Protein-polymer amphiphilic chimeras: recent advances and future challenges
Kelly
Velonia
*
Department of Materials Science and Technology, University of Crete, University Campus Voutes, 71003, Heraklion, Crete, Greece. E-mail: velonia@materials.uoc.gr; Fax: +30 2810 394 273; Tel: +30 2810 394 036
First published on 18th March 2010
Abstract
During the past decades numerous studies have focused on the bridging of components derived from both the natural and the synthetic world with the aim of creating new biomaterials with improved properties and functions. This review illustrates the range of well-defined protein-polymer amphiphiles—Giant Amphiphiles—prepared in very recent years. Giant Amphiphiles are classified in terms of the conceptually different approaches developed to tackle their synthesis. The variety of high turnover methodologies, the expression of the intrinsic properties of the synthetic and/or the biological component and the aggregation/activity profile of each individual system are discussed with the aim of providing a basis for the rational design of new generations of biomacromolecular chimeras aimed for high-impact applications.
 Kelly Velonia | Kelly Velonia received her PhD in chemistry from the University of Crete in 1999 where she worked with Profs I. Smonou and G. J. Karabatsos. In 2000 she joined the group of Prof. R.J.M. Nolte at the Radboud University Nijmegen as a post-doctoral fellow. In 2001 she worked with Prof. F.C. De Schryver at the University of Leuven and in 2002 she returned to Nijmegen as a Marie Curie Individual Fellow. In 2004 Kelly joined the Department of Organic Chemistry of the University of Geneva as an Assistant Professor. In 2007 she was appointed Assistant Professor at the Department of Materials Science of the University of Crete. Kelly's current research is focused on the construction of multifunctional protein-polymer assemblies. |
Introduction
Among the most stimulating challenges for contemporary materials science is the construction of ordered nanoscale assemblies that are designed to exploit the best of what both Nature and chemistry have evolved to offer. Along these lines, the combination of biological with polymeric materials to create chimeric biomacromolecules has been an area of flourishing research during the past decades.Protein-polymer biomacromolecular chimeras aim to merge and express the intrinsic properties, functions and advantages of both their natural and synthetic components. Ideally, such biohybrids should surpass individual component limitations by expressing their characteristics in a synergistic and combinatorial manner and acquire new functions with increased complexity. In the past decades, a wide variety of polymer-protein adducts was synthesized by essentially random coupling of the biological component to one or more polymeric matrices.1,2 The most successful, up to date, expression of the objectives of bioconjugation can be found in the area of polymer therapeutics where the covalent attachment of single or multiple synthetic polymeric tails to proteins or drugs has often resulted in stabilization and enhancement of their biological activity and even found practical application.3–6 Protein PEGylation in particular has unambiguously led to the most pronounced examples of such functional biomacromolecules.7,8 Profiting from the impressive advances in polymer science, molecular biology and characterization and imaging techniques, protein polymer bioconjugates are at the focal point of contemporary research for the development of innovative supramolecular biomaterials and fundamental components for bio-sensors, artificial enzymes, lab-on-a-chip, photonics, nano-electronic and bioprocessing applications.9–13
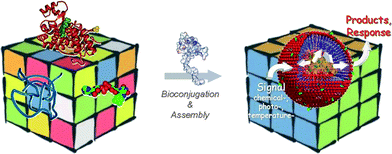
This review focuses on the rather new class of protein-polymer chimeras that are designed to mimic the hierarchical self-assembly displayed both in biological material systems13,14 and in synthetic block copolymers15 over a range of lengths (Scheme 1). The Giant Amphiphiles which are highlighted here are protein-polymer bioconjugates with a global amphiphilic character introduced by the hydrophobic polymer moiety. These bioconjugates differ from other bioadducts in the sense that the bioconjugation position is precisely known and defines the exact polymer-to-protein ratio. Since the chemistries concerning both specific bioconjugation16–23 and functionalisation of proteins with polymers15,24–28 have been comprehensively reviewed in the very recent years, this report rather focuses on the conceptually different strategies that have successfully applied these chemistries to construct Giant Amphiphiles and aims to provide insight for their further development. The different subclasses of Giant Amphiphiles are presented in the light of the developed synthetic methodologies and along with architectures, catalytic/response profiles and possible applications of the developed nanostructured biomaterials.
Characteristics and synthetic considerations
From the chemical point of view, Giant Amphiphiles are diblock copolymers which by design have considerably higher molecular weights than synthetic molecular or polymeric soaps. Nature synthesizes proteins in a bionomical, highly efficient and precise manner, thus the protein block offers the intrinsic structural advantage of monodispersity over the synthetic block in addition to its functional and structural characteristics. However, the bioblock is also expected to introduce several disadvantages and restrictions to the chimeric biomacromolecules, especially when these are intended for biomedical applications. Proteins can be toxic or elicit negative immune responses, they are susceptible to enzymatic and chemical degradation, have limited solubility in non-aqueous solvents and restricted stability with temperature and pH. Since ideal biomaterials should have minimal immune responses and preserve important protein bioactivating and biopassivating functions, the incorporation of a polymer on the protein structures offers the perspective of reducing the protein induced undesired biological responses and reinforcing stability. Moreover, the synthetic polymer is expected to contribute its chemical and physicochemical characteristics to the biomacromolecular chimeras with the perspective of modifying biodistribution and solubility of the biological component along with the possibility of introducing new functionalities.Polymers, on the other hand, provide broad choices in both chemistry and morphologies. Conventional chemical synthesis of polymers often results in polydisperse materials with a broad distribution of molar masses that present a challenge for characterization, affect biological activity and in vivo toxicity and limit applicability in biomedicine.30 However, the advances in controlled radical polymerization techniques (CRP) such as atom transfer radical polymerization (ATRP),31 reversible addition-fragmentation transfer polymerization (RAFT)32 and nitroxide mediated polymerization,33 have introduced unprecedented possibilities for the polymerization of an unlimited variety of monomers to afford polymers with low polydispersity indices and highly defined structures bearing diverse functional and end functional groups.
The synthesis of the Giant Amphiphiles themselves, similar to all specific bioconjugation approaches, requires methods that are specific to a unique functionality within the complex protein structure and proceed with a high degree of specificity and efficiency under mild conditions.1–13 While no such universal technique exists to date, significant progress has been made toward the development of a wide range of bioconjugation reactions that fulfil the above criteria.1–13,16–23 Nevertheless, the selection of the appropriate strategy for a specific coupling reaction remains a challenge for the majority of proteins. Bioconjugation is highly dependent on the structure of the protein, the synthetic availability of complementary reactive group(s) on the polymer or heterobifunctional spacer to be conjugated and practical limitations such as reaction conditions, chemical compatibility, solubility or even economic aspects that may often result in rejecting certain bioconjugate routes. Knowledge of the sequence of a protein can provide significant information concerning the possible conjugation site. Besides the obvious selection of the α-N terminus (α-N) of the peptide backbone, the most useful functionalities for specific protein modification have proven to be amino acids side chains of cysteine (Cys), lysine (Lys), tyrosine (Tyr), and glutamine (Gln).16–23 The chemical approach to be used is often dictated by the availability of a unique target amino acid that can act as a single docking point on the protein. When such an amino acid is available, “click” bioconjugations are usually pursued as they are designed to proceed in high yields, under mild conditions and create inoffensive products (Fig. 1).34,35 Moreover the target docking point (amino acid or cofactor) needs to be reasonably exposed on the surface of the ternary protein structure and not buried in its interior, to facilitate the bioconjugation reaction. However, proteins, and especially the larger ones, are multivalent and likely to contain several copies of the twenty canonical amino acids within their structure. In such a case, several alternatives can be considered. Specific modification might be possible when only one of multiple residue copies is exposed on the protein surface, while in the case that two or more amino acids are exposed, bioconjugation becomes very difficult to direct. Since various methods to engineer natural and non-natural amino acids within protein structures have been developed, genetic modification might also provide an alternative route.36
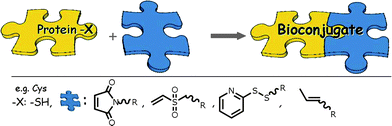 | ||
| Fig. 1 Specific bioconjugation on unique cysteine residues. | ||
Finally, an ideal bioconjugation should have little or no effect on the protein conformation and function, e.g., catalytic activity, specificity, receptor recognition or transport properties. In order to augment the possibility of such a modification, the bioconjugation point should ideally be situated far from the active site. Knowledge and assessment of the protein structure is therefore a prerequisite for successful selective functionalisation and requires information about the protein ternary structure acquired either via single-crystal X-ray or by solution NMR studies.
This report focuses on the conceptually different strategies that have been developed to synthesize Giant Amphiphiles and broadly classifies them in to three major categories in respect to the protein: A the “grafting to” approach, B the “grafting to” through non covalent interactions approach and, C the “grafting from” approach. Giant Amphiphiles are presented with emphasis on the individualities of each system in order to provide insight into the methodologies and constraints that should be applied when designing novel generations of such protein-polymer bioconjugates.
Synthetic approaches
A “Grafting to” approach
The “grafting to” approach involves direct conjugation of end-functionalized polymer chains on a suitable functionality exposed on the protein surface (Fig. 2). The method is conceptually simple, but faces several experimental restrictions in the specific case of Giant Amphiphiles. In addition to the limitations that are expected in reactions between macromolecules and especially multifunctional ones such as the proteins and to steric crowding, the synthesis of Giant Amphiphiles is also hampered by the different solubility of the two blocks. In order to achieve a reaction between monomeric species that is unhindered by aggregation, either a careful selection of the reaction medium has to be performed or the amphiphilic character should be introduced in a post-conjugation step.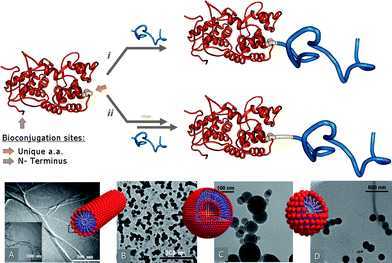 | ||
| Fig. 2 A schematic representation of the “grafting to” synthesis of Giant Amphiphiles through the i direct and ii indirect bioconjugation approach. Morphologies of Giant amphiphiles: A CALB-PS micellar rods (reprinted with permission from ref. 37, copyright 2002 ACS Publications), B BSA-PS vesicles (reprinted with permission from ref. 52, copyright 2005 The Royal Society of Chemistry), C BSA-PA@Bz and D BSA-PA@C10 spherical structures (reprinted with permission from ref. 60, copyright 2007 The Royal Society of Chemistry). | ||
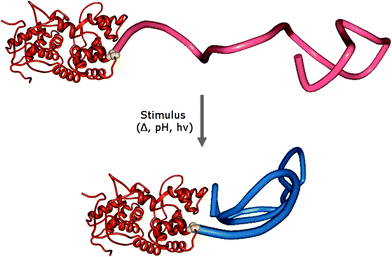
The specific conjugation of “smart” polymers to proteins has also led to the creation of Giant Amphiphiles in the sense that “smart”/responsive polymers can become insoluble and induce amphiphilicity to their bioconjugates upon an external stimulus such as temperature and/or pH, light, electric field, chemicals and ionic strength (Fig. 3). Since stimuli-responsive polymers undergo reversible changes in size and hydrophobicity in response to external stimuli, such “smart” macromolecular chimeras could find application in medical and biotechnological processes that rely on controlling and manipulating the recognition capabilities of proteins and potentially also on the programmed formation of superstructures for biotechnology and medicine. During their pioneering studies in this area, Hoffman and collaborators extensively used the tetrameric protein streptavidin (SAv) and engineered SAv mutants as model proteins.40–45 The bioconjugation with “smart” polymers such as poly(N-isopropylacrylamide) (pNIPAAm) or poly(N,N-diethylacrylamide) (pDEAAm) was performed by either affinity bioconjugation (vide infra) or in a specific covalent manner at temperatures below the lower critical solution temperature, LCST, of the polymer. More specifically in the case of pDEAAm, the polymer was coupled to a SAv mutant approximately 20 Å from the biotin binding site through the typical mercaptyl-reactive vinyl sulfone group.41,45 It was shown that below its LCST, the polymer exists in an extended state acting as a “blocking shield” against the binding of large biotinylated proteins to SAv. In contrast, above the LCST the polymer collapses causing an exposure of the binding sites and thereby allowing interactions. The degree of gating was found to be sensitive to the size of the interacting biotinylated protein since the immuno-γ-globulin, IgG (150 kDa) was unable to bind both below and above the LCST, the protein G (6.2 kDa) was found to bind at all temperatures while the intermediate-sized bovine serum albumin, BSA (67 kDa) could only bind at temperatures above LCST.41 The shielding was also found to depend on the size of PDEAAm suggesting that “smart” polymer shields could be tailored to achieve a wide range of size-dependent ligand discrimination with use in affinity separations, biosensors and diagnostics technologies.
The specific bioconjugation of the temperature and light responsive DMA-co-4-phenylazophenyl acrylate (DMAA) and DMA-co-N-4-phenylazophenyl acrylamide (DMAAm) to a mutant of endogluconase 12A from Trichoderma reesei (EG 12A) led to smart bioconjugates with intriguing behavior.46 In the developed system, the photo-responsive polymers serve both as antennae and actuators that reversibly respond to distinct optical signals and photo-regulate bioconjugate activity. DMAA and DMAAm display opposite photoinduced phase transitions with the temperature-responsive LCST behavior shifted in opposite directions under UV vs. VIS illumination. This responsiveness profile was utilized to engineer molecular switches that turn EG 12A “on” as DMAA expands with light, and “off” when DMAA collapses together with an opposite response from DMAAm. The bioconjugates proved to possess approximately 60% of the activity of the native enzyme when the light stimulus caused “on” response and complete lack of activity when either of the polymers was in its collapsed state. More importantly the photoswitching properties proved to be reversible and could be cycled.
Maynard and co-workers described a alternative generic method for the preparation of protein-polymer conjugates through a reversible disulfide bond without the need for postsynthesis modification of the polymers.47,48 Their studies involved grafting 2-hydroxyethyl methacrylate from a pyridyl disulfide modified initiator via ATRP.31 The resulting polymers (Mw/Mn ≈ 1.25 or less) were specifically coupled to the free cysteine residue of BSA through the preserved pyridyl disulfide end group. Maynard also recently reported on the synthesis of a “smart” ABA triblock copolymer, bearing enzymes as end A blocks.49 This synthetic strategy involved RAFT polymerization from a bistrithiocarbonate chain transfer agent (CTA) followed by radical exchange of the trithiocarbonates with a protein-reactive azo-initiator to produce difunctional pNIPAAm. The V131C mutant T4 of lysozyme (T4L) which contains one free thiol was reacted with the difunctional pNIPAAm in the presence of tris(2-carboxyethyl)phosphine hydrochloride (TCEP) and ethylenediaminetetraacetic acid (EDTA) and the homodimer structure was verified using standard bioconjugate chemistry techniques. This methodology was more recently extended to star pNIPAAm protein conjugates.50 Multimeric protein conjugates could potentially exhibit biological activities superior to that of the monomeric conjugates or the unmodified proteins.
Davis et al. constructed branched “smart” bioconjugates from mid-chain-functional poly(N-(2-hydroxypropyl) methacrylamide (pHPMA).51 A novel mid-chain-functional chain transfer agent was synthesized and used as a RAFT polymerization agent to prepare branched pHPMA containing a mid-chain thiol-reactive pyridyldisulfide group, predesigned molecular weights, narrow polydispersities and high functionalization efficiencies. Upon incubation with BSA well-defined bioconjugates with the potential to improve biomolecule stability and enhance circulation time were formed.
Following this general approach Nolte and collaborators designed a maleimide functionalized spacer bearing a 1-alkyne function at the second functional end.52 The heterobifunctional linker was initially coupled to the free cysteine of BSA through a “click” maleimide-thiol conjugation step. Subsequently a PS of well defined length (Mn = 4150 Da, Mw/Mn = 1.15) bearing a terminal azide was conjugated to the BSA-1-alkyne biohybrid via a “click”, Cu(I) catalyzed, [3 + 2] Huisgen cycloaddition reaction in a THF/phosphate buffer reaction medium.53–55 The resulting BSA-PS Giant Amphiphiles were characterized with size exclusion fast performance liquid chromatography (FPLC) while their assembly architecture was studied with TEM, revealing the formation of micellar aggregates in the range of 30–70 nm. In later experiments Nolte and co-workers applied the same methodology to prepare a BSA-lipase heterodimer56 and even followed the “clicking” reaction through fluorescence using 3-azido coumarin terminated PEG.57 “Click” chemistry mediated direct ligation was also employed by Lutz and collaborators for the bioconjugation of PS (Mn = 2200 Da, Mw/Mn = 1.21) to the sequence-defined oligopeptide TAT. The reaction proceeded in high yields when excess PS was used.58
A combination of RAFT polymerization and copper catalyzed azide-alkyne “click” cycloaddition was utilized by Sumerlin and collaborators to prepare “smart” BSA bioconjugates.59 Initially, propargyl maleimide was coupled to the free cysteine residue of BSA and in a second step a azido-terminated pNIPAM, which was prepared via RAFT, was “clicked” to the functionalized BSA under the mild copper catalyzed Huisgen cycloaddition conditions. Dynamic light scattering verified the formation of large pNIPAM-BSA bioconjugates forming stable nanoparticles above the LCST.
Finally, the post functionalization approach developed by Velonia and collaborators conceptually differs from the previously employed synthetic pathways of this category in the sense that the protein is initially coupled to a multifunctional hydrophilic polymer instead of a low molecular weight spacer.60 This coupling leads to a water soluble protein-polymer biohybrid on which hydrophibicity is introduced in a subsequent step. More specifically, a hydrophilic polymer containing a terminal maleimide functionality, a gradient of glycerol and propargyl repeat units and Hostasol as fluorescent tracer (Mn = 9.5 kDa, Mw/Mn = 1.15), was synthesized via ATRP polymerization.61 Bioconjugation to the carrier protein BSA led to hydrophilic bioconjugates which were isolated using standard protein purification techniques. In the subsequent post-functionalization step, multiple hydrophobic groups were coupled to the grafted alkyne moieties of the polymer backbone by a single multi-“clicking” [3 + 2] Huisgen cycloaddition reaction inducing an amphiphilic character to the final product.53 Different aggregation behaviour was observed on the resulting Giant Amphiphiles, depending on the nature of the hydrophobic moiety which introduced the amphiphilicity (Fig. 2C and D). Following this approach, the creation of new generations of multifunctional Giant Amphiphiles bearing multiple active or responsive functionalities on the polymer backbone can easily be envisioned. It is worth mentioning that the proven compatibility of the, “click”, Huisgen [3 + 2] cycloaddition reaction with bioconjugation is of significant importance as the azide function can be engineered to recombinant proteins through non-natural aminoacids.62,53 To this end it was demonstrated that in such a CALB mutant, one of the five engineered residues of azidohomoalanine was exposed to the solvent and could undergo a [3 + 2] cycloaddition reaction.63 On the other hand, though many of the above-mentioned strategies have profited from the efficiency and compatibility of the “click” [3 + 2] Huisgen cycloaddition with bioconjugation, the effect of copper on the active site or the multiple surface functionalities of proteins has not yet been studied and should be considered especially when aiming for biomedical applications.
B “Grafting to” through non-covalent interactions approach
The non-covalent interactions found in Nature between proteins and their cofactors, the natural and synthetic host–guest interactions and metal-coordination were recruited in an attempt to construct “tethered” biomacromolecules and the aspiration to use these biohybrids as modular/responsive systems.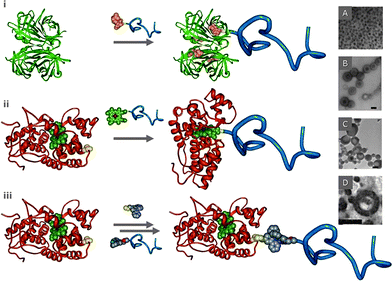 | ||
| Fig. 4 A schematic representation of the “grafting to” through non-covalent interactions, synthesis of Giant Amphiphiles viai. affinity binding, ii. cofactor reconstitution and iii. metal-to-ligand coordination. Morphologies of Giant amphiphiles: A. SAV-PS monolayer (reprinted with permission from ref. 65, copyright 2001 Wiley-VCH), B. HRP-PS (bar 200 nm, reprinted with permission from ref. 74, copyright 2006 Wiley-VCH), C. Mb-PS vesicles (bar 100 nm, reprinted with permission from ref. 74, copyright 2006 Wiley-VCH), D. Mb–PS144-b-PEG113 toroids (bar 100 nm, reprinted with permission from ref. 75, copyright 2007 ACS Publications). | ||
As previously mentioned, extensive studies on the construction of SAv bioconjugates were performed by Hoffman and collaborators.40–42 These studies demonstrated that “smart” polymer–SAv bioconjugates could act as molecular switches (gates) since small external changes in temperature or pH could alter the physical properties of the conjugated polymer and reversibly control access and/or release of biotin and biotinylated macromolecules.40–42,66,67 For example control over the aggregation of SAv-“smart” bioconjugates was demonstrated through a biotin-terminated pNIPAAm-b-pAA (poly(acrylic acid), 17.4 kDa, Mw/Mn = 1.09) which was conjugated to SAv through biotin affinity.68 It was found that the typical aggregation and phase separation of the pNIPAAm–SAv conjugates which follows the thermally induced collapse and dehydration of pNIPAAm was prevented through the shielding action of the pAA block. In addition, the cloud point and aggregation properties of the [(pNIPAAm)-b-(pAA)]-SAv conjugate were found to depend on pH, verifying that the aggregation properties of “smart” protein-polymer conjugates significantly differ from those of the free “smart” block copolymers. Uludag and collaborators studied the thermoreversible pNIPAAm comprising protein-reactive N-acryloxysuccinimide (NASI) and hydrophobic alkylmethacrylates (AMAs).69 The LCSTs of such polymers were shown to be effectively controlled by the AMA mole content. AMAs with longer side-chains were found to be more effective in lowering the LCST while, polymers without NASI exhibited a stable LCST in phosphate buffer. The enormous application potential of the SAv-biotin approach was demonstrated in several studies. For example, Stayton and coworkers reversibly immobilized immunoassay components within microfluidic channels and controlled them by integrated heating elements providing a “smart” mobile affinity matrix for microfluidic immunoassays.70 The technique involved co-modification of latex beads with the temperature-responsive polymer pNIPAAm and biotinylated PEG. The reversible transition of pNIPAAm was utilized to drive the aggregation and dis-aggregation of the modified beads in heated zones within the microchannels. When biotinylated monoclonal antibodies for the drug digoxin were bound via streptavidin to the biotin-PEG-coated beads, the beads were reversibly immobilized to the surface of PET microfluidic channels in response to a thermal stimulus. In a quantitative competitive assay the antibodies were shown to reversibly bind digoxin from a flow stream. In a separate study, the feasibility of localizing a therapeutic protein, the recombinant human bone morphogenetic protein-2 (rhBMP-2), to a polymer application site was explored in a rat intramuscular injection model. The results indicated that polymers capable of conjugating to rhBMP-2 were most effective in localizing the protein irrespective of the LCST (13–25 °C) proving that thermosensitive polymers can be engineered for delivery to improve therapeutic efficacy of proteins. It was further demonstrated that fibroblast treatment with the polymer-conjugated form of IFN-γ induced and prolonged the expression of indoleamine 2,3-dioxygenase (IDO) mRNA and protein in a three-dimensional cell culture system in vitro, a method proposed for developing non-rejectable skin substitutes for wound coverage and wound healing promoting factors.71 Though the latter cases do not strictly fall in the category of well defined Giant Amphiphiles, as the responsive polymers were more or less randomly coupled to the proteins, they are included in this review as examples of the application potential of biomacromolecular chimeras.
The cofactor reconstitution approach was also used for the construction of ABC triblock copolymers in which one of the ABC blocks is the enzyme.75 For this purpose, a synthetic diblock, polystyrene-b-polyethylene glycol (PSm-b-PEG113, with m varied), was end functionalized with the heme cofactor through a “click” [3 + 2] Huisgen cycloaddition reaction.53 The reconstitution of apo-HRP and apo-Mb around the polymers led to the biotriblock copolymers which assembled in a large variety of superstructures in aqueous solutions depending on the protein and/or the PS block length.
C “Grafting from” approach
The specific “grafting” of polymers directly “from” protein macroinitiators is a newly introduced, extremely promising method as it bypasses all the restrictions caused by component incompatibility, the need for tedious purifications, the efficiency of necessary postsynthesis modifications and is applicable to a variety of proteins (Fig. 5). This approach was very elegantly introduced by Maynard and collaborators.23,79,80 In a first demonstration of the efficiency of this method, SAv was coupled with a biotinylated ATRP initiator to produce a biomacroinitiator which was then exposed to an aqueous solution of CuBr/2,2′-bipyridine containing either NIPAAm or poly(ethylene glycol) methyl ether methacrylate (PEGMA) as monomers.79 The polymerization reaction was conducted in the presence of 2-bromoisobutyrate-functionalized Wang resin acting as sacrificial initiator to minimize the amount of protein used. The formation of the “smart” protein-polymer bioconjugates was confirmed by size exclusion chromatography (SEC) while the grafted polymer was characterized using standard polymer chemistry techniques upon dissociation from SAv. In a separate study by the Maynard group, the free cysteines of BSA (Cys-34) and of the mutant T4 lysozyme (V131C) were functionalized with heterobifuctional spacers possessing an ATRP initiator and a thiol specific end functionality which could link to the protein in either a reversible disulfide linkage (pyridyl disulfide) or an irreversible bond (maleimide).80 Polymerization of NIPAAm from these biomacroinitiators, led to the thermoresponsive BSA-pNIPAAm and lysozyme-pNIPAAm with yields larger than 65%. Interestingly, the lytic activity of lysozyme was retained in the bioconjugate, while it was also demonstrated that a “sacrificial” resin-bound initiator was not necessary for large amounts of protein macroinitiator.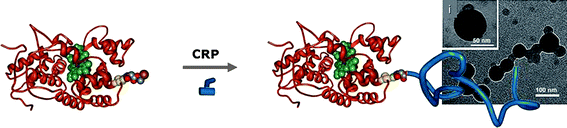 | ||
| Fig. 5 A schematic representation of the “grafting from” synthesis of Giant Amphiphilesi. Morphology of BSA-PS Giant amphiphiles (reprinted with permission from ref. 84, copyright 2008 Wiley-VCH). | ||
Near-uniform (Mw/Mn = 1.05) “smart” protein–polymer conjugates were created by Matyjaszewski and co-workers by chymotrypsin initiated ATRP of monomethoxy poly(ethylene glycol)–methacrylate.81 During this study, an increase in the molecular weights and polydispersity indices of the bioconjugates was observed when the number of ATRP initiating positions per molecule of protein was increased. Various other nonionic, cationic and anionic monomers were investigated to demonstrate the generic nature of the ATRP “grafting from” methodology while, all bioconjugates were found to retain 50 to 80% of the original enzyme activity.
Well-defined “smart” protein-polymer bioconjugates were prepared also via in situ RAFT polymerization.82 A hydrophilic RAFT agent was initially conjugated to the free thiol of BSA to form a BSA-macroRAFT agent which was then used to control the polymerization of two different hydrophilic monomers i.e. NIPAAm and hydroxyethyl acrylate (HEA) in aqueous medium. The controlled character of the RAFT polymerizations was found to be unaffected during the polymerization while the “smart” BSA bioconjugates were found to retain esterase activity. Hybrid temperature-dependent phase separation and aggregation behaviour was expressed in the bioconjugates with the LCST increasing with the decrease of the molecular weight of the pNIPAAm block.
Very recently, Sumerlin and collaborators reported on a novel approach to create “smart” protein-polymer biohybrids with RAFT agent immobilization via the “R-group” approach.83 In this case, the protein was initially modified with a RAFT agent and subsequently subjected to polymerization to afford reductively stable, high molecular weight pNIPAAm bioconjugates without adverse effects on the protein structure. The responsive behaviour of the smart bioconjugates allowed isolation and, even more interestingly, environmental modulation of their bioactivity.
Finally, Giant Amphiphiles were also prepared by Velonia and collaborators via BSA-initiated ATRP of styrene.84 During these studies a maleimide appended ATRP initiator was initially conjugated to the free thiol of BSA or HSA (human serum albumin), while the ATRP polymerization of styrene was subsequently performed in aqueous solutions using standard ATRP reaction conditions. The resulting Giant Amphiphiles were fully characterized and found to display significantly low polydispersities and the characteristic aggregation properties of amphiphilic biomacromolecules while the degree of polymerization was found to be controlled by the excess of the hydrophobic monomer. More importantly the ability to form multi-enzyme nanoreactors was demonstrated by performing the BSA-initiated ATRP polymerization in the presence of a guest protein. The protein, HRP, was found to be statistically included within the BSA-PS superstructures and retain catalytic activity.
The method of specifically “grafting” polymers directly “from” protein macroinitiators is extremely promising as it bypasses most of the restrictions paused by the amphiphilic character of Giant Amphiphiles. In fact, it was recently demonstrated that when hydrophilic pOEGMA was “grafted from” the N-terminus of myoglobin via ATRP polymerization, low polydispersity biopolymers with significantly improved pharmacological properties as compared to those of the native Mb, were produced.85 Nevertheless, as in the case of the copper catalyzed Huisgen cycloaddition, possible adverse interactions of the residual transition metal CRP catalysts with the protein active site or surface functionalities should be carefully considered, especially when aiming for biorelated applications. Equally as important is the evaluation of the protein macroinitiator stability under the polymerization reaction conditions.
Conclusions
This review illustrates the range of well-defined protein-polymer amphiphiles that were prepared in very recent years following significant advances in polymer and biological chemistry. Giant Amphiphiles were designed to express the well controlled molecular masses, topology and “high end” functionality (recognition, catalysis, information processing, responsiveness) of both their biological and synthetic moiety on the molecular and supramolecular level. The prototype studies leading to such biomacromolecular chimeras were presented and classified here in terms of the conceptually different approaches developed to tackle their synthesis. The variety of high turnover methodologies which led to specific bioconjugation of natural and engineered proteins to a plethora of synthetic polymers was presented in respect to the individuality of each system. The establishment of these new, generic synthetic approaches probably represents the most significant outcome of the pilot studies to bridge the synthetic to the natural world as it opens up routes to novel generations of molecularly engineered biomaterials and biomaterial scaffolds for specific applications. Notably the macromolecular Giant Amphiphiles were shown to exhibit assembling properties similar to those observed in amphiphiles of other categories while, the intrinsic properties of both their synthetic and biological component were expressed in several of the studies which were discussed. Future studies are now challenged to combine the acquired knowledge and comply with the increased demand for high added value biomaterials. To achieve this, the fundamental limitations of either the chosen synthetic route or the stability of the biocomponent itself need to be addressed and surpassed. Among the challenges leading to future applications is the establishment of universal synthetic approaches, the elimination of the factors affecting protein integrity in order to profit from the full spectrum of their functionalities and the combination of several proteins with biocompatible, biodegradable and/or (multi)functional polymers. Rational design of new generations of biomacromolecular chimeras based on the knowledge acquired during the pilot studies presented here is therefore expected to have a profound effect on the programmed formation of ideal scaffolds or bioactive components aimed for high-impact biophysical, medicinal and biotechnological applications.Notes and references
- A. S. Hoffman and P. S. Stayton, Macromol. Symp., 2004, 207, 139 CrossRef CAS.
- B. Gallot, Prog. Polym. Sci., 1996, 21, 1035 CrossRef CAS.
- R. Langer and D. A. Tirrell, Nature, 2004, 428, 487 CrossRef CAS.
- R. Duncan, Nat. Rev. Drug Discovery, 2003, 2, 347 CrossRef CAS; E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974 CrossRef CAS.
- F. F. Davis, Adv. Drug Delivery Rev., 2002, 54, 457 CrossRef CAS.
- A. Abuchowski, T. van Es, N. C. Palczuk and F. F. Davis, J. Biol. Chem., 1977, 252, 3578 CAS.
- F. M. Veronese, Biomaterials, 2001, 22, 405 CrossRef CAS; P. Caliceti and F. M. Veronese, Adv. Drug Delivery Rev., 2003, 55, 1261 CrossRef CAS; H. Maeda, Adv. Drug Delivery Rev., 2001, 46, 169 CrossRef CAS.
- S. Zalipsky, Adv. Drug Delivery Rev., 1995, 16, 157 CrossRef CAS; K. Ulbrich, J. Strohalm, V. Subr, D. Plocova, R. Duncan and B. Rihova, Macromol. Symp., 1996, 103, 177 CAS.
- G. M. Whitesides, Small, 2005, 1, 172 CrossRef CAS.
- Nanobiotechnology: Concepts, Applications and Perspectives, ed. C. M. Niemeyer, C. A. Mirkin, Wiley-VCH, Weinheim, 2004 Search PubMed.
- D. S. Goodsell, Bionanotechnology: Lessons from Nature, Wiley-Liss, Hoboken, NJ, 2004 Search PubMed.
- R. S. Tu and M. Tirrell, Adv. Drug Delivery Rev., 2004, 56, 1537 CrossRef CAS.
- S. Zhang, Nat. Biotechnol., 2003, 21, 1171 CrossRef CAS.
- M. Gross, Travels to the Nanoworld: Miniature Machinery in Nature and Technology, Plenum, New York, 1999 Search PubMed; C.-I. Branden and J. Tooze, Intronduction to Protein Structure, Garland, New York, 2nd edn, 1999 Search PubMed; J. M. Benyus, Biomimicry: Innovation Inspired by Nature, Quill-William Morrow, New York, 1997 Search PubMed.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267 CrossRef CAS; D. Y. Chen and M. Jiang, Acc. Chem. Res., 2005, 38, 494 CrossRef CAS; H. W. Shen and A. Eisenberg, Angew. Chem., Int. Ed., 2000, 39, 3310 CrossRef CAS; L. F. Zhang and A. Eisenberg, Polym. Adv. Technol., 1998, 9, 677 CrossRef CAS; G. J. Liu, Curr. Opin. Colloid Interface Sci., 1998, 3, 200 CAS.
- G. T. Hermanson, Bioconjugate Techniques, 1996, Academic, San Diego, CA Search PubMed.
- F. Rusmini, Z. Zhong and J. Feijen, Biomacromolecules, 2007, 8, 1775 CrossRef CAS.
- P. Thordarson, B. Le Droumaguet and K. Velonia, Appl. Microbiol. Biotechnol., 2006, 73, 243 CrossRef CAS.
- G. Pasut and F. M. Veronese, Prog. Polym. Sci., 2007, 32, 933 CrossRef CAS.
- M. A. Gauthier and H.-A. Klok, Chem. Commun., 2008, 2591 RSC.
- I. S. Carrico, Chem. Soc. Rev., 2008, 37, 1423 RSC.
- L. S. Wong, F. Khan and J. Micklefield, Chem. Rev., 2009, 109, 4025 CrossRef CAS.
- K. L. Heredia and H. D. Maynard, Org. Biomol. Chem., 2007, 5, 45 RSC.
- H.-A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 1 CrossRef CAS.
- H. G. Börner and H. Schlaad, Soft Matter, 2007, 3, 394 RSC; J.-F. Lutz and H. G. Börner, Prog. Polym. Sci., 2008, 33, 1 CrossRef CAS.
- C. de las Heras Alarcon, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276 RSC; D. Cunliffe, S. Pennadam and C. Alexander, Eur. Polym. J., 2004, 40, 5 CrossRef CAS.
- D. W. P. M. Löwik, L. Ayres, J. M. Smeenk and J. C. M. van Hest, Adv. Polym. Sci., 2006, 202, 19; A. (T). J. Dirks, R. J. M. Nolte and J. J. L. M. Cornelissen, Adv. Mater., 2008, 20, 3953 CrossRef CAS.
- A. S. Hoffman and P. S. Stayton, Prog. Polym. Sci., 2007, 32, 922 CrossRef CAS.
- Protein models created using the Protein Workshop software. J. L. Moreland, A. Gramada, O. V. Buzko, Q. Zhang and P. E. Bourne, BMC Bioinformatics, 2005, 6, 21 Search PubMed.
- L. W. Seymour, R. Duncan, J. Strohalm and J. Kopecek, J. Biomed. Mater. Res., 1987, 21, 1341 CAS; A. Godwin, M. Hartenstein, A. H. E. Muller and S. Brocchini, Angew. Chem., Int. Ed., 2001, 40, 594 CrossRef CAS.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721 CrossRef CAS; J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614 CrossRef CAS; M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689 CrossRef CAS.
- A. B. Lowe and C. L. McCormick, Handb. RAFT Polym., 2008, 235 Search PubMed; E. Rizzardo, G. Moad and S. H. Thang, Handb. RAFT Polym., 2008, 189 Search PubMed.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661 CrossRef CAS.
- M. D. Best, Biochemistry, 2009, 48, 6571 CrossRef CAS.
- C. Barner-Kowollik and A. J. Inglis, Macromol. Chem. Phys., 2009, 210, 987 CrossRef CAS.
- Q. Wang, A. R. Parrish and L. Wang, Chem. Biol., 2009, 16, 323 CrossRef CAS; R. E. Connor and D. A. Tirrell, Polym. Rev., 2007, 47, 9 Search PubMed; N. Hino, Y. Okazaki, T. Kobayashi, A. Hayashi, K. Sakamoto and S. Yokoyama, Nat. Methods, 2005, 2, 201 CrossRef CAS; N. Budisa, Angew. Chem., Int. Ed., 2004, 43, 6426 CrossRef CAS; D. S. Yeo, R. Srinivasan, G. Y. Chen and S. Q. Yao, Chem.–Eur. J., 2004, 10, 4664 CrossRef CAS; K. Rose, J. Am. Chem. Soc., 1994, 116, 30 CrossRef; H. F. Gaertner, R. E. Offord, R. Cotton, D. Timms, R. Camble and K. Rose, J. Biol. Chem., 1994, 269, 7224 CAS.
- K. Velonia, A. E. Rowan and R. J. M. Nolte, J. Am. Chem. Soc., 2002, 124, 4224 CrossRef CAS.
- Y. Yu, L. Zhang and A. Eisenberg, Macromolecules, 1998, 31, 1144 CrossRef CAS.
- G. L. Ellman, Arch. Biochem. Biophys., 1959, 82, 70 CrossRef CAS.
- Z. Ding, C. J. Long, Y. Hayashi, E. V. Bulmus, A. S. Hoffman and P. S. Stayton, Bioconjugate Chem., 1999, 10, 395 CrossRef CAS.
- Z. Ding, R. B. Fong, C. J. Long, P. S. Stayton and A. S. Hoffman, Nature, 2001, 411, 59 CrossRef CAS.
- V. Bulmus, Z. Ding, C. J. Long, P. S. Stayton and A. S. Hoffman, Bioconjugate Chem., 2000, 11, 78 CrossRef CAS.
- A. G. Chilkoti, G. Chen, P. S. Stayton and A. S. Hoffman, Bioconjugate Chem., 1994, 5, 504 CrossRef CAS.
- Z. Ding, G. Chen and A. S. Hoffman, J. Biomed. Mater. Res., 1998, 39, 498 CAS.
- P. S. Stayton, T. Shimoboji, C. Long, A. Chilkoti, G. Chen, J. M. Harris and A. S. Hoffman, Nature, 1995, 378, 472 CrossRef CAS.
- T. Shimoboji, E. Larenas, T. Fowler, S. Kulkarni, A. S. Hoffman and P. S. Stayton, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16592 CrossRef CAS.
- D. Bontempo, K. L. Heredia, B. A. Fish and H. D. Maynard, J. Am. Chem. Soc., 2004, 126, 15372 CrossRef CAS.
- H. D. Maynard, K. L. Heredia, R. C. Li, D. P. Parra and V. Vázquez-Dorbatt, J. Mater. Chem., 2007, 17, 4015 RSC.
- L. Tao, C. S. Kaddis, R. R. Ogorzalek Loo, G. N. Grover, J. A. Loo and H. D. Maynard, Chem. Commun., 2009, 2148 RSC.
- L. Tao, C. S. Kaddis, R. R. Ogorzalek Loo, G. N. Grover, J. A. Loo and H. D. Maynard, Macromolecules, 2009, 42, 8028 CrossRef CAS.
- L. Tao, J. Liu and T. P. Davis, Biomacromolecules, 2009, 10, 2847 CrossRef CAS.
- A. J. Dirks, S. S. van Berkel, N. S. Hatzakis, J. A. Opsteen, F. L. van Delft, J. J. L. M. Cornelissen, A. E. Rowan, J. C. M. van Hest, F. P. J. T. Rutjes and R. J. M. Nolte, Chem. Commun., 2005, 4172 RSC.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS; C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef CAS; V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS; R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 565 CrossRef; R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 633 CrossRef; R. Huisgen, in 1,3-Dipolar Cycloaddition Chemistry, ed. A. Padwa, Wiley, New York 1984, p. 1 Search PubMed.
- M. D. Best, Biochemistry, 2009, 48, 6571 CrossRef CAS.
- B. Le Droumaguet and K. Velonia, Macromol. Rapid Commun., 2008, 29, 1073 CrossRef.
- N. S. Hatzakis, H. Engelkamp, K. Velonia, J. Hofkens, P. C. M. Christianen, A. Svendsen, S. A. Patkar, J. Vind, J. C. Maan, A. E. Rowan and R. J. M. Nolte, Chem. Commun., 2006, 2012 RSC.
- A. J. Dirks, J. J. L. M. Cornelissen and R. J. M. Nolte, Bioconjugate Chem., 2009, 20, 1129 CrossRef CAS.
- J.-F. Lutz, H. G. Börner and K. Weichenhan, Aust. J. Chem., 2007, 60, 410 CrossRef CAS.
- M. Li, P. De, S. R. Gondi and B. S. Sumerlin, Macromol. Rapid Commun., 2008, 29, 1172 CrossRef CAS.
- B. Le Droumaguet, G. Mantovani, D. M. Haddleton and K. Velonia, J. Mater. Chem., 2007, 17, 1916 RSC.
- V. Ladmiral, G. Mantovani, G. J. Clarkson, S. Cauet, J. L. Irwin and D. M. Haddleton, J. Am. Chem. Soc., 2006, 128, 4823 CrossRef CAS; J. Geng, G. Mantovani, L. Tao, J. Nicolas, G. Chen, R. Wallis, D. A. Mitchell, B. R. G. Johnson, S. D. Evans and D. M. Haddleton, J. Am. Chem. Soc., 2007, 129, 15156 CrossRef CAS.
- K. L. Kiick, E. Saxon, D. A. Tirrell and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 19 CrossRef CAS; E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007 CrossRef CAS.
- S. Schoffelen, M. H. L. Lambermon, M. B. van Eldijk and J. C. M. van Hest, Bioconjugate Chem., 2008, 19, 1127 CrossRef CAS.
- C. M. Niemeyer, Chem.–Eur. J., 2001, 7, 3188 CrossRef CAS; C. M. Niemeyer, Int. J. Environ. Anal. Chem., 2005, 85, 639 CrossRef CAS; C. M. Niemeyer, Nano Today, 2007, 2, 42 CrossRef.
- J. M. Hannink, J. J. L. M. Cornelissen, J. A. Farrera, P. Foubert, F. C. De Schryver, N. A. J. M. Sommerdijk and R. J. M. Nolte, Angew. Chem., Int. Ed., 2001, 40, 4732 CrossRef CAS.
- P. S. Stayton, T. Shimoboji, C. Long, A. Chilkoti, G. Chen, J. M. Harris and A. S. Hoffman, Nature, 1995, 378, 472 CrossRef CAS.
- T. Shimoboji, Z. Ding, P. S. Stayton and A. S. Hoffman, Bioconjugate Chem., 2001, 12, 314 CrossRef CAS.
- S. Kulkarni, C. Schilli, B. Grin, A. H. E. Müller, A. S. Hoffman and P. S. Stayton, Biomacromolecules, 2006, 7, 2736 CrossRef CAS.
- H. Uludag, B. Norrie, N. Kousinioris and T. Gao, Biotechnol. Bioeng., 2001, 73, 510 CrossRef CAS.
- N. Malmstadt, A. S. Hoffman and P. S. Stayton, Lab Chip, 2004, 4, 412 RSC.
- K. Sarkhosh, E. E. Tredget, H. Uludag, R. T. Kilani, A. Karami, Y. Li, T. Iwashina and A. Ghahary, J. Cell. Physiol., 2004, 201, 146 CrossRef CAS.
- L. Fruk, C.-H. Kuo, E. Torres and C. M. Niemeyer, Angew. Chem., Int. Ed., 2009, 48, 1550 CrossRef CAS.
- M. J. Boerakker, J. M. Hannink, P. H. H. Bomans, P. M. Frederik, R. J. M. Nolte, E. M. Meijer and N. A. J. M. Sommerdijk, Angew. Chem., Int. Ed., 2002, 41, 4239 CrossRef CAS.
- M. J. Boerakker, N. E. Botterhuis, P. H. H. Bomans, P. M. Frederik, E. M. Meijer, R. J. M. Nolte and N. A. J. M. Sommerdijk, Chem.–Eur. J., 2006, 12, 6071 CrossRef CAS.
- I. C. Reynhout, J. J. L. M. Cornelissen and R. J. M. Nolte, J. Am. Chem. Soc., 2007, 129, 2327 CrossRef CAS.
- K. Velonia, P. Thordarson, P. R. Andres, U. S. Schubert, A. E. Rowan and R. J. M. Nolte, Polym. Prepr., 2003, 44, 648 CAS.
- J. R. Peterson and P. Thordarson, Chiang Mai J. Sci., 2009, 36, 236 Search PubMed.
- J. R. Peterson, T. A. Smith and P. Thordarson, Chem. Commun., 2007, 1899 RSC.
- D. Bontempo and H. D. Maynard, J. Am. Chem. Soc., 2005, 127, 6508 CrossRef CAS.
- K. L. Heredia, D. Bontempo, T. Ly, J. T. Byers, S. Halstenberg and H. D. Maynard, J. Am. Chem. Soc., 2005, 127, 16955 CrossRef CAS.
- B. S. Lele, H. Murata, K. Matyjaszewski and A. J. Russell, Biomacromolecules, 2005, 6, 3380 CrossRef CAS.
- C. Boyer, V. Bulmus, J. Liu, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, J. Am. Chem. Soc., 2007, 129, 7145 CrossRef CAS.
- P. De, M. Li, S. R. Gondi and B. S. Sumerlin, J. Am. Chem. Soc., 2008, 130, 11288 CrossRef CAS.
- B. Le Droumaguet and K. Velonia, Angew. Chem., Int. Ed., 2008, 47, 6263 CrossRef CAS.
- W. Gao, W. Liu, J. A. Mackay, M. R. Zalutsky, E. J. Toone and A. Chilkoti, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15231, DOI:10.1073/pnas.0904378106.
| This journal is © The Royal Society of Chemistry 2010 |