrossRef CAS.
- (a) P. K. Bhowmik, R. A. Burchett, H. Han and J. J. Cebe, Macromolecules, 2001, 34, 7579 CrossRef CAS; (b) P. K. Bhowmik, R. A. Burchett, H. Han and J. J. Cebe, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2710 CrossRef CAS; (c) P. K. Bhowmik, H. Han, J. J. Cebe, I. K. Nedeltchev, S.-W. Kang and S. Kumar, Macromolecules, 2004, 37, 2688 CrossRef CAS; (d) P. K. Bhowmik, H. Han and A. K. Nedeltchev, Polymer, 2006, 47, 8281 CrossRef CAS; (e) P. K. Bhowmik, H. Han and A. K. Nedeltchev, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1028 CrossRef CAS; (f) P. K. Bhowmik, S. Kamatam, H. Han and A. K. Nedeltchev, Polymer, 2008, 49, 1748 CrossRef CAS; (g) P. K. Bhowmik, H. Han, A. K. Nedeltchev, H. D. Mandal, J. A. Jimenez-Hernandez and P. M. McGannon, Polymer, 2009, 50, 3128 CrossRef CAS.
- (a) G. Decher, Science, 1997, 277, 1232 CrossRef CAS; (b) Z. Tang, N. A. Kotov, S. Maganov and B. Ozturk, Nat. Mater., 2003, 2, 413 CrossRef CAS; (c) C. Jiang, S. Markutsya, Y. Pikus and V. V. Tsukruk, Nat. Mater., 2004, 3, 721 CrossRef CAS; (d) C. J. Lefaux, J. A. Zimberlin, A. V. Dobrynin and P. T. Mather, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 3654 CrossRef CAS.
- (a) Q. Pei, G. Yu, C. Zhang, Y. Yang and A. J. Heeger, Science, 1995, 269, 1086 CrossRef CAS; (b) J. Slinker, D. Bernards, P. L. Houston, H. D. Abruña, S. Bernhard and G. G. Malliaras, Chem. Commun., 2003, 2392 RSC; (c) D. A. Bernards, S. Flores-Torres, H. D. Abruña and G. G. Malliaras, Science, 2006, 313, 1416 CrossRef CAS.
- A. R. Katritzky, R. T. C. Brownlee and G. Musumarra, Tetrahedron, 1980, 36, 1643 CrossRef CAS.
- (a) P. D. Sybert, W. H. Beever and J. K. Stille, Macromolecules, 1981, 14, 493 CrossRef CAS; (b) E. K. Zimmermann and J. K. Stille, Macromolecules, 1985, 18, 321 CrossRef CAS; (c) A. K. Agrawal and S. A. Jenekhe, Chem. Mater., 1993, 5, 633 CrossRef CAS.
- (a) D. Tanner, J. A. Fitzgerald and B. R. Phillips, Angew. Chem., Int. Ed. Engl., 1989, 28, 649 CrossRef; (b) J. Lin and D. Sherrington, Adv. Polym. Sci., 1994, 111, 177 CAS.
- (a) A. K. Agrawal and S. A. Jenekhe, Chem. Mater., 1992, 4, 95 CrossRef CAS; (b) A. K. Agrawal and S. A. Jenekhe, Chem. Mater., 1993, 5, 633 CrossRef CAS; (c) A. K. Agrawal and S. A. Jenekhe, Chem. Mater., 1996, 8, 579 CrossRef CAS; (d) S. A. Jenekhe, X. Zhang, X. L. Chen, V. E. Choong, Y. Gao and B. R. Hsieh, Chem. Mater., 1997, 9, 409 CrossRef CAS; (e) X. Zhang, A. S. Shetty and S. A. Jenekhe, Acta Polym., 1998, 49, 52 CrossRef CAS; (f) X. Zhang, A. S. Shetty and S. A. Jenekhe, Macromolecules, 1999, 32, 7422 CrossRef CAS; (g) X. Zhang and S. A. Jenekhe, Macromolecules, 2000, 33, 2069 CrossRef CAS.
- (a) Y. Shirota, J. Mater. Chem., 2000, 10, 1 RSC; (b) Y. Shirota, J. Mater. Chem., 2005, 15, 75 RSC; (c) Y. Shirota and H. Kageyama, Chem. Rev., 2007, 107, 953 CrossRef CAS; (d) P. Strohriegl and J. V. Grazulevicius, Adv. Mater., 2002, 14, 1439 CrossRef CAS; (e) S. F. Swallen, K. L. Kearns, M. K. Mapes, Y. S. Kim, R. J. McMahon, M. D. Ediger, T. Wu, L. Yu and S. Satija, Science, 2007, 315, 353 CrossRef CAS; (f) G.-S. Liou, Y.-L. Yang and Y. O. Su, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2587 CrossRef CAS.
- T. L. Greaves and C. J. Drummond, Chem. Rev., 2008, 108, 206 CrossRef CAS.
- (a) H. Hong, R. Sfez, E. Vaganova, S. Yitzchaik and D. Davidov, Thin Solid Films, 2000, 366, 260 CrossRef CAS; (b) C. Wang, M. Kilitziraki, J. A. H. MacBride, M. R. Bryce, L. E. Horsburgh, A. K. Sheridan, A. P. Monkman and I. D. W. Samuel, Adv. Mater., 2000, 12, 217 CrossRef CAS; (c) J. M. Hancock and S. A. Jenekhe, Macromolecules, 2008, 41, 6864 CrossRef CAS.
- S. Fery-Forgues and D. Lavabre, J. Chem. Educ., 1999, 76, 1260 CrossRef CAS.
Footnotes |
| † This article is dedicated to Professor Vernon F. Hodge with best wishes on the occasion of his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available: Schemes S1 and S2, NMR spectra and FTIR spectra. See DOI: 10.1039/c0py00037j |
| This journal is © The Royal Society of Chemistry 2010 |