Chemistry, chances and limitations of the radical ring-opening polymerization of cyclic ketene acetals for the synthesis of degradable polyesters
Seema
Agarwal
*
Philipps-Universität Marburg, Fachbereich Chemie, Hans-Meerwein Strasse, D-35032, Marburg, Germany. E-mail: seema@chemie.uni-marburg.de; Fax: +00 49 6421 2825785; Tel: +00 49 6421 2825755
First published on 23rd March 2010
Abstract
This article highlights the progress in the field of radical ring-opening polymerization (RROP) of cyclic ketene acetals for the synthesis of functionalised polyesters, (bio)degradable vinyl polymers, and speciality designed degradable polymers. The method of radical ring-opening polymerization offers the advantage in either making special functional polyesters which are not possible by conventional methods or for making polyesters for special applications where low volume shrinkage is of utmost importance. Furthermore, the beauty of radical ring-opening polymerization is in making a new class of degradable materials called poly(vinyl-co-ester)s. Having ester linkages distributed onto the vinyl polymer backbone is emphasised, no other method can combine vinyl polymer backbone with ester units in a random way. The method has opened new opportunities of using the known biomaterials for degradable applications. Other possibilities and challenges in using this method are making specialised degradable materials like ionomers and polymeric-inorganic hybrid materials, which are also discussed in detail.
 Seema Agarwal | Seema Agarwal has worked in the department of chemistry at Philipps-University, Marburg Germany since the year 2000. She did a double masters in chemistry and polymers science and technology and later obtained her PhD from the Indian Institute of Technology, Delhi, India. Her scientific professional career started in India by working as Scientist for about 9 years and then shifted to Germany. She was a Humboldt Fellow from 1997–1999 in the laboratory of Prof. Andreas Greiner, Germany. She finished habilitation in Macromolecular Chemistry in 2007 (Philipps-University, Marburg, Germany). Her research interests are: biodegradable and bio polymers, structure-property-relationship studies of polymers, NMR of polymers, smart polymers, controlled polymerizations and electrospinning. |
1 Introduction
Physical and chemical stability of the polymers is one of the properties that makes them suitable for many different applications. But in recent times the stability of conventional synthetic polymers to the degrading action of different agencies like living system, chemicals or light is considered as problem in several domains where they are used for a limited period of time before becoming waste. Furthermore, the importance and relevance of (bio)degradable polymers is reinforced due to the potential applications in various medical and non-medical fields like agriculture, medicine, pharmacy, biomedical, and also as environmentally friendly materials.1 This scenario makes the studies related to the design, synthesis and properties of biodegradable polymers extremely important and relevant. In fact, a large amount of research has been conducted all over the world to find out methods either to convert synthetic plastic waste into biodegradable and compostable material or to make new or modify the existing biodegradable polymers.Large number of commercially available biodegradable materials for medical and non-medical applications are based on polyesters like homopolymers or copolymers of glycolide, lactide, ε-caprolactone, and dioxanone etc.2–8 In general, the conventional synthesis of these polyesters is either by ring-opening polymerization of cyclic esters or by condensation polymerization (condensation of the diacids/diacid derivatives with diols) (Scheme 1). Lot of literature is already available and still increasing day by day on the use of different initiator systems like cationic, anionic and metal catalysts, as well as on mechanisms of ring-opening polymerization (ROP) of cyclic esters for the synthesis of polyesters. Recent advances in ROP of lactones and related compounds can be seen in details in review articles.9,10
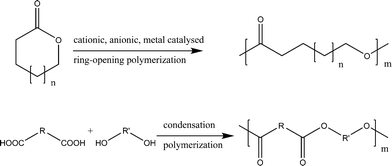 | ||
| Scheme 1 Conventional synthetic routes to polyesters: ring-opening polymerization of cyclic esters and condensation polymerization. | ||
Furthermore, the synthesis of aliphatic polyesters by condensation polymerization has also been well explored.11,12 The corresponding high molecular weight polyesters cannot be obtained by direct condensation of the related carboxylic acids and diols because of the reversibility of the condensation reactions, cyclization reactions, and the high conversion of the reaction required.
Another route for the synthesis of polyesters, which is not very well explored, is by radical ring-opening polymerization (RROP) of cyclic ketene acetals. Although the method offers the advantage of using free radical chemistry, low volume shrinkage etc. it could not attract many researchers in the past. After the pioneering work of Bailey et al.,13 there was slow progress in this field. Recently, the synthesis of degradable polymers has raised interest in this promising synthetic method. This is the only method that allows the combination of ester units with the C–C back bone of vinyl polymers in a random way to generate a new class of materials (poly(vinyl-co-ester)s). This article highlights the importance of RROP including its chemistry for the synthesis of functionalised polyesters, (bio)degradable vinyl polymers, and speciality designed degradable polymers. The resulting polymers besides being (bio)degradable could also be useful for variety of speciality applications like precision materials, adhesives, ionomers and thermoplastic elastomers etc. This review is not a comprehensive collection of related literature but an overview of concept and progress in the field of RROP for the synthesis of polyesters and polyester based new materials.
2 Chemistry of polyester synthesis by radical ring-opening polymerization
First, Bailey and co-workers, and later others, studied the free radical ring-opening polymerization (RROP) of different cyclic ketene acetals (CKAs) using conventional radical initiators like azobisisobutyronitrile (AIBN) and t-butyl peroxide (TBPO) generating polyesters.13 Cyclic ketene acetals are the isomers of the corresponding cyclic lactones and can undergo radical addition at the vinyl double bond with subsequent possibilities of either ring-opening or ring retaining or a combination of the two (Scheme 2).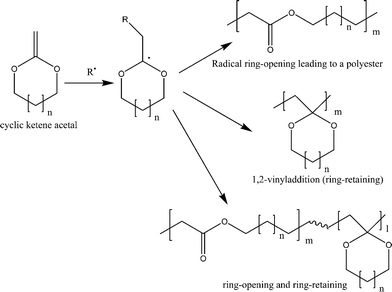 | ||
| Scheme 2 Radical ring-opening polymerization (RROP) of cyclic ketene acetals: different possibilities during polymerization. | ||
The ring-opening after radical attack gives the polyester that is the same as that from the ring-opening of the corresponding isomeric cyclic ester. For example, the radical ring-opening polymerization of CKA 2-methylene-1,3-dioxepane gives aliphatic degradable polyester, poly(caprolactone) (PCL), which is conventionally obtained by metal catalysed ring-opening of caprolactone (Scheme 3). During the free radical reaction of cyclic ketene acetals, the probability of ring-opening over a 1,2-vinyl addition reaction depends on the monomer concentration, temperature, ring-size, substituents etc. The driving force for the ring-opening, in general, is the relief of ring strain or the formation of ester linkages, as the carbon oxygen bond is about 40 Kcal mol−1 more stable than a carbon–carbon double bond.14
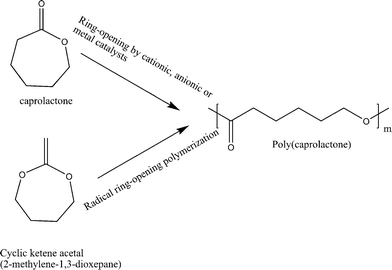 | ||
| Scheme 3 Synthetic routes to the aliphatic biodegradable polymer poly(caprolactone) (PCL) by radical ring-opening polymerization of cyclic ketene acetal and metal/anionic/cationic ring-opening polymerization of caprolactone. | ||
3 Radical ring-opening polymerization of different CKAs
Many different cyclic ketene acetals (Table 1) with varied ring sizes and substituents were tried for radical ring-opening polymerization either with an aim of studying the effect of structure, ring size etc. on the ring-opening reaction or to make a functional polyester.| Cyclic ketene acetal | Reference(s) | |
|---|---|---|
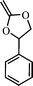
|
2-methylene-4-phenyl-1,3-dioxalane (1) | 15,20 |
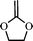
|
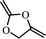
2-methylene-1,3-dioxalane (2) | 15 |
|
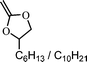
2–methylene-4-hexyl/decyl-1,3-dioalane (3) | 16 |
|
2,4-dimethylene-1,3-dioxalane (4) | 17 |
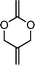
|
2,5-dimethylene-1,3-dioxane (5) | 17 |
|
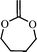
2-methylene-1,3-dioxepane (6) | 18,20,21,22,23 |
|
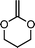
2,5-methylene-1,3-dioxane (7) | 18 |
|
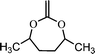
4,7-dimethyl-2-methylene-1,3-dioxepane (8) | 19 |
|
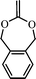
5,6-benzo-2-methylene-1,3-dioepane (9) | 19 |
|
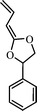
4-phenyl-2-propenylene-1,3-dioxalane (10) | 24 |
|
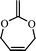
2-methylene-1,3-dioxe-5-pene (11) | 25 |
|
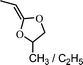
2-ethylidene-4-methyl/ethyl-1,3-dioxane (12) | 26 |
|
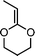
2-ethylidene-1,3-dioxane (13) | 26 |
|
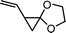
1-vinyl-4,7-dioxaspiro[2,4]heptane (14) | 27 |
|
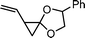
1-vinyl-5-phenyl-4,7-dioxaspiro[2,4]heptane (15) | 27 |
|
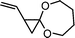
1-vinyl-4,9-dioxaspiro-[2.6]nonane (16) | 27 |
|
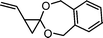
1-vinyl-6,7-benzo-4,9-dioxaspiro[2.6]nonane (17) | 27 |
|
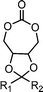
9,9-disubstituted-4-methylene-3,5,8,10-tetraoxabicyclo[5.3.0]decane (18) | 28 |
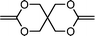
|
3,9-bis-methylene-2,4,8,10-tetraoxa-spiro[5,5]undecane (19) | 29,30 |
|
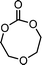
2-methylene-1,3,6-trioxocane (20) | 31 |
The CKA 2-methylene-4-phenyl-1,3-dioxolane (1), a substituted five membered ring is shown to undergo quantitative and regioselective radical ring-opening at all temperatures from 60 to 150 °C to give polyester, poly((γ-β-phenyl) butyrolactone).15 The driving force for quantitative ring-opening was the formation of a stable secondary benzyl radical. This ring-opening reaction can be nicely compared with the parent unsubstituted five membered CKA 2-methylene-1,3-dioxolane (2) which underwent ring-opening depending upon the temperature of polymerization. It was never a quantitative ring-opening which could be due to the formation of a primary unstable radical after ring-opening reaction. The high temperature favored the ring-opening reaction to give polyester (50% ring-opening at 60 °C and 83% at 125 °C) and the extent of ring-opening could be increased by decreasing the concentration of the monomer. The case with alkyl substituted five membered CKA (like hexyl and decyl substituted derivatives, 2-methylene-4-hexyl/decyl-1,3-dioxalane (3)), was similar with respect to the effect of temperature.16 Also, unlike phenyl substituted CKA (1), alkyl substituents provided no regioselectivity for ring-opening. These facts indicated that a radical stabilizing group onto the ring has a tremendous role in quantitative ring-opening giving polyesters. Furthermore, Endo and co-workers reported the preparation and ring-opening polymerization of exo-methylene substituted five membered cyclic ketene acetals like 2,4-dimethylene-1,3-dioxolane (4) and six-membered 2,5-dimethylene-1,3-dioxane (5) which are expected to undergo radical ring-opening polymerization through stable allyl radicals.17 The resulting polyesters would be highly desirable as reactive polymers since the exo methylene group could be used for cross-linking, grafting and addition of other groups etc. but the results of polymerization were not very encouraging. At low temperature (60 °C, AIBN initiator) 2,4-dimethylene-1,3-dioxolane (4) gave a mixture of ring-opened and ring retained structures and at 120 °C in DMF produced quantitative ring-opening but very low molecular weight (Mn = 1650) and branched structures. 2,5-Dimethylene-1,3-dioxane (5) only underwent partial ring-opening, forming a cross-linked structure, which may be due to the participation of allyl bonds during radical polymerization.17
The extent of ring-opening is also dependent upon the ring size and the final polymer structure is decided by the relative rates of ring-opening isomerization and propagation (Scheme 4). For a seven membered ring, 2-methylene-1,3-dioxepane (6), 100% ring-opening was observed at low temperatures (50 °C) giving a very well known biodegradable polyester poly(caprolactone) PCL.18 The reason was the relief of the ring strain in the seven membered ring, compared with the five and six membered rings and also the increase in steric hinderance of the non-ring opened radical of the seven membered ring in relation to its five- and six-membered homologs (7). The other seven membered rings like cis- and trans-4,7-dimethyl-2-methylene-1,3-dioxepane (8) and 5,6-benzo-2-methylene-1,3-dioxepane (9) also gave quantitative ring-opening at 120 °C.19 The increased stability of the ring-opened radical and the increase in steric hinderance to direct non-ring-opened polymerization are believed to promote the extent of free-radical ring-opening during polymerization.
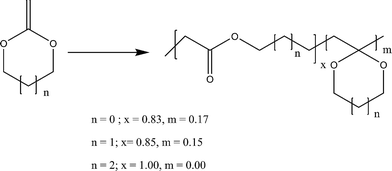 | ||
| Scheme 4 The effect of ring size on radical ring-opening polymerization.18 | ||
Attempts were also carried out for photopolymerization of CKAs for the synthesis of polyesters and a quantitative ring-opening is shown for the polymerizations of 2-methylene-1,3-dioxepane (6)20a and 2-methylene-4-phenyl-1,3-dioxolane (1)20b using 2-ethylanthraquinone (λmax = 255,327 nm) and benzoin isopropylether (λmax = 247,326 nm). This could be of great interest for low shrinkage photoresist polyesters. In general, a primary cause of contraction in volume during polymerization is due to the reduction of van der Waals distances between monomer molecules due to covalent bond lengths. When bond formation is accompanied by bond breaking, the shrinkage of the process is substantially reduced. During the ring-opening polymerization of cyclic monomers, one ring is opened in each monomeric unit, and thus one bond is broken for each new bond formed thereby showing low volume shrinkages. In general, the development of systems that shows low shrinkage during polymerizations is highly relevant with respect to applications as matrix resins or dental restoration, adhesives, coatings, and precision castings etc. The volume shrinkage of 2-methylene-1,3-dioxepane on photopolymerization was only 7.2% (one can easily compare with very high volume shrinkage of vinyl polymers like poly(methyl methacrylate) which is about 21%).
Most previous studies on RROP of CKAs have concentrated only on the synthetic aspects of monomer and polymer synthesis without commenting on the detailed characterization of the polymers. The detailed structural characterization of the polyester (poly(caprolactone)) made by RROP of 2-methylene-1,3-dioxepane (6) was provided first by Jin and Gonsalves in 1997.21 They showed the presence of 1,4- and 1,7-H-transfer reactions leading to branched structures with about 20% branch density for RROP of (6) at 50 °C for 72 h using AIBN as the initiator (Mn = 42,000) (Scheme 5). The growing active center at the chain end of the polymer is a primary radical (similar to the unstable polyethylene growing radical) and therefore led to backbiting reactions of the type seen during free radical polymerization of ethylene also. The branches affected the crystal structure and unlike semicrystalline PCL made by conventional metal catalysed ROP of caprolactone, the PCL made by RROP (named as PCLB) was amorphous with glass transition temperature of −57 °C. The branching in the polymer structure causes higher concentration of the end-groups thereby leading to the increased dislocations in the lamellar thickening process, a known effect, and hence the imperfect crystals or no crystallization. The branching is dependent upon the temperature of polymerization and later the RROP of (6) at 120 °C showed the absence of 1,4-H-abstraction reactions. The presence of only 1,7-H-abstraction reactions were confirmed using 2D-NMR techniques and this PCL (Mw = 30,000) showed a Tg of −61 °C and two melting peaks around 25 °C and 36 °C.22 Recently we have made use of these properties of branched PCL in modifying and improving the properties of conventional semicrystalline PCL.23 Semi-crystalline PCL and amorphous PCLB made by RROP of (6) have similar chemical repeat unit structures and the solution blending of the two led to some interesting property change in PCL keeping its glass transition temperature and melting temperature almost the same. The blending of semi-crystalline PCL and the amorphous PCLB showed synergistic effect on crystallinity leading to the increased crystallinity of PCL and induced crystallinity of amorphous PCLB, minima in the % elongation and maxima in spherulite size depending upon the ratio of amorphous and crystalline part. There was a drastic increase in the compostability as compared to pure PCL on adding even low % of amorphous PCLB in the blends without affecting the thermal stability (Fig. 1). The addition of PCLB also affected the transparency and it increased exponentially from about 8% at 650 nm for PCL to about 45% for a blend with a composition of PCL![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PCLB 30:70. These new materials could be of high interest for the packaging industry or other applications requiring fast degrading transparent polymer.
:PCLB 30:70. These new materials could be of high interest for the packaging industry or other applications requiring fast degrading transparent polymer.
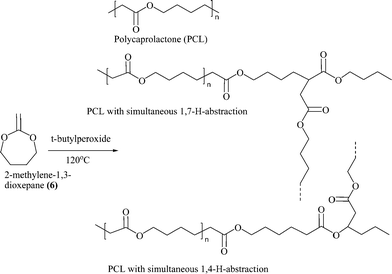 | ||
| Scheme 5 Back-bitting leading to the branched structures in the poly(caprolactone) (PCL) made by radical ring-opening polymerization (RROP) of 2-methylene-1,3-dioxepane (6). | ||
 | ||
| Fig. 1 An increased degradation rate at higher poly(caprolactone) made by radical ring-opening polymerization (PCLB) content in the blends of poly(caprolactone) (PCL) and PCLB was observed due to the reduced overall crystallinity.23 | ||
4 Functional polyesters by radical ring-opening polymerization
The use of substituted designed CKAs for RROP could lead to the synthesis of functional polyesters. RROP has made possible the synthesis of many functional polyesters which otherwise are either not possible or difficult to make by the conventional routes. An interesting CKA tried for radical ring-opening polymerization was the phenyl substituted vinylketene cyclic acetal (4-phenyl-2-propenylene-1,3-dioxolane) (10).24 Moderate molecular weight (Mn = 26,900) functional trans-α,β-unsaturated polyester was prepared in benzene at 55 °C (Scheme 6), which otherwise is not accessible by state-of-the-art synthesis.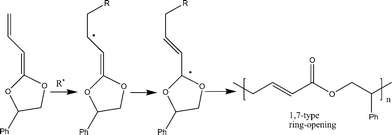 | ||
| Scheme 6 Radical ring-opening polymerization (RROP) mechanism of 4-phenyl-2-propenylene-1,3-dioxolane.24 | ||
Another attempt to synthesize a similar kind of polyesters with double bonds in the main chain using double bond containing cyclic ketene acetal like 2-methylene-1,3-dioxe-5-pene (11) was not successful.25 Other monomers tried for the synthesis of functional polyesters were: 2-ethylidene substituted CKAs (12,13)26 (which resulted in partial ring-opening) and vinylcyclopropane CKAs by Endo and co-workers (14–17).27 The new hybrid monomers (vinyl cyclopropanes) having both vinylcyclopropane and CKA structure were expected to have double ring opening in presence of the radical initiator and leading to the formation of unsaturated polyesters (Scheme 7) but contrary to it the end products were a mixture of single ring-opened structure, double ring opened structures cyclobutane and lactone ring containing units.
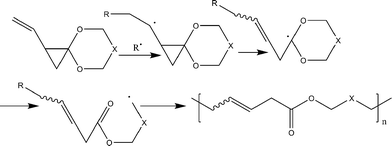 | ||
| Scheme 7 Radical ring-opening polymerization of vinylcylopropane cyclic ketene acetals.27 | ||
Some interesting bicyclic CKAs like bicyclic 2-methylene-1,3-dioxepane (18) were also polymerised by radical initiators at different temperatures by Moszner et al.28 The idea of using these specialised monomers was to have negative volume shrinkage during polymerization. Such materials are always desired for adhesive and composite applications as explained earlier. Also, bifunctional cyclic ketene acetals like 3,9-bis-methylene-2,4,8,10-tetraoxa-spiro[5,5]undecane (19) were studied for RROP as low shrinkage cross-linkers.29,30 Its copolymerization with 9,9-dimethyl-4-methylene-3,5,8,10-tetraoxabicycyclo[5.3.0]decane (18) produced cross-linked rubbery gel polyesters having shrinkage in the range 0.2–9.9% depending upon the amount of the cross-linker.
Synthesis of poly(ester ether)s is also possible by radical polymerization of 2-methylene-1,3,6-trioxocane (MTC) (20). It is made up of alternating butyrolactone and ethyleneoxide units (Scheme 8).31 The synthesis of this polyester ether is not possible by any other route in a simple way.
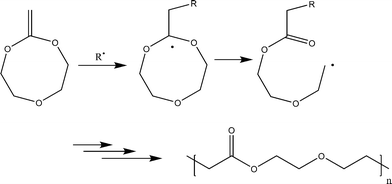 | ||
| Scheme 8 Radical-ring-opening polymerization of 2-methylene-1,3,6-trioxocane (MTC) (20) for the synthesis of poly(ester ether).31 | ||
Partially fluorinated and perfluorinated dioxolane and dioxane derivatives (Fig. 2) were studied for RROP by Okamoto and co-workers.32 This study was carried out to study the influence of fluorine on RROP and to make fluorinated polyesters in a simple way. The partially fluorinated monomer 2-difluoromethylene-1,3-dioxolane yielded only vinyl addition product as compared to the hydrocarbon analogue which provided 50% ROP product at 60 °C. The same was true for the fully fluorine substituted perfluoro-2-methylene-4-methyl-1,3-dioxolane. In general, the monomers with fluoro-substituents have fewer tendencies toward ring-opening.
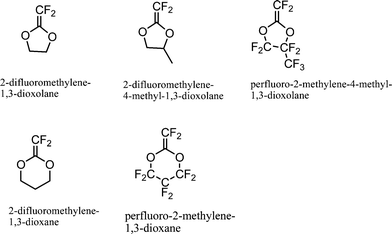 | ||
| Fig. 2 Fluorinated cyclic ketene acetals studied for radical ring-opening polymerization.32 | ||
5 Polyesters with controlled chain lengths by living radical ring-opening polymerization
Controlled radical polymerization techniques like nitroxide mediated polymerization,33 atom transfer radical polymerization34 and reversible addition fragmentation transfer polymerization, RAFT,35 in general, provide the opportunity to control the polymer chain lengths, polydispersities and gives the possibility of making different macromolecular architectures like block, graft and star polymers. These common controlled polymerization techniques could be extended to radical ring-opening polymerization providing polyesters with controlled chain lengths and in different macromolecular architectures. It is not possible to form polyesters with controlled chain lengths by conventional condensation polymerization. Although there are methods known for the living ring-opening polymerization of cyclic esters for the synthesis of aliphatic polyesters but controlled RROP provides the advantage of not requiring very stringent conditions of anionic or metal catalyzed polymerizations of cyclic esters. The first example of a living RROP of 2-methylene-1,3-dioxepane (6) for the synthesis of poly(caprolactone) (PCL) was provided by Wei et al. using TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy)36,37 as chain transfer agent. It underwent quantitative ring-opening to afford PCL with polydispersity less than 1.5. Later ATRP and RAFT of various CKAs were carried out by different authors and led to ring-opening/ring-retaining depending upon the CKA structure. For example, three CKAS, 5,6-benzo-2-methylene-1,3-dioxepane (9), 2-methylene-4-phenyl-1,3-dioxolane (1) and 4,7-dimethyl-2-methylene-1,3-dioxepane (8) were compared for their ring-opening and controlled polymerization behavior using ATRP method by Yuan and Pan38a at different temperatures. The polymerizations were carried out using an α-bromobutyrate/CuBr/2,2′-bipyridine ATRP system. 5,6-Benzo-2-methylene-1,3-dioxepane (9) resulted in complete ring-opening, forming polyester with a controlled molecular weight and low polydispersities. Five membered 2-methylene-4-phenyl-1,3-dioxolane (1) and seven membered 4,7-dimethyl-2-methylene-1,3-dioxepane (8) gave a mixed structure with ring-opened and ring retained structures by ATRP in contrast to the quantitative ROP under conventional radical polymerization. The proportion of ring-opened structure increased with the increase in the temperature of atom transfer radical polymerization for 2-methylene-4-phenyl-1,3-dioxolane and the ring-opening was regioselective. The higher temperatures also provided more control with low polydispersity, for example the polydispersities of 1.18 at 140 °C and 1.42 at 80 °C were observed for 2-methylene-4-phenyl-1,3-dioxolane.38b Interestingly the catalyst system for RROP i.e. ATRP vs. conventional radical initiator changed the course of polymerization. Although there are no systematic studies reported in this direction but it gives a hint of participation of the monomers (CKAs) in the coordination process during ATRP polymerization. The mechanistic details regarding this have still not been investigated. Like ATRP, reversible addition fragmentation transfer polymerization (RAFT) of 5,6-benzo-2-methylene-1,3-dioxepane (9) also provided a completely ring-opened structure using 1-(ethoxycarbonyl)prop-1-yl-dithiobenzoate and dicumylperoxide.39Although the few attempts in the literature regarding the controlled polymerization of CKAs are promising, the field is still open and there is a need to provide a systematic study regarding the formation of high molecular weight polyesters with controlled chain lengths and chain ends.
One of the major advances in polymer syntheses has been the development of different new materials using a combination of various polymerization techniques also called multimode polymerizations. One way to modify the properties of the vinyl polymers and to introducing functionalities on the polymer backbone is the formation of the block copolymers with polyesters as one of the blocks. The block copolymers of aliphatic degradable polyesters like PCL or polylactide (PLA) and vinyl polymers are generally made by a combination of living ionic polymerization for the vinyl monomers followed by co-ordination-insertion ring-opening polymerization of cyclic esters like caprolactone/lactide has been mainly utilized. The method first requires the synthesis of hydroxyl terminated vinyl polymer, which is then converted to aluminium alkoxide macroinitiator for example, for ring-opening polymerization of cyclic esters (Scheme 9).40 Ring-opening polymerization of cyclic esters combined with atom transfer radical polymerization (ATRP) can also produce similar linear block copolymers.41 The use of polymeric initiators is another method of synthesizing block copolymers. In this class, polymeric azoinitiators42 gained considerable importance and they are generally made by the reaction of the growing polymer chain/coupling of end functionalized polymeric chain with suitably modified azo compounds generating polymeric initiators for further polymerization of a second monomer giving block copolymers. This method can also generate highly attractive polymeric materials composed of a condensation polymer segment (e.g. polyether, polyester, polyamide, polyurethane, and polysiloxane) with another polymer segment from vinyl monomers.
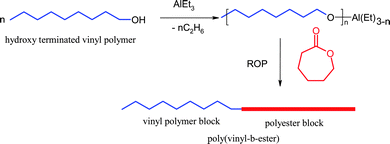 | ||
| Scheme 9 Synthetic protocol for the preparation of block copolymers (poly(vinyl-b-ester)s) by conventional ROP. | ||
Recently controlled radical ring-opening polymerization of cyclic ketene acetals have also been tried and utilized for the formation of block copolymers (poly(vinyl-b-ester)s) (Scheme 10). This method of formation of block copolymers is much simpler than the previous methods discussed above.
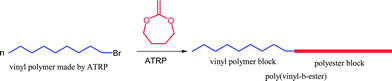 | ||
| Scheme 10 Synthesis of block copolymers (poly(vinyl-b-ester)s) using RROP. | ||
Using this method block copolymers of 5,6-benzo-2-methylene-1,3-dioxepane (9) with styrene, MMA and MA were also shown by Yuan and Pan43,44
6 Degradable vinyl addition polymers by copolymerization with CKAs
Generally, a high molecular weight polymer based on a C–C backbone, like vinyl polymers (poly(methyl methacrylate), polystyrene, polyethylene etc.), tends to be resistant to hydrolysis, oxidative cleavage, enzymatic attack, etc. and therefore is characterized as a non-(bio)degradable polymer. One of the methods of engineering degradability into these non-degradable vinyl polymers could be by designing and the judicious addition of unstable chemical linkages like ester units. However, synthesis of random copolymers of vinyl polymers having ester linkages is not a straightforward and easy task using conventional synthetic methods due to the entirely different polymerization techniques involved in their formation. Free radical ring-opening co-polymerization of cyclic ketene acetals (CKAs) with commercial vinyl polymers could be utilized for the random introduction of an ester group onto the C–C backbone of an addition polymer. It is expected to make vinyl polymers degradable due to the presence of ester linkages, besides having impact on the other physical and chemical properties, thermal stability etc. The detailed exploration of this approach is highly promising for generating an entirely new class of degradable vinyl polymers with a range of physical and chemical properties which could be considered a (bio)degradable substitute for conventional polymers in many applications depending on the end properties achieved and could also be considered a step forward in solving the problem of waste management.There are some attempts in the literature to gain the basic understanding of the formation of these new materials i.e. ester incorporated vinyl polymers using both conventional radical initiators and controlled radical polymerizations like ATRP and RAFT. Aryl (phenyl) substituted 2-methylene-1,3-dioxolane (1) with many different vinyl monomers like styrene (St), methyl methacrylate (MMA), vinyl acetate (VAc), 4-vinylpyridine gave 100% ring-opening but after long reaction times of 24 h, the % yield was in the range 51–78% under conventional radical polymerization conditions. The amount of ester units in the copolymers was always less than that in the feed as determined by NMR.15 The copolymerization of alkyl (hexyl and decyl) substituted 2-methylene-1,3-dioxolane with styrene at different temperatures is shown by Bailey et al.16 Copolymerization was carried out at 110 °C using 2 mol% of di-t-butyl peroxide for a long time (65 h) and resulted in 4 mol% of non-ring-opened structures and non-regioselective ring-opening. The seven membered CKA, 2 methylene-1,3-dioxepane (6) opens quantitatively in copolymerizations with various vinyl monomers like St, MMA, vinylanisole, vinylaceate at 120 °C.18 Like homopolymerization studies, these initial reports of Bailey et al. showed the copolymerizability but lacked the detailed copolymer characterization in terms of the microstructure and copolymerization parameters. The only copolymerization parameters reported that time were for the copolymerization of 2 methylene, 1,3-dioxepane (6) with styrene r6 = 0.021 and rst = 22.6.45 The hydrolysis of these copolymers led to an oligomer with hydroxyl and carboxyl end groups which could be further used as telechelics for the synthesis of polyesters or polyurethanes. Later Davis and co-workers46 in an attempt to study the details about copolymerization of 2 methylene, 1,3-dioxepane (6) and styrene, found no copolymerizability between the two (2 methylene-1,3-dioxepane with styrene) at 30 and 40 °C. The experimental data indicated the homopolymerization of styrene, with (6) acting as diluent. Also, the first attempt to provide propagation rate coefficients for the RROP of 2 methylene, 1,3-dioxepane (6) and MMA were done by Davis and co-workers47 They have reported r6 = 0.057 and rMMA = 34.12. The low reactivity of cyclic ketene acetals as compared to vinyl monomers is due to the presence of two electron donating substituents which can not stabilize the resulting radical and thus makes the copolymerization difficult. Detailed copolymerization characteristics of (6) and methyl acrylate in benzene were studied by Zhou and co-workers.48 Although the ring-opening was quantitative 1,7-H-transfer reactions led to branched polymers. The molecular weights were in the range 104–105 and broad polydispersities ranging from 1.3–3.0 were obtained. The reactivity parameters were r6 = 0.0235 and rMA = 26.535. The copolymers could enzymatically be degraded using proteinase K or crude enzyme extracted from earthworms.
Also, the copolymerization of other CKAs like 4,7-dimethyl-2-methylene-1,3-dioxepane with St and MMA and 4-vinylanisole gave 100% ring-opening but low yields after long reaction times.19 Compared to the conventional radical polymerization, ATRP of 4,7-dimethyl-2-methylene-1,3-dioxepane (8) with conventional vinyl monomers like styrene, acrylonitrile and methyl acrylate was studied by Yuan and Pan.49 The initiator system used was ethyl α-bromobutyrateCuBr/2,2′-bipyridyl. For all molar ratios of (8)/St in feed, the contents of (8) in the copolymers were relatively low, like conventional radical polymerization, due to much higher reactivity of styrene. A maximum of 7.3 mol% of (8) could be incorporated with about 67 mol% of ring-opened structures. With AN, an alternate system was formed with about 70 mol% ring-opened units but the polydispersity index were high (1.53–1.90). High polydispersities 1.54–1.84 were also seen during copolymerization of 5,6-benzo-2-methylene-1,3-dioxepane (9) with n-butyl acrylate using ethyl-2-bromoisobutyrate and N,N,N′,N′′-pentamethyldiethylenetriamine/CuBr as the initiator and catalyst respectively by Matyjaszewski and coworkers. The reactivity ratios determined were r9 = 0.08 and rBA = 3.7.50 We provided the details of copolymerization of 5,6-benzo-2-methylene-1,3-dioxepane (9) and St and MMA using benzyl bromide, CuBr and pentamethyldiethylenediamine ligand system.51,52 Under ATRP reaction conditions the copolymerization parameters were r9 = 1.08 and rSt = 8.53 as compared to that of r9 = 0.53 and rMMA = 1.96. The same trend was seen during conventional radical ring-opening copolymerization of (9) with styrene and MMA in our laboratory. MMA, an electrophilic monomer is found to have increased copolymerizability tendency with nucleophilic cyclic ketene acetals like (9) in our case and the amount of ester linkages incorporated are more on PMMA backbone as compared to that on the PS backbone starting from the same feed composition. The enhanced reactivity of MMA during its copolymerization with BMDO could lead to the synthesis of copolymers at low temperatures using conventional radical initiator like AIBN with quantitative ring-opening. At 65 °C using AIBN as initiator, incorporation of about 23 mol% of (9) onto the PMMA backbone took place as compared to the formation of only homo polystyrene under similar conditions starting with 50 :50 mol% of (9):MMA or (9):St (Scheme 11).
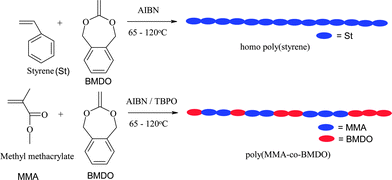 | ||
| Scheme 11 Increased reactivity of MMA during copolymerization with 5,6-benzo-2-methylene-1,3-dioxepane (9) leading to random copolymers having ester units. | ||
Low dielectric constant, permittivity, optical loss, and low flammability besides high water and oil resistance are some of the properties that make fluorine containing polymers attractive. To study the effect of substituents on vinyl monomers on copolymerizability behavior with cyclic ketene acetals, pentafluorostyrene (PFS) was chosen as the comonomer by us. PFS appeared to be an interesting monomer. The resulting PFS radical is expected to be more electrophilic as compared to the corresponding styrene radical because of the presence of 5 inductively electron withdrawing fluorine atoms making the copolymerization of PFS with electron rich CKAs more feasible. Also, the ‘F’ atoms can donate electron density by conjugation making the situation almost similar as that of styrene during their copolymerization with cyclic ketene acetals. This study was expected to answer this question and the resulting new poly(fluorinated vinyl-co-ester)s were expected to combine after use degradability with hydrophobicity with better adhesion at various surfaces for different applications like hydrophobic coatings etc.
Our actual studies showed the enhanced reactivity of PFS during its copolymerization with 5,6-benzo-2-methylene-1,3-dioxepane (9) as compared to styrene monomer under conventional free radical polymerization conditions and copolymerization took place with quantitative ring-opening for any feed composition in the temperature range of 65–120 °C. The reactivity ratios for PFS and (9) are determined to be r9 = 0.35 and rPFS = 9.9 and the microstructure characterization showed a gradient structure with random distribution of ester units at the start of the copolymerization and then the formation of blocks of 5,6-benzo-2-methylene-1,3-dioxepane with increased reaction time.53
The copolymers with alternating caprolactone and fluoroalkene units (3,3,4,4,5,5,6,6,6-nonafluoro-1-hexene, tridecafluoro-1-octene, heptadecafluoro-1-decene) in the chain have been synthesised by Sen and co-workers.54 In films, the perfluoroalkyl tails self assemble and segregate to the air side, thereby presenting a hydrophobic surface with water contact angle as high as 130°, while the ester functionality makes the material degradable. The previous related literature also aimed to make biodegradable and water repellant new partially fluorinated polymers and reported the formation of alternate copolymers of CKAs with fluorinated olefins.55 For example the copolymer of tetrafluoroethylene and 2-methylene-1,3-dioxepane (6) gave a polyester with 51.6 mol% of (6) (Mn = 19,800; MW = 144,000; Tm = 95 °C and Tg = −28.4 °C), on the other hand, a copolymer of chlorotrifluoroethylene with (6) had 53.6 mol% of (6) and Tm = 70.2 °C and Tg = −14 °C (Mn = 30,200; Mw = 121,000).
Some basic studies without much detail are also reported in the literature regarding the copolymerization of ethylene and 2-methylene-1,3-dioxepane.56 This was done with an aim to provide degradable ester units in the polyethylene backbone. They were synthesised at 120 °C for 30 min at a pressure of 1800 psi. Very low conversion of monomers was achieved with incorporation of about 2.1 to 10.4 mol% of ester units.
The value of n depends upon the mol% of ester incorporated and was approximately 47 and 9 for 2.1 and 10.4 mol% of ester units, respectively. There is no data provided for the copolymers with higher yields. There was a decrease in the melting point of PE from 117 to 85 on increasing the ester content from 2 to 15%.
The free-radical chemistry of introducing degradable ester linkages onto the vinyl polymers backbone was also utilised by us in providing materials with the combined properties like degradability, non-toxicity and low glass transition temperatures. Such materials could be proposed for different applications like degradable gums, coatings etc. For this purpose, the ester units having PCL structure were successfully introduced onto poly(vinyl acetate) (PVAc) backbone by ring-opening polymerization of (6) and vinyl acetate (VAc).57 1D and 2D-NMR studies gave an insight into the polymer microstructure and shown to have random distribution of ester linkages onto the PVAc backbone. It was possible to control the properties of new materials such as glass transition temperature, viscosity, mechanical properties etc. by controlling the amount of ester units from (6) in the copolymers. The degraded product after hydrolysis was non-toxic (Fig. 3).
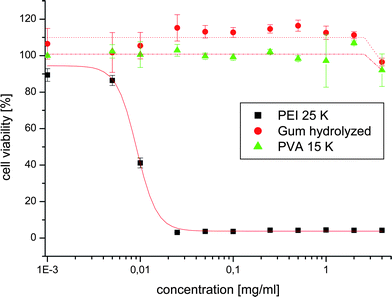 | ||
| Fig. 3 Cell viability studies for poly(vinyl acetate-co-ester) (copolymer of 2-methylene-1,3-dioxepane (6) with vinyl acetate (25:75 molar ratio).57 | ||
Furthermore, interesting use of copolymerization of CKAs with vinyl monomers was made in tackling the main limitation of non-biodegradability of biorelevant polymers like thermoresponsive poly(N,N′-isopropylacrylamide) (PNIPAAM) and poly oligo(ethylene glycol) methacrylates.58a The method of making polyesters by radical-ring-opening polymerization provided the easy solution to this problem. The bulk ATRP of 2-(2-methoxyethoxy)ethyl methacrylate (MEO2MA), oligo(ethylene glycol) methacrylate (OEGMA) and CKA 5,6-benzo-2-methylene-1,3-dioxepane (9) was carried out at 90 °C in the presence of homogenous catalyst system copper(I)chloride/2-2′bipyridyl. The aim was to make ALL-IN-ONE (biocompatible, biodegradable and thermoresponsive) biopolymers. High yields were obtained in a few hours. The incorporation of ester units led to biocompatible polymers with the degradability and with varied lower critical solution temperature (LCST values) (Fig. 4). Smart PEG based degradable polymers could be prepared via a simple one pot atom transfer radical polymerization.58b Also, degradable ester linkages were successfully introduced onto the water soluble thermoresponsive vinyl polymer poly(NIPAAM) by copolymerizing with 5,6-benzo-2-methylene-1,3-dioxepane (9). The polymers were hydrolytically degradable and the properties like glass transition temperature (Tg) and LCST could be easily varied by changing the amount of ester linkages in the copolymers.59 Further work involved making the cross-linked degradable, thermoresponsive gels and immobilisation with GRGDS peptide sequences, improving cell attachment for tissue engineering applications.60 Studies on the synthesis and properties of temperature responsive and biodegradable hydrogels were also carried out by Zhou and co-workers.61 Cross-linked poly(NIPAAm-co-CL) hydrogels have been prepared by cross-linking of copolymers of NIPAAM and 2-methylene-1,3-dioxepane (6) in toluene using N,N′-methylenebisacrylamide as a cross-linking agent. Introduction of CL units led to a temperature dependent biodegradability i.e. the weight loss was about 30% after 14 days at 22 °C but reduced to 20.9% at 37 °C.
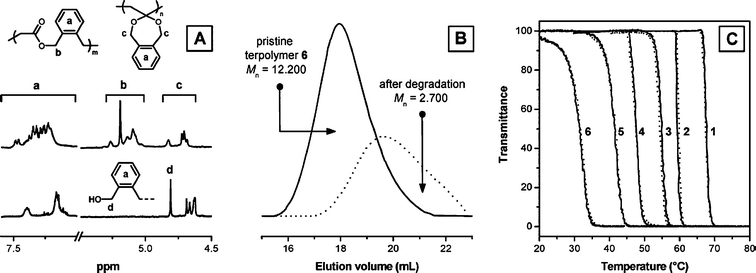 | ||
| Fig. 4 (A) 1H NMR before and after degradation, (B) SEC chromatograms before and after degradation, (C) plots of transmittance as a function of temperature (670 nm, 1 °C min−1) measured for aqueous solutions (3 mg ml−1) of terpolymers P(MEO2MA-co-OEGMA-5,6-benzo-2-methylene-1,3-dioxepane (9)) of various compositions.58b | ||
The formation of such ALL-IN-ONE polymers have opened completely new application possibilities and the chemistry can be extended further for grafting surfaces for the development of functional coatings or degradable thermoresponsive surfaces. There is recent literature using the same chemistry for making hydrolytically degradable polymer brushes by copolymerization of 5,6-benzo-2-methylene-1,3-dioxepane (9) and poly(ethylene glycol)methacrylate (Fig. 5).62
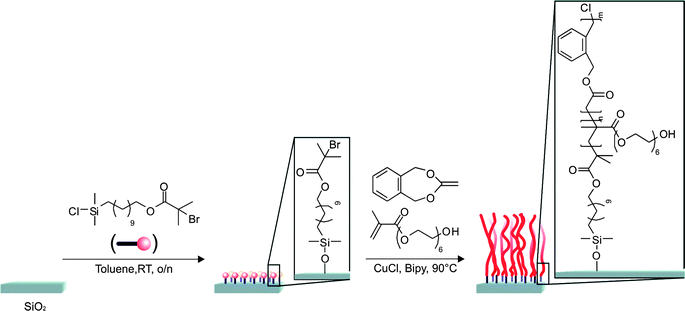 | ||
| Fig. 5 Hydrolytically degradable polymer brushes by copolymerization of 5,6-benzo-2-methylene-1,3-dioxepane (9) and poly(ethylene glycol)methacrylate.62 | ||
Also, we have shown the use of this chemistry in making degradable polycations. Cationic polyelectrolyte like poly(2-dimethylaminoethyl methacrylates) (PDMAEMA) are explored as vectors for gene delivery due to efficient DNA complexation. The details about the use of cationic polymers as gene delivery systems can be read in the review articles by De Smedt et al.63 and Anderson and co-workers.64 Toxicity and non-degradability of the polycations is an important factor that limits the use for many applications. The recent trend is to design new degradable and non-toxic carrier systems. We successfully showed the formation of less toxic and degradable polycations which made highly stable polyplexes with DNA (Fig. 6).65 Degradable ester linkages were introduced randomly onto the vinyl polymer poly(DMAEMA) backbone by a combination of radical ring-opening polymerization of 5,6-benzo-2-methylene-1,3-dioxepane (9) and conventional vinyl polymerization of DMAEMA (Scheme 12).
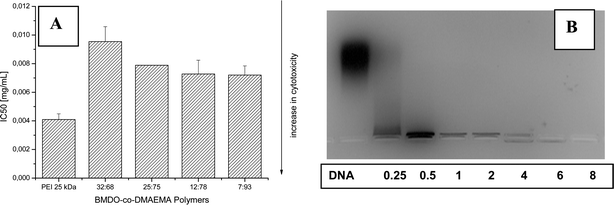 | ||
| Fig. 6 (A) IC50 doses for different 5,6-benzo-2-methylene-1,3-dioxepane (9)-co-DMAEMA polymers and the standard PEI (polyethyleneimine) 25 kDa. (Means + SD are shown.) (B) Agarose gel electrophoresis of polymer/herring testes DNA complexes with different N/P ratios. Free DNA (lane 1) was compared with the polyplexes.65 | ||
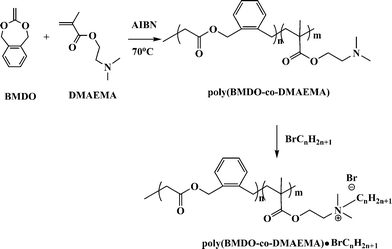 | ||
| Scheme 12 A reaction scheme for the synthesis of degradable polycations.65 | ||
Interestingly, in our attempt to make pH responsive water soluble and biodegradable polymers the copolymerization of methacrylic acid and 5,6-benzo-2-methylene-1,3-dioxepane (9) led to different copolymer structures depending upon the amount of (9) in the feed.66 The copolymerization proceeded in an unconventional way, i.e., by in situ formation of a new vinyl monomer on mixing (9) and MAA. This copolymerization does not follow any of the conventional routes of copolymerization of cyclic ketene acetals with vinyl monomers known until now, i.e., 1,2-vinyl addition at the double bond of (9) giving poly acetal rings and/or free-radical ring-opening polymerization generating ester linkages in the backbone. Instead, this work showed a new route to copolymerization first by addition of MAA to the double bond of (9) generating a new vinyl monomer (3-methyl- 1,5-dihydrobenzo[e][1,3]dioxepin-3-yl methacrylate), which is unstable at higher temperatures like 120 °C and then rearranging into a new structure (2-(acetoxymethyl)benzyl methacrylate) with vinylic double bond and ester linkages in the side chain. Different monomer ratios in the feed generated different structures with ester linkages either in the backbone or as the side chain (Scheme 13). The new polymers depending on their structure could be used as pH sensitive functional materials.66
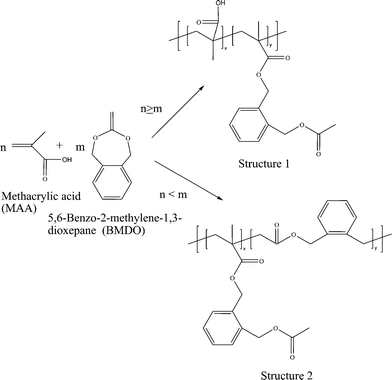 | ||
| Scheme 13 Unconventional copolymerization routes for 5,6-benzo-2-methylene-1,3-dioxepane (9) and methacrylic acid (MAA).66 | ||
The technique of making degradable microspheres of 2-methylene-1,3-dioxepane (6) with water soluble polymer, 2-vinylpyrrolidone was shown by Lee and co-workers using dispersion polymerisation in supercritical carbon dioxide (scCO2) using siloxane based polymeric surfactant and fluorinated polymeric dispersants.67 Poly(vinylpyrrolidone) is an important polymer for the cosmetic industry.
The free radical ring-opening polymerization of 2-methylene-1,3-dioxepane (6) with vinyl monomers is also used to make specialized materials. We reported the first successful synthesis of poly(caprolactone) (PCL) based degradable ionomers using free-radical chemistry. Degradable cationic ionomers were synthesized by random radical terpolymerization of 2-methylene-1,3-dioxepane (6), methyl methacrylate (MMA) and N,N-dimethylaminoethyl methacrylate (DMAEMA) and followed by quaternization of amine with alkyl bromide. The terpolymerization of (6), MMA and DMAEMA lead to a polymer with ester groups randomly distributed in the polymer chains. Small angle X-ray scattering (SAXS) analysis, transmission electron microscopy (TEM) and dynamic mechanical analysis (DMA) showed ionic aggregates existing in ionomers.68
Furthermore, new polymeric-inorganic-hybrid materials were made using RROP chemistry. A series of functionalised poly(caprolactone) copolymers, poly(caprolactone-co-vinylphosphonic acid) and poly(CL-co-dimethyl vinylphosphonate) were synthesized by copolymerization of 2-methylene-1,3-dioxepane (6) with vinylphosphonic acid and dimethyl vinylphosphonate. The copolymers had a random structure as proved by DSC. The aim was to make polymeric–inorganic hybrid materials. The introduction of dimethoxyphosphinyl groups can provide nucleation sites for hydroxyapatite (HAP) through Ca2+ ion accumulation.69
7 Conclusions
It is evident from the literature that the radical ring-opening polymerization of CKAs led to the synthesis of homopolyesters with low to moderate molecular weights with branched structures due to 1,4- and 1,7-hydrogen transfer reactions. Although the detailed properties evaluation of the polyesters made by RROP is missing in the literature, very few examples have been extensively studied, like radical ring-opening of 2-methylene-1,3-dioxepane, which clearly shows that this free radical based chemistry cannot be considered as substitute for conventional methods like condensation and ionic/metal catalysed polymerizations of cyclic esters for the synthesis of known degradable homopolyesters because of limited molecular weights and transfer reactions. The present need for overcoming this limitation is to carry out research for achievement of high molecular weights, and structure–property relationship studies—the effect of microstructure, amount of ester linkages—on the resulting polymer properties. The present method of radical ring-opening polymerization offers the advantage in either making special functional polyesters which are not possible by conventional methods or for making polyesters for special applications where low volume shrinkage is of utmost importance like matrix resins or dental restoration, adhesives, coatings and precision castings etc. This method also makes the synthesis of polyesters with controlled chain lengths and controlled chain ends using living radical polymerization techniques possible. This would make the synthesis of various macromolecular architectures like block and star polymers etc. based on polyester repeat units simple. Furthermore, the beauty of radical ring-opening polymerization is its use in making a new class of degradable materials with ester linkages distributed onto the vinyl polymer backbone called poly(vinyl-co-ester)s. No other method can combine vinyl polymer backbone with ester units in a random way. Using this strategy, the method of radical ring-opening polymerization has also opened new possibilities of using the known biomaterials for degradable applications. Other opportunities using this method exploited up to now are in making specialised degradable structures like ionomers and polymeric–inorganic hybrid materials which otherwise are not at all possible or are difficult to make by state-of-the-art synthetic methods. The field is still open and could provide many different degradable structures and new properties for a wide range of applications.Acknowledgements
I would like to thank Deutsche Forschungsgemeinschaft (DFG) for the financial support of free radical ring-opening polymerization chemistry in my group. I acknowledge the work carried out by my co-workers Holger Wickel, Liqun Ren, Rimpu Kumar and Christian Speyerer for the progress of this field.References
- D. Goldberg, J. Environ. Polym. Degrad., 1995, 3(2), 61 Search PubMed.
- H. R. Kricheldorf, T. Mang and J. M. Jonte, Macromolecules, 1984, 17, 2173 CrossRef CAS.
- C. Jaimes, A. Collet, O. Giani-Beaune, F. Schue, W. Amass and A. Amass, Polym. Int., 1998, 45, 5 CrossRef CAS.
- J. W. Pack, S. H. Kim, I. W. Cho, S. Y. Park and Y. H. Kim, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 544 CrossRef CAS.
- S. Agarwal, M. Karl, K. Dehnicke, G. Seybert, W. Massa and A. Greiner, J. Appl. Polym. Sci., 1999, 73, 1669 CrossRef CAS.
- S. Agarwal, M. Karl, K. Dehnicke and A. Greiner, e-polymers, 2001, 12 Search PubMed.
- S. Agarwal, N. Naumann and X. Xie, Macromolecules, 2002, 35, 7713 CrossRef CAS.
- W. J. Evans and H. Katsumata, Macromolecules, 1994, 27, 4011 CrossRef CAS.
- A. C. Albertsson and I. K. Varma, Adv. Polym. Sci., 2002, 157, 1 CAS.
- M. Labet and W. Thielemans, Chem. Soc. Rev., 2009, 38(12), 3484 RSC.
- G. Z. Papageorgiou, D. S. Achilias and D. N. Bikiaris, Macromol. Chem. Phys., 2009, 210, 90 CrossRef CAS.
- J. Su, Y. Chen and L. Tan, J. Biomater. Sci., Polym. Ed., 2009, 20, 99 CrossRef CAS.
- W. J. Bailey, P. Y. Chen, W. B. Chiao, T. Endo, L. Sidney, N. Yamamoto, N. Yamazaki and K. Yonezawa, Contemporary Topics Polym. Sci., 1979, 3, 29 Search PubMed.
- W. J. Bailey, J. L. Chou, P. Z. Feng, V. Kuruganti and L. L. Zhou, Acta Polym., 1988, 39, 335 CrossRef.
- W. J. Bailey, S. R. Wu and Z. Ni, Makromol. Chem., 1982, 183, 1913 CrossRef CAS.
- W. J. Bailey, S. R. Wu and Z. Ni, J. Macromol. Sci., Part A: Pure Appl. Chem., 1982, 18(6), 973 Search PubMed.
- T. Yokozawa, R. Hayashi and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 3739 CrossRef CAS.
- W. J. Bailey, Z. Ni and S. R. Wu, J. Polym. Sci. Polym. Chem., 1982, 20, 3021 Search PubMed.
- W. J. Bailey, Z. Ni and S. R. Wu, Macromolecules, 1982, 15, 711 CrossRef CAS.
- (a) T. Endo, M. Okawara, W. J. Bailey, K. Azuma, K. Nate and H. Yokono, J. Polym. Sci., Polym. Lett. Ed., 1983, 21, 373 CrossRef CAS; (b) T. Endo, N. Yako, K. Azuma and K. Nate, Makromol. Chem., 1985, 186, 1543 CrossRef CAS.
- S. Jin and K. E. Gonsalves, Macromolecules, 1997, 30, 3104 CrossRef CAS.
- S. Agarwal, Polym. J., 2007, 39, 163 CrossRef CAS.
- S. Agarwal and C. Speyerer, Polymer, 2010, 51(5), 1024 CrossRef CAS.
- I. Cho and S. K. Kim, J. Polym. Sci., Part C: Polym. Lett., 1990, 28, 417 CrossRef CAS.
- P. Plikk, T. Tyson, A. F. Wistrand and A. C. Albertsson, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4587 CrossRef CAS.
- D. Bhatia, D. Braun and V. Choudhary, Indian J. Chem. Technol., 2003, 10, 548 Search PubMed.
- F. Sanda, T. Takata and T. Endo, Macromolecules, 1994, 27, 1099 CrossRef CAS.
- N. Moszner, T. V. ölkel, V. Rheinberger and E. Klemm, Macromol. Chem. Phys., 1997, 198, 749 CrossRef CAS.
- T. Schulze and E. Klemm, Polym. Bull., 1993, 31, 409 CrossRef CAS.
- T. Schulze and E. Klemm, Macromol. Chem. Phys., 1995, 196, 567 CrossRef CAS.
- N. Yako and T. Endo, Polym. Prepr. Jpn., 1985, 34, 154 Search PubMed.
- W. Liu, F. Mikes, Y. Guo, Y. Koike and Y. Okamoto, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5180 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101(12), 3661 CrossRef CAS.
- (a) M. Ouchi, T. terashima and M. sawamoto, Chem. Rev., 2009, 109, 4963 CrossRef CAS; (b) W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32(1), 93 CrossRef CAS.
- (a) C. Barner-Kowollik and S. Perrier, J. Polym. Sci., Part A: Polym. Chem., 2008, 46(17), 5715 CrossRef CAS; (b) G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49(5), 1079 CrossRef CAS.
- Y. Wei, E. J. Connors, X. Jia and B. Wang, Chem. Mater., 1996, 8, 604 CrossRef CAS.
- Y. Wei, E. J. Connors, X. Jia and C. Wang, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 761 CrossRef CAS.
- (a) J. Y. Yuan and C. Y. Pan, Chinese J. Polym. Sci., 2001, 20(1), 9; (b) C. Y. Pan and X. D. Lou, Macromol. Chem. Phys., 2000, 201, 1115 CrossRef CAS.
- T. He, Y. F. Zou and C. Y. Pan, Polym. J., 2002, 34(3), 138 CrossRef CAS.
- (a) Y. Wang and M. A. Hillmyer, Macromolecules, 2000, 33, 7395 CrossRef CAS; (b) E. M. Frick and M. A. Hillmyer, Macromol. Rapid Commun., 2000, 21, 1317 CrossRef CAS; (c) S. K. Varshney and J. X. Zhang, Polymer preprints, 2000, 41(2), 1715 CAS.
- (a) J. M. Messman, A. D. Scheuer and R. F. Storey, Polymer, 2005, 46, 3628 CrossRef CAS; (b) I. Ydens, P. Degee, P. Dubois, J. Libiszowski, A. Duda and S. Penczek, Macromol. Chem. Phys., 2003, 204, 171 CrossRef CAS.
- (a) T. C. Chang, H. B. Chen, Y. C. Chen and S. Y. Ho, J. Polym. Sci., Part A: Polym. Chem., 1996, 34, 2613 CrossRef CAS; (b) R. Walz, B. Bömer and W. Heitz, Makromol. Chem., 1977, 178, 2527 CrossRef CAS.
- J. Y. Yuan and C. Y. Pan, Eur. Polym. J., 2002, 38, 1565 CAS.
- J. Y. Yuan and C. Y. Pan, Chinese J. Polym. Sci., 2002, 20(2), 171 CAS.
- W. J. Bailey, T. Endo, B. Gapud, Y. N. Lin, Z. Ni, C. Y. Pan, S. E. Shaffer, S. R. Wu, N. Yamazaki and K. Yonezawa, J. Macromol. Sci., Part A: Pure Appl. Chem., 1984, 21(8), 979 Search PubMed.
- L. M. Morris, T. P. Davis and R. P. Chaplin, Polymer, 2001, 42, 495 CrossRef CAS.
- G. E. Roberts, M. L. Coote, J. P. A. Heuts, L. M. Morris and T. P. Davis, Macromolecules, 1999, 32, 1332 CrossRef CAS.
- L. F. Sun, R. X. Zhou and Z. L. Liu, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 2898 CrossRef CAS.
- J. Y. Yuan and C. Y. Pan, Eur. Polym. J., 2002, 38, 2069 CrossRef CAS.
- J. Huang, R. Gil and K. Matyjaszewski, Polymer, 2005, 46, 11698 CrossRef CAS.
- H. Wickel and S. Agarwal, Macromolecules, 2003, 36, 6152 CrossRef CAS.
- H. Wickel, S. Agarwal and A. Greiner, Macromolecules, 2003, 36, 2397 CrossRef CAS.
- S. Agarwal, J. Polym. Res., 2007, 13(5), 403 Search PubMed.
- S. Borkar, A. Sen and J. R. Shallenberger, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1225 CrossRef CAS.
- H. N. Cripps, patent number 1990, 4,906,714 March 6.
- B. Wu and R. W. Lenz, J. Environmental Polym. Deg., 1998, 6(1), 23 Search PubMed.
- S. Agarwal, R. Kumar, T. Kissel and R. Reul, Polym. J., 2009, 41(8), 650 CrossRef CAS.
- (a) J. F. Lutz, O. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046 CrossRef CAS; (b) J. F. Lutz, J. Andrieu, S. Üzgün, C. Rudolph and S. Agarwal, Macromolecules, 2007, 40(24), 8540 CrossRef CAS.
- L. Ren and S. Agarwal, Macromol. Chem. Phys., 2007, 208, 245 CrossRef CAS.
- D. J. Siegwatrt, S. A. Bencherif, A. Srinivasan, J. O. Hollinger and K. Matyjaszewski, J. Biomed. Mater. Res., Part A, 2008, 87a, 345 CrossRef.
- L. F. Sun, R. X. Zhou and Z. L. Liu, Macromol. Biosci., 2003, 3, 725 CrossRef CAS.
- C. Riachi, N. Schüwer and H. A. Klok, Macromolecules, 2009, 42(21), 8076 CrossRef CAS.
- S. C. De Smedt, J. Demeester and W. E. Hennik, Pharm. Res., 2000, 17(2), 113 CrossRef.
- D. N. Nguyen, J. J. Green, J. M. Chan, R. Langer and D. G. Anderson, Adv. Mater., 2008, 21(8), 847.
- S. Agarwal, L. Ren, T. Kissel and N. Bege, Macromol. Chem. Phys., 2010 DOI:10.1002/macp.200900579.
- L. Ren, C. Speyerer and S. Agarwal, Macromolecules, 2007, 40, 7834 CrossRef CAS.
- S. Kwon, K. Lee, H. Kim, Y. W. Lee and W. Bae, Colloid Polym. Sci., 2008, 286, 1181 CrossRef CAS.
- S. Agarwal and L. Ren, Macromolecules, 2009, 42, 1574 CrossRef.
- S. Jin and K. E. Gonsalves, Macromolecules, 1998, 31, 1010 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |