The development of poly(dendrimer)s for advanced processing
Jack P.
Gunning
a,
Jack W.
Levell
b,
Mark F.
Wyatt
c,
Paul L.
Burn
*d,
Jeremy
Robertson
a and
Ifor D. W.
Samuel
*b
aDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, Mansfield Rd, Oxford, UK OX1 3TA
bOrganic Semiconductor Centre, SUPA, School of Physics and Astronomy, University of St. Andrews, North Haugh, St. Andrews, Fife, UK KY16 9SS. E-mail: idws@st-andrews.ac.uk
cEPSRC National Mass Spectrometry Service Centre (NMSSC), Institute of Mass Spectrometry (IMS), School of Medicine, Swansea University, Singleton Park, Swansea, UK SA2 8PP
dCentre for Organic Photonics & Electronics, School of Molecular and Microbial Sciences, University of Queensland, Chemistry Building, Queensland, 4072, Australia. E-mail: p.burn2@uq.edu.au
First published on 5th March 2010
Abstract
A norbornenyl-based homopolymer that has a dendronised iridium(III) complex attached to every monomer unit has been synthesized. The dendronised iridium(III) complex is comprised of three facially arranged 2-phenylpyridyl ligands. Two of the ligands bear first generation biphenyl-based dendrons with 2-ethylhexyloxy surface groups attached and the third ligand is attached to the polymer backbone via a benzyloxy ester. The polymer was formed by ring opening metathesis using the Grubbs III catalyst and was found to have an Mp of 130 kDa by MALDI-TOF mass spectrometry. At a concentration of 25 mg cm−3 the polymer solution had a viscosity of 1.09 mPa s, which was 34% higher than a solution containing a dendrimer of same weight per volume. The dendrimer had the same core, dendrons, and surface groups but differed from the polymer in that it had dendrons attached to three of the ligands rather than the two of the polymer. The solution photoluminescence quantum yield (PLQY) of the poly(dendrimer) was found to be 57%, indicating that intra-polymer chromophore interactions were not leading to strong quenching of the luminescence. However, in the solid-state the PLQY dropped significantly, indicating that inter-polymer chromophore interactions were significant. The presence of the dendrons allowed the simple blending of the polymer with 4,4′-bis(N-carbazolyl)-2,2′-biphenyl (CBP), and the blended film had a PLQY of 50%. Simple bilayer devices with a blended emissive layer and an electron injection and transport layer had an external quantum efficiency of 6.2% at a brightness of 100 cd m−2, showing that poly(dendrimer)s are a promising class of OLED material.
Introduction
Solution processable phosphorescent complexes are of interest for application in organic light-emitting diodes (OLEDs) and bulk heterojunction organic solar cells (OSCs). Heavy metals such as osmium, ruthenium, rhenium, platinum, and iridium all give rise to phosphorescent materials, and of these iridium(III) complexes have been the most widely studied.1 More recently it has been found that iridium(III) complexes, when blended with traditional binary mixtures in bulk heterojunction solar cells, can lead to more efficient devices.2 However, simple small molecule iridium(III) complexes tend not be very soluble, which limits their processing to evaporation under high vacuum. Whilst excellent devices can be formed by this method there is now concern that the evaporation process can in some cases lead to isomerisation during the deposition,3 and devices tend to be limited to smaller sizes. To overcome this there has been significant effort to develop solution processable iridium(III) complexes. While ‘monochrome’ devices can be formed by simple processes such as spin-coating, the formation of more complex pixelated structures requires techniques such as inkjet printing or photopatterning. An advantage of inkjet printing over photopatterning is that it makes better use of the materials as each material is only deposited where it is required. However, while in principle inkjet printing is a simple process, the need to deposit materials in the desired position with the correct pixel shape requires careful control over the materials properties and processing conditions. The printing of polymers is dependent on many factors including solvent(s), temperature, surface tension, and ink viscosity with latter dependent on concentration, polymer molecular weight and polydispersity.4 The two primary strategies that have been adopted to achieve solution processable iridium(III) complexes that could lead to inkjet printable materials have been to attach the complex to a polymer backbone,5 or to encapsulate it within a dendritic architecture.6 In the cases where the complexes are attached to the polymer backbone, the polymers have been primarily copolymers, with a very low percentage of the monomer composed of the complex, with the remaining units variably containing hole and electron transport moieties. However, a homopolymer containing a simple iridium(III) complex has been recently reported.7 The polymer consisted of bis(2-phenylpyridyl)iridium(III) acetylacetonate complexes attached to a poly(styrene) backbone (1 in Fig. 1). Remarkably, the polymer was solution processable in spite of there being no solubilising groups. However, the solid-state photoluminescence quantum yield of neat and blended films was low. Hence there is a need to be able to control the intermolecular interactions of the emissive chromophores within such homopolymers in the solid-state. Dendrimer architectures that have the phosphorescent complexes at their core can control the intermolecular interactions that govern the optoelectronic properties of the materials, but the viscosity of the solutions used for processing is quite low. It would therefore be advantageous to have the control over the intermolecular interactions provided by the dendritic architecture, combined with the viscosity associated with polymers to allow patterning by inkjet printing.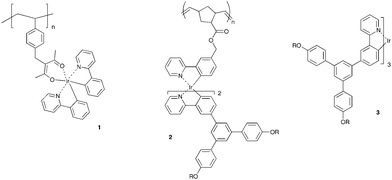 | ||
| Fig. 1 Structures of a polystyrene with small molecule iridium(III) complex side chains 1, the poly(dendrimer) of this work 2, and a first generation dendrimer 3 that has the same ligands and dendrons as 2, R = 2-ethylhexyl. | ||
In this manuscript we describe the preparation and properties of a poly(dendrimer) that contains phosphorescent iridium(III) complexes at the core of side chain dendrimers (2 in Fig. 1). The design concept of the poly(dendrimer) was to have the polymer backbone buried inside the dendrimer coating with the dendrons on the outer surface responsible for controlling the intermolecular interactions of the emissive cores of neighboring chains. The polymer backbone is formed by a ring opening polymerisation of a norbornene monomer. We chose pure exo-norbornene isomers for the polymerization as they have been shown to exhibit significantly higher rates of propagation under milder conditions, with higher conversions, and more control than the 80 : 20 endo–exo isomeric mixtures available from a Diels–Alder reaction of cyclopentadiene and a non-symmetric dienophile.8 The higher rate of propagation is primarily due to reduced steric interactions between the growing polymer chains and the incoming monomer.9 The dendronised iridium(III) complex side-chain moiety consists of three 2-phenylpyridyl ligands. Attached to the phenyl rings of two of the 2-phenylpyridyl ligands are first generation dendrons that are composed of biphenyl units and 2-ethylhexyloxy surface groups. The third 2-phenylpyridyl ligand provides the attachment point to the polymer backbone. The advantage of using three cyclometallated ligands is that the stability of the complex should be improved due to the larger binding constant of the 2-phenylpyridyl moiety relative to the acetylacetonate ligand. We describe the synthesis of the homopolymer and its physical, photophysical and device properties, and compare them to the monomer, and a dendrimer that has the same core, dendrons, and surface groups but with a dendron attached to each ligand (3 in Fig. 1).
Results and discussion
The synthetic strategy for the formation of the poly[iridium(III) dendrimer] 2 is shown in Scheme 1. The first step in the synthesis involves cleaving tetra[2-(5-{3,5-bis[4-(2-ethylhexyloxy)phenyl]phenyl}phenyl)pyridyl]diiridium(III) dichloride 410 with 3-(pyridin-2-yl)benzaldehyde.11 One of the key problems with making heteroleptic iridium(III) complexes with ligands that have similar binding constants is that ligand scrambling can occur.10 That is, while the dimer 4 may have two dendritic ligands, it is possible that during the process of cleaving the dimer and forming the desired heteroleptic complex, one or more of the dendronised ligands could be replaced by a 3-(pyridin-2-yl)benzaldehyde ligand. Given the relatively small quantities used, it was necessary to carry out the cleaving of the dimer in solution. Although 2-ethoxyethanol has been successfully used as a solvent for this type of reaction, it was not successful for the current work. We found that the highest yields were achieved when dry diethylene glycol dimethyl ether was used as the solvent. When dimer 4 was reacted with 3-(pyridin-2-yl)benzaldehyde in the presence of silver triflate at 110 °C for 16 hours, after careful purification it was possible to isolate the desired heteroleptic complex 5 in a 42% yield. Iridium(III) complexes can have either a facial or a meridional structure. In the case of homoleptic complexes the isomer assignment by 1H NMR is straightforward due to the symmetry, that is, the facial isomer has one set of signals for each type of proton whereas the meridional has three.12 The asymmetric nature of the heteroleptic complex 5 caused by the introduction of the 3-(pyridin-2-yl)benzaldehyde ligand made the determination of whether it was the facial or meridional isomer more difficult. However, the 1H NMR of 5 showed that there was a single isomer and comparison with that of fac- and mer-tris(2-phenylpyridyl)iridium(III)11 and the homoleptic dendrimer 313 indicated that 5 was the facial isomer. The next step in the synthesis involved the reduction of aldehyde 5 to the benzylalcohol 6, and this was achieved in a quantitative yield using lithium aluminium hydride in diethylether at 0 °C. For the coupling of dendrimer 6 to the norbornenyl monomer, commercially available exo-bicyclo[2.2.1]hept-5-ene-2-carboxylic acid14 was converted to the corresponding acid chloride with oxalylchloride. The acid chloride was then coupled to 6 to form the ester-linked monomer 7 in an 87% yield. Given that double bonds have been reported to provide pathways for the quenching of the luminescence, we were interested to see whether the double bond in the monomer would have an effect on the photophysical properties of the monomer. We therefore treated 7 with hydrogen and a palladium on carbon catalyst to give 8. Monomer 7 was then polymerized with Grubbs III catalyst15 to give polymer 2 in ∼98% yield. Finally, polymer 2 was hydrogenated to remove the double bonds in the backbone to give the poly[iridium(III) dendrimer] 9. Importantly, infrared spectroscopy showed that the ester functionality remained intact through the polymerization and hydrogenation steps, with 8 and 2 having carbonyl stretches in the range 1724–1725 cm−1. For polymer 9 there was no significant change in the molecular weight indicating that the dendrimers were still attached to the polymer backbone.![Reagents and conditions: (i) AgOSO2CF3, MeO(CH2CH2O)2Me, 110 °C, 16 h, Ar; (ii) LiAlH4, diethylether, 0 °C, 1 h, N2; (iii) exo-bicyclo[2.2.1]hept-5-ene-2-carboxylic acid chloride, py, 0 °C to rt, 16 h, N2; (iv) Pd/C, H2; (v) Grubbs III catalyst, CH2Cl2, 18 h then EtOCHCH2.](/image/article/2010/PY/c0py00039f/c0py00039f-s1.gif) | ||
Scheme 1
Reagents and conditions: (i) AgOSO2CF3, MeO(CH2CH2O)2Me, 110 °C, 16 h, Ar; (ii) LiAlH4, diethylether, 0 °C, 1 h, N2; (iii) exo-bicyclo[2.2.1]hept-5-ene-2-carboxylic acid chloride, py, 0 °C to rt, 16 h, N2; (iv) Pd/C, H2; (v) Grubbs III catalyst, CH2Cl2, 18 h then EtOCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2. CH2. | ||
The first step in the analysis of the physical properties of polymer 2 was to determine its molecular weight. Gel permeation chromatography against polystyrene standards (Fig. 2) gave an ![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) w = 4.7 × 104 and n = 3.0 × 104 and hence a polydispersity of 1.5. A polydispersity of 1.5 is larger than might be expected from a living polymerization, however, this divergence might be due to the size of the monomer that needs to be added to the chain end; that is, steric factors may play a much more important role in chain growth. The molecular weight achieved for the homopolymer is similar to that reported for random norbornenyl-based copolymers comprising a 9 : 1 ratio of carbazolyl containing monomer units and small molecule fac-tris(2-phenylpyridyl)iridium(III) complexes.16 That is, the bulky dendritic units are not detrimental to the polymerisation. In fact, the use of a dendrimer-based monomer has solved the solubility problem observed for the norbornene-based homopolymers that had small molecule iridium(III) complexes on every unit. While the poly(dendrimer) 2 had excellent solubility in a range of solvents, the equivalent small molecule-containing polymers were insoluble.16 In the latter case the insolubility of the homopolymer was ascribed to large dipole interactions causing aggregation and hence precipitation of the growing polymer chains. Interestingly we were also able to get a matrix-assisted laser desorption/ionisation time-of-flight (MALDI-TOF) mass spectrum of the polymer (Fig. 3). In the full spectrum, Mp was approximately 130 kDa (Fig. 3a), and the distribution appeared bimodal, with a secondary maximum at approximately 250 kDa (this latter peak may be due to the combined mass of two chains due to the high concentrations used in the MALDI-TOF measurement). This clearly shows that the GPC underestimates the molecular weight of the poly(dendrimer) 2. In the low mass range (Fig. 3b) a series of peaks is observed, separated by an amount corresponding to the addition of one monomer unit. Importantly, polymer 9, which does not have any remaining double bonds in the backbone, has a similar w and n as the starting material, indicating that no polymer degradation occurs during the hydrogenation. Thermogravimetric analysis of polymer 2 showed that it had high thermal stability with a 5% weight loss recorded at 378 °C. Differential scanning calorimetry showed that polymer 2 did not exhibit any thermal transitions in the range −20 to 250 °C (over three heating and cooling cycles at 20 °C min−1). Finally, we measured the viscosity of 2 at different concentrations in chlorobenzene (Fig. 4) at 20 °C. Chlorobenzene has a viscosity of 0.75 mPa s and it was found that the viscosity of the polymer solution increased with increasing concentration up to 1.09 mPa s for 25 mg cm−3. A viscosity of 1.09 mPa s is 34% higher than the first generation dendrimer 3, which had a viscosity of 0.81 mPa s.
w = 4.7 × 104 and n = 3.0 × 104 and hence a polydispersity of 1.5. A polydispersity of 1.5 is larger than might be expected from a living polymerization, however, this divergence might be due to the size of the monomer that needs to be added to the chain end; that is, steric factors may play a much more important role in chain growth. The molecular weight achieved for the homopolymer is similar to that reported for random norbornenyl-based copolymers comprising a 9 : 1 ratio of carbazolyl containing monomer units and small molecule fac-tris(2-phenylpyridyl)iridium(III) complexes.16 That is, the bulky dendritic units are not detrimental to the polymerisation. In fact, the use of a dendrimer-based monomer has solved the solubility problem observed for the norbornene-based homopolymers that had small molecule iridium(III) complexes on every unit. While the poly(dendrimer) 2 had excellent solubility in a range of solvents, the equivalent small molecule-containing polymers were insoluble.16 In the latter case the insolubility of the homopolymer was ascribed to large dipole interactions causing aggregation and hence precipitation of the growing polymer chains. Interestingly we were also able to get a matrix-assisted laser desorption/ionisation time-of-flight (MALDI-TOF) mass spectrum of the polymer (Fig. 3). In the full spectrum, Mp was approximately 130 kDa (Fig. 3a), and the distribution appeared bimodal, with a secondary maximum at approximately 250 kDa (this latter peak may be due to the combined mass of two chains due to the high concentrations used in the MALDI-TOF measurement). This clearly shows that the GPC underestimates the molecular weight of the poly(dendrimer) 2. In the low mass range (Fig. 3b) a series of peaks is observed, separated by an amount corresponding to the addition of one monomer unit. Importantly, polymer 9, which does not have any remaining double bonds in the backbone, has a similar w and n as the starting material, indicating that no polymer degradation occurs during the hydrogenation. Thermogravimetric analysis of polymer 2 showed that it had high thermal stability with a 5% weight loss recorded at 378 °C. Differential scanning calorimetry showed that polymer 2 did not exhibit any thermal transitions in the range −20 to 250 °C (over three heating and cooling cycles at 20 °C min−1). Finally, we measured the viscosity of 2 at different concentrations in chlorobenzene (Fig. 4) at 20 °C. Chlorobenzene has a viscosity of 0.75 mPa s and it was found that the viscosity of the polymer solution increased with increasing concentration up to 1.09 mPa s for 25 mg cm−3. A viscosity of 1.09 mPa s is 34% higher than the first generation dendrimer 3, which had a viscosity of 0.81 mPa s.
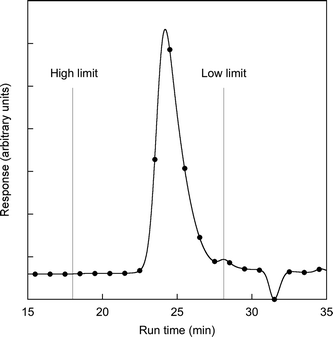 | ||
| Fig. 2 Gel permeation chromatography trace of 2 (high and low limits are the calibration limits). | ||
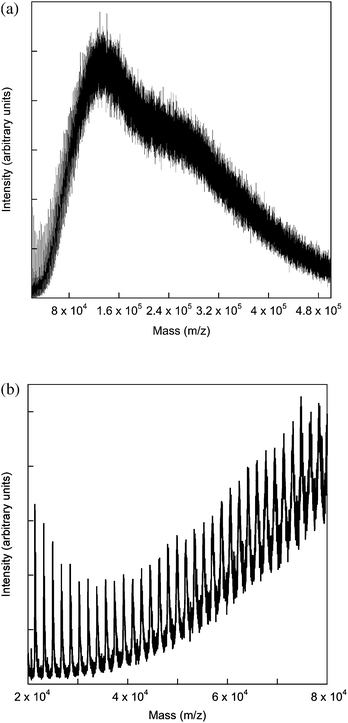 | ||
| Fig. 3 (a) MALDI-TOF mass spectrum of the high molecular weight section of polymer 2 and (b) the low molecular weight section. | ||
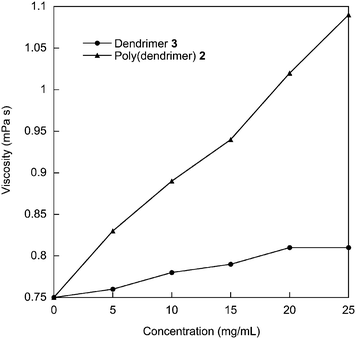 | ||
| Fig. 4 Viscosity versus concentration of 2 and dendrimer 3. | ||
Photophysical and device properties
We first investigated the photophysical properties of the materials. The solution UV-visible and photoluminescence (PL) spectra for monomer 7 and polymer 2 are shown in Fig. 5. The corresponding spectra for the hydrogenated equivalents are identical to their double bond-containing counterparts. The UV-visible spectra of the monomer and polymer comprise two regions. The strong absorptions at shorter wavelengths, below 320 nm, arise from π–π* transitions of the ligands and the biphenyl units in the dendrons. The weaker, longer wavelength absorptions are normally assigned to a series of overlapping ‘metal-to-ligand’ transitions. The solution PL spectra of the monomer 7 and model compound 8 had maxima at around 512–513 nm with polymers 2 and 9 having slightly longer wavelength maxima at 516–517 nm. The Commission Internationale de l'Eclairage (CIE) co-ordinates of the polymer 2 were (0.30, 0.62), which are essentially the same as the homoleptic dendrimer 3 (0.31, 0.63).13 Importantly, there was no strong red tail to the emission, indicating that in solution the dendrimers along the polymer backbone were not strongly interacting; that is, there was no significant emission due to excimers.17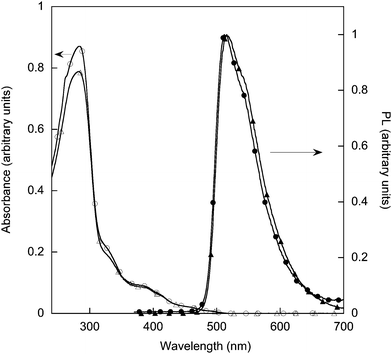 | ||
| Fig. 5 Solution UV-visible spectra of 7 (open circles) and 2 (open triangles), and PL spectra 7 (closed circles) and 2 (closed triangles) (excitation at 360 nm). | ||
We compared the solution PL quantum yields (PLQYs) of the monomers and poly(dendrimer)s. Monomer 7 and model compound 8 had PLQYs of 32% and 52%, respectively, while polymers 2 and 9 had quantum yields of 57% and 38%, respectively. An important point to note is that the PLQY of polymer 2 is significantly higher than the previously reported fac-tris(2-phenylpyridyl)iridium(III) containing norbornenyl copolymers, which had maximum PLQYs of 20–30% even though the iridium(III) complex was ‘diluted’ along the polymer backbone by other monomer units.16,18
We have investigated the photophysics further by measuring time-resolved phosphorescence of the monomer, model compound and polymers in dichloromethane solution, and the results are shown in Fig. 6. Monomer 7 has a near monoexponential decay with a lifetime of 1.5 µs, implying a radiative rate constant of 2.1 × 105 s−1. In spite of its higher PLQY, model compound 8 has a faster decay with a lifetime of 1.2 µs, giving a radiative rate constant of 4.4 × 105 s−1. The results show that the periphery of the molecule, even though not conjugated to the ligand, has a substantial effect on the rate of radiative decay of the excited state. The situation in the polymers is different—though more conventional in the sense that polymer 9, which has a lower PLQY, also has a faster decay of its phosphorescence than polymer 2. Both polymers have biexponential decays, suggesting the possibility of two different environments for the emitting species. For polymer 2 the lifetime components (pre-exponential factors) are 0.7 µs (0.31) and 1.5 µs (0.69). For polymer 9, the lifetime components are 0.6 µs (0.38) and 1.5 µs (0.62). The different effect of double bonds in the polymers compared with the monomer and model compound could arise either from the different positioning of the double bonds relative to the ligand or to a different conformation of the molecule per repeat unit. The lower PLQY and faster decay of polymer 9 compared with polymer 2 may arise from increased intra-polymer inter-chromophore interactions due to the greater flexibility of the saturated backbone of polymer 9.
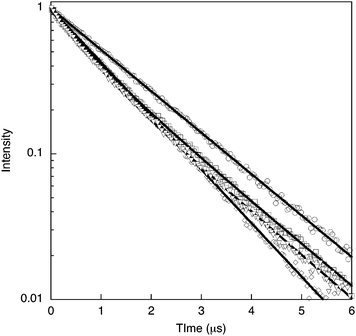 | ||
| Fig. 6 Solution time resolved luminescence of 7 (circles), 8 (diamonds), 2 (squares), and 9 (inverted triangles) (excitation at 393 nm). | ||
Thin films of polymer 2 were formed by spin-coating from a dichloromethane solution and the PL spectrum is shown in Fig. 7. The films were uniform and there was no evidence of strong aggregation with the films being clear to the eye. The film PLQY of the polymer 2 was 9% and that for monomer 7, 6%. The decrease in PLQY is significantly less than that seen in a study on a styrene-based homopolymer 1 (Fig. 1) that had bis(2-phenylpyridyl)iridium(III)-3-(4-ethylbenzyl)acetylacetonate units, which had a PLQY of less than 1%.7 In this latter case there were no dendrons attached to the ligands, and hence intermolecular interactions that lead to the quenching of the luminescence could easily occur. However, the film PLQY of monomer 7 is less than the film PLQY (65%) of the dendritic iridium(III) complex 3 that has the same dendrons and surface groups but a dendron attached to each of the 2-phenylpyridyl ligands.19 There is therefore a clear advantage of having a greater number of dendrons attached to the core complex. The quenching of the luminescence in the film of poly(dendrimer) 2 shows that the polymer backbone is less effective in controlling the intermolecular chromophore interactions in the solid state than a dendron. To understand the role the inter-polymer chromophore interactions played in the quenching of the luminescence in the solid-state, poly(dendrimer) 2 was blended with 4,4′-bis(N-carbazolyl)-2,2′-biphenyl (CBP), a host that is commonly used with small iridium(III) complexes. It was found that when the polymer was blended as a 30 wt% mixture in CBP the solid-state PLQY increased to 50%, which is similar to the solution PLQY. Importantly the emission spectrum (Fig. 7) was that of the iridium(III) complex, indicating that energy transfer from the CBP host was complete. A 30 wt% blend of polymer 2 in CBP corresponds to around 12 wt% of the iridium(III) complex in the film. For the polymer 1 with bis(2-phenylpyridyl)iridium(III)-3-(4-ethylbenzyl)acetylacetonate units a 6 wt% film showed incomplete energy transfer from the CBP host, and a 19 wt% blended film only had a PLQY of 7%.7 Therefore, not only did the addition of the dendrons improve the solution processablity of the homopolymer, but it improved the neat film performance, and the ability to blend with a host, giving a higher PLQY.
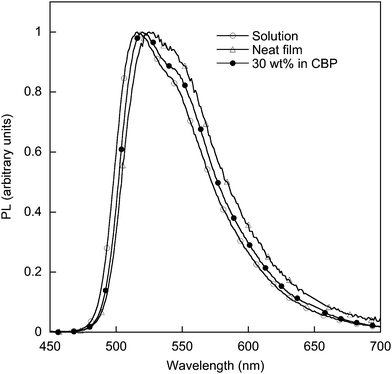 | ||
| Fig. 7 PL spectra of 2 in solution, neat film, and 30 wt% in CBP. | ||
Finally, given that the film PLQY of the 30 wt% poly(dendrimer) 6 blended with CBP was 50%, we prepared a simple bilayer device with a 1,3,5-tris(2-N-phenylbenzimidazolyl)benzene (TPBI) electron transport layer [ITO/poly(dendrimer) 2:CBP/TPBI/LiF/Al] and the device characteristics are shown in Fig. 8. The OLED characteristics (Fig. 8a and b) show that the device had a maximum external quantum efficiency (EQE) of 7.6% at low brightness. A brightness of 100 cd m−2 was achieved at 12.4 V with an EQE of 6.2%, a luminance efficiency of 5.0 lm W−1 and a current efficiency of 19.7 cd A−1, respectively. The electroluminescence and PL spectra were essentially the same with the device giving green emission with CIE co-ordinates of (0.37,0.59).
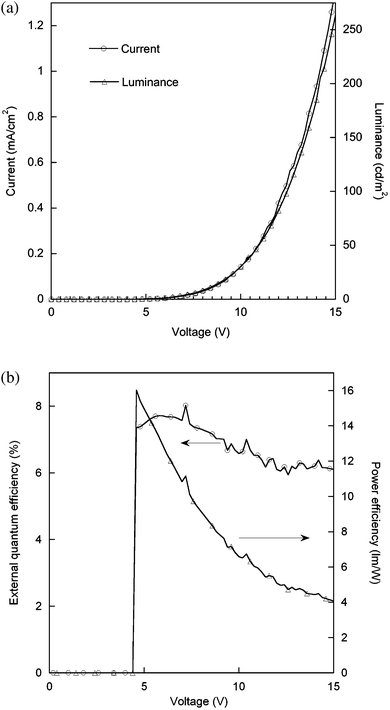 | ||
| Fig. 8 Characteristics of the bilayer device (ITO/2:CBP/TPBI/LiF/Al). (a) Voltage versus current and luminance. (b) Voltage versus external quantum and power efficiency. | ||
Conclusions
We have prepared the first fac-tris(2-phenylpyridyl)iridium(III) norbornenyl-based homopolymer. The attachment of first generation biphenyl based dendrons with 2-ethylhexyloxy surface groups gave good solubility to the polymer, and the chromophores being held closely together along the polymer backbone did not lead to quenching of the luminescence in solution. Neat films of the polymer had a very low PLQY, indicating that inter-polymer chromophore interactions were not controlled by the dendrons. However, the dendrons did allow the formation of good blends in which the emission was from the iridium(III) complex, with the PLQY being similar to that observed in solution. Efficient bilayer devices were formed and, importantly, the viscosity of the polymer solutions was increased showing the way to dendritic materials that could be inkjet printed.Experimental section
General methods
All commercial reagents were used as received unless otherwise noted. Tetrahydrofuran was distilled from sodium and benzophenone under a nitrogen atmosphere before use. For the polymerisations the dichloromethane and ethylvinyl ether were distilled off calcium hydride and then deoxygenated before use. 1H NMR spectra were recorded using a 500 MHz Bruker spectrometer, in deuterated chloroform or dichloromethane solution. For the 1H NMR 7.27 ppm and 5.32 ppm were used as the residual peaks for deuterated chloroform and dichloromethane, respectively. Abbreviations used in the assignment of the spectra are: G1-bpH = first generation branching phenyl H, spH = surface phenyl H, BnH = benzylH, LH = ligand H, and NbH = norbornenyl, norbornyl or polymer backbone H. Coupling constants are quoted to the nearest 0.5 Hz. Gel permeation chromatography was carried out using PLgel Mixed-A columns (600 mm + 300 mm lengths, 7.5 mm diameter) from Polymer Laboratories calibrated with polystyrene narrow standards (Mp = 580–3.2 × 106) in tetrahydrofuran. The tetrahydrofuran was deoxygenated with helium and pumped at a rate of 1 cm3 min−1 at 30 °C. Microanalyses were carried out at London Metropolitan University, UK. UV-visible absorption spectra were recorded as solutions in HPLC grade dichloromethane with a Perkin-Elmer UV-vis Lambda 25 spectrometer or a Varian Cary 300 spectrophotometer; sh = shoulder. Infrared spectra were recorded on a Perkin Elmer Paragon 1000 FT-IR spectrometer. Melting points were measured in a glass capillary on a BUCHI Melting Point B-545 and are uncorrected. Thermogravimetric analysis (TGA) was carried out on a Perkin Elmer STA6000 and differential scanning calorimetry was performed using a Perkin Elmer Diamond DSC by ANNF-Q (the Australian National Fabrication Facility—Queensland Node, University of Queensland, QLD, Australia). Mass spectra were recorded on an Applied Biosystems Voyager DE-STR MALDI-TOF instrument, using 2-[(2E)-3-(4-tert-butylphenyl)-2-methylprop-2-enylidene]malononitrile (DCTB) matrix in positive reflection or linear mode, at the EPSRC National Mass Spectrometry Service Centre (NMSSC), Swansea, UK.Solution PLQYs were measured by a relative method using quinine sulfate in 0.5 M sulfuric acid as the standard, which has a PLQY of 0.546.20 The materials were dissolved in dichloromethane and freeze-thaw degassed. Photoluminescence spectra were recorded in a JY Horiba Fluoromax 2 fluorimeter, with the solutions excited at 360 nm. The optical densities of the standard and sample were similar and small (∼0.1). The accuracy of these measurements is estimated to be ±10% of the stated value. The time-resolved luminescence measurements were performed using the time-correlated single photon counting technique, with excitation at 393 nm from a pulsed Picoquant GaN diode laser and an instrument response of ∼200 ps. The signal was detected by a cooled Hamamatsu microchannel plate photomultiplier tube behind a monochromator set to 520 nm. For the solid-state PLQY measurements the sample was placed in an integrating sphere under a nitrogen purge and excited with the 325 nm line of a He–Cd laser at a power of ∼0.3 mW. The luminescence was detected from a calibrated photodiode behind a UV filter and the PLQY was calculated in accordance with the method of Greenham et al.21
OLEDs were prepared on ITO substrates that had been etched with concentrated hydrochloric acid and zinc powder. The ITO was then cleaned by sonication in acetone and 2-propanol followed by oxygen plasma ashing. The organic layer was spin-cast from a dichloromethane solution containing a blend of poly(dendrimer) 2 with 70% CBP by weight. The solution concentration was 20 mg cm−3 and the spin speed of 2000 rpm resulted in ∼100 nm thick films. The samples were then placed in an evaporator operating at a pressure of 2 × 10−6 mbar. A 60 nm hole-blocking layer of TPBI, followed by a 0.7 nm layer of LiF and a >100 nm layer of aluminium were deposited by thermal evaporation to complete the device. A shadow mask was used to give a final active area of 6 mm2. The electroluminescence spectrum was recorded using a cooled Andor DV420-BV CCD spectrometer. The current and voltage characteristics were recorded using a Keithley source-measure unit and the light emission was measured using a calibrated photodiode.
[Bis(2-{5-[3,5-bis(4-{2-ethylhexyloxy}phenyl)phenyl]phenyl}pyridyl)-2-(5-formylphenyl)pyridyl]iridium(III) 5
A solution of tetra[2-(5-{3,5-bis[4-(2-ethylhexyloxy)phenyl]phenyl}phenyl)pyridyl]diiridium(III) dichloride 410 (917 mg, 0.305 mmol), 3-(pyridin-2-yl)benzaldehyde11 (575 mg, 3.14 mmol), and silver trifluoromethanesulfonate (165 mg, 0.642 mmol) in diethylene glycol dimethyl ether (9 cm3), was deoxygenated with argon for 20 min and then heated at 110 °C for 16 h under argon. The reaction was allowed to cool to room temperature and diluted with diethylether (100 cm3). The organic layer was washed with water (3 × 100 cm3) and brine (100 cm3), dried over anhydrous magnesium sulfate, filtered through Celite®, and the solvent removed. The residue was purified by preparative thin layer chromatography with diethylether as eluent to give the desired 5 contaminated with 3-(pyridin-2-yl)benzaldehyde. The dendrimer 5 and 3-(pyridin-2-yl)benzaldehyde were separated using size exclusion chromatography over Sephadex™ LH-20 using methanol : dichloromethane (1 : 3) as eluent (10 iterations) to give 5 as a green luminescent yellow solid (422 mg, 42%). Found: C, 74.1; H, 6.8; N, 2.5. C102H112IrN3O5 requires C, 74.15; H, 6.8; N, 2.5%; λmax (CH2Cl2)/nm 267 [log ε/dm3 mol−1 cm−1 (4.94)]; νmax (KBr)/cm−1 1683 (CO); δH (500 MHz; CD2Cl2) 0.89–0.97 (24 H, m, CH3), 1.31–1.58 (32 H, m, CH2), 1.72–1.79 (4 H, m, CH), 3.88–3.94 (8 H, m, OCH2), 6.86 (1 H, d, J = 8, LH), 7.04–7.69 (11 H, m, spH and LH), 7.12 (1 H, m, LH), 7.21 (2 H, m, LH), 7.26 (2 H, m, LH), 7.60–7.69 (14 H, m, spH, and LH and/or G1-bpH), 7.72–7.80 (6 H, m, G1-bpH and/or LH), 8.50 (2 H, d, J = 2, LH), 8.10–8.14 (3 H, m, LH), 8.19 (1 H, d, J = 1, LH) and 9.89 (1 H, s, HCO); m/z (MALDI-TOF) found: 1655.8 (4%), 1654.8 (23%), 1653.8 (53%), 1652.8 (93%), 1651.8 (100%), 1650.8 (54%) and 1649.8 (49%) (M+˙). C102H112IrN3O5 requires 1655.8 (5%), 1654.8 (18%), 1653.8 (50%), 1652.8 (92%), 1651.8 (100%), 1650.8 (49%) and 1649.8 (43).
[Bis(2-{5-[3,5-bis(4-{2-ethylhexyloxy}phenyl)phenyl]phenyl}pyridyl)-2-(5-methanolphenyl)pyridyl]iridium(III) 6
A solution of 5 (453 mg, 0.274 mmol) in diethylether (5 cm3) was added to a stirred suspension of lithium aluminium hydride (52 mg, 1.37 mmol) in diethylether (5 cm3) under a nitrogen atmosphere at 0 °C. After 1 h the reaction was quenched by careful addition of water (10 cm3). Diethylether (100 cm3) was added and the organic layer was washed with water (3 × 50 cm3), hydrochloric acid (1 M, 25 cm3), water (50 cm3) and brine (50 cm3), dried over anhydrous magnesium sulfate, and filtered. The solvents were removed to give 6 as a green luminescent yellow solid (574 mg, 100%). Found: C, 74.1; H, 7.0; N, 2.45. C102H114IrN3O5 requires C, 74.1; H, 6.95; N, 2.5%; λmax (CH2Cl2)/nm 284 [log ε/dm3 mol−1 cm−1 (4.51)]; νmax (KBr)/cm−1 3447 (OH); δH (500 MHz; CDCl3) 0.92–0.98 (24 H, m, CH3), 1.32–1.60 (33 H, m, CH2, OH), 1.74–1.82 (4 H, m, CH), 3.88–3.93 (8 H, m, OCH2), 4.62 (2 H, d, J = 5.5, BnH), 6.89–6.95 (4 H, m, LH), 6.98–7.02 (10 H, m, spH and LH), 7.07 (1 H, d, J = 8, LH), 7.24–7.28 (2 H, m, LH), 7.58–7.64 (16 H, m, spH, G1-bpH and/or LH), 7.73 (1 H, d, J = 1, LH), 7.75 (4 H, d, J = 1.5, G1-bpH or LH), 7.95 (1 H, d, J = 8, LH), 7.99 (2 H, m, LH) and 8.00–8.03 (2 H, m, LH); m/z (MALDI-TOF) found: 1651.7 (53%), 1652.7 (63%), 1653.7 (100%), 1654.7 (94%), 1655.7 (49%), 1656.7 (18%) and 1657.7 (7%) (M+√). C102H114IrN3O5 requires 1651.8 (43%), 1652.8 (49%), 1653.8 (100%), 1654.8 (92%), 1655.8 (49%), 1656.8 (17%) and 1657.9 (5%).[Bis(2-{5-[3,5-bis(4-{2-ethylhexyloxy}phenyl)phenyl]phenyl}pyridyl)-2-(5-methanolylphenyl-exo-bicyclo[2.2.1]hept-5-ene-2-carboxylic acid ester)pyridyl]iridium(III) 7
Oxalylchloride (211 µL, 15.1 mmol) and N,N-dimethylformamide (2 drops, cat.) were added to a solution of exo-bicyclo[2.2.1]hept-5-ene-2-carboxylic acid14 (166 mg, 1.21 mmol) in dichloromethane (1 cm3) under a nitrogen atmosphere at 0 °C. After 1.5 h the volatiles were removed and the resultant crude acid chloride was dissolved in pyridine (500 µL, 0.907 mmol) under a nitrogen atmosphere at 0 °C. A solution of 6 (200 mg, 121 µmol) in pyridine (500 µL, 0.91 mmol) and dichloromethane (500 µL) was added and the reaction was allowed to warm to room temperature over 16 h. The reaction was diluted with diethylether (50 cm3), and the organic layer was washed with water (3 × 30 cm3) and brine (30 cm3), dried over anhydrous magnesium sulfate, filtered, and the solvents were removed. The residue was purified by size exclusion chromatography over Sephadex™ LH-20 using methanol : dichloromethane (1 : 3) as eluent to give 7 as a green luminescent yellow solid (187 mg, 87%). Found: C, 74.5; H, 7.0; N, 2.2. C110H122IrN3O6 requires C, 74.5; H, 6.9; N, 2.4%; λmax (CH2Cl2)/nm 273sh, 283, 328sh, 386sh, 415sh, 460sh and 494sh; νmax (KBr)/cm−1 1724 (CO); δH (500 MHz; CDCl3) 0.92–1.00 (24 H, m, CH3), 1.32–1.60 (35 H, m, CH2, NbH), 1.74–1.81 (4 H, m, CH), 1.94–1.99 (1 H, m, NbH), 2.26–2.29 (1 H, m, NbH), 2.92 (1 H, br. s, NbH), 3.07 (1 H, br. s, NbH), 3.88–3.94 (8 H, m, OCH2), 5.09 (2 H, m, BnH), 6.08–6.14 (2 H, m, NbH), 6.90–6.95 (4 H, m, LH), 6.97–7.03 (10 H, m, spH and LH), 7.04 (1 H, d, J = 8, LH), 7.24–7.28 (2 H, m, LH), 7.58–7.66 (16 H, m, spH, G1-bpH and/or LH), 7.68 (1 H, d, J = 1.5, LH), 7.75 (4 H, s, G1-bpH or LH), 7.94 (1 H, d, J = 8.5, LH), 7.98 (2 H, br. t, LH) and 8.00–8.03 (2 H, m, LH); m/z (MALDI-TOF) found: 1771.9 (44%), 1772.9 (60%), 1773.9 (100%), 1774.9 (91%), 1775.9 (44%), 1776.9 (25%) and 1777.9 (12%) (M+√). C110H122IrN3O6 requires 1771.9 (41%), 1772.9 (50%), 1773.9 (100%), 1774.9 (97%), 1775.9 (56%), 1776.9 (22%) and 1777.9 (7%).
[Bis(2-{5-[3,5-bis(4-{2-ethylhexyloxy}phenyl)phenyl]phenyl}pyridyl)-2-(5-methanolylphenyl-exo-bicyclo[2.2.1]heptane-2-carboxylic acid ester)pyridyl]iridium(III) 8
Palladium on carbon (10%, 1 mg) was added to a solution of 7 (10 mg, 5.6 µmol) in ethyl acetate (1.0 cm3). The reaction was deoxygenated with three pump/fill (argon) cycles, and then placed under an atmosphere of hydrogen with three pump/fill (hydrogen) cycles. The reaction mixture was stirred for 3 h under hydrogen. The suspension was then filtered through Celite® (which was disposed carefully by keeping moist with ethyl acetate) and the solvents were removed to give 8 as a green luminescent yellow solid (9.9 mg, 99%). λmax (CH2Cl2)/nm 272sh, 283, 331sh, 387sh, 411sh, 459sh and 493sh; νmax (KBr)/cm−1 1724 (CO); δH (500 MHz; CDCl3) 0.90–0.97 (24 H, m, CH3), 1.14–1.60 (39 H, m, CH2, NbH), 1.71–1.80 (4 H, m, CH), 1.86–1.89 (1 H, bm, NbH), 2.29 (1 H, br. m, NbH), 2.34–2.37 (1 H, m, NbH), 2.52 (1 H, br. s, NbH), 3.88–3.93 (8 H, m, OCH2), 5.05 (2 H, m, BnH), 6.88–7.06 (15 H, m, spH and LH), 7.24–7.28 (2 H, m, LH), 7.58–7.68 (16 H, m, spH, G1-bpH and/or LH), 7.74 (5 H, bm, G1-bpH and/or LH), 7.94 (1 H, d, J = 8.5, LH), 7.98 (2 H, bt, LH) and 7.98–8.03 (2 H, m, LH); m/z (MALDI-TOF) found: 1773.8 (48%), 1774.8 (55%), 1775.8 (100%), 1776.8 (92%), 1777.8 (60%), 1778.8 (26%) and 1779.8 (10%) (M+√). C110H124IrN3O6 requires 1773.9 (41%), 1774.9 (50%), 1775.9 (100%), 1776.9 (97%), 1777.9 (56%), 1778.9 (22%) and 1779.9 (7%).
Poly[({bis[2-(5-{3,5-bis[4-(2-ethylhexyloxy)phenyl]phenyl}phenyl)pyridyl]-2-(5-methanolylphenyl)pyridyl}iridium(III))-1,4-vinylcyclopentane-2-carboxylic acid ester] 2
Using standard Schlenk techniques, a solution of Grubbs III catalyst15 in dichloromethane (1.0 mg cm−3, 1.0 cm3, 1.1 µmol) was added to a vigorously stirred solution of 7 (102 mg, 57.5 µmol) in dichloromethane (4.0 cm3) under argon. Ethylvinyl ether (3 drops, excess) was added after 18 h, and after a further 5 min the solvents were removed. The residue was purified by size exclusion chromatography over Sephadex™ LH-20 using methanol : dichloromethane (1 : 3) as eluent to give 2 (100 mg). λmax (CH2Cl2)/nm 270sh, 282, 327sh, 386sh, 414sh, 456 and 491sh; νmax (KBr)/cm−1 1725 (CO); δH (500 MHz; CD2Cl2) 0.76–1.06 (24 H, br. m, CH3), 1.16–1.76 (35 H, br. m, CH2, CH, NbH), 1.84–2.11 (1 H, br. m, NbH), 2.26–2.56 (1 H, br. s, NbH), 2.76–3.24 (1 H, br. m, NbH), 3.66–3.96 (8 H, br. m, OCH2), 4.74–5.26 (4 H, br. m, BnH and alkene-H) and 6.36–8.28 (43 H, br. m, spH, G1-bpH and LH); m/z (MALDI-TOF; DCTB) Mp = 130 kDa; GPC w = 4.7 × 104, n = 3.0 × 104, PDI = 1.5.
Poly[({bis[2-(5-{3,5-bis[4-(2-ethylhexyloxy)phenyl]phenyl}phenyl)pyridyl]-2-(5-methanolylphenyl)pyridyl}iridium(III))-1,4-ethylcyclopentane-2-carboxylic acid ester] 9
Compound 8 (3.9 mg,w = 4.4 × 104, n = 2.9 × 104) was dissolved in ethyl acetate (5 cm3) and reduced using a Thales Nanotechnology H-cube® containing a 10% palladium on charcoal CatCart® catalyst, hydrogen at a pressure of 6 bar and flow rate 0.5 cm3 min−1 for 10 min, and a catalyst temperature of 50 °C to give 9 as a green luminescent yellow solid (3.9 mg, 100%). λmax (CH2Cl2)/nm 272sh, 283, 330sh, 390sh, 412sh, 460 and 494; δH (500 MHz; CD2Cl2) 0.76–0.96 (24 H, br. m, CH3), 1.14–1.76 (39 H, br. m, CH2, CH, NbH), 1.84–2.13 (1 H, br. s, NbH), 2.19–2.56 (1 H, br. s, NbH), 2.76–3.23 (1 H, br. m, NbH), 3.66–3.90 (8 H, br. m, OCH2), 4.69–5.16 (2 H, br. m, BnH) and 6.36–8.05 (43 H, br. m, spH, G1-bpH and LH); GPC w = 4.2 × 104, n = 2.7 × 104, PDI = 1.5.
Acknowledgements
Jack P. Gunning was funded by an EPSRC Doctoral Training Account. Professor Paul L. Burn is recipient of an Australian Research Council Federation Fellowship (project number FF0668728). Professor Ifor Samuel holds an EPSRC Senior Research Fellowship (grant number EP/C542398). Dr Mark F. Wyatt is supported by the EPSRC NMSSC (grant number EP/F014341/1).References
- Y. You and S.-Y. Park, Dalton Trans., 2009, 1267 RSC.
- Z. Xu, B. Hu and J. Howe, J. Appl. Phys., 2008, 103, 043909 CrossRef.
- E. Baranoff, S. Suàrex, P. Bugnon, C. Barolo, R. Buscaino, R. Scopelliti, L. Zuppiroli, M. Grätzel and Md. K. Nazeeruddin, Inorg. Chem., 2008, 47, 6575 CrossRef CAS.
- M. Singh, H. M. Haverinen, P. Dhagat and G. E. Jabbour, Adv. Mater., 2010, 22, 673 CrossRef CAS.
- (a) A. J. Sandee, C. K. Williams, N. R. Evans, J. E. Davies, C. Boothby, A. Kohler, R. H. Friend and A. B. Holmes, J. Am. Chem. Soc., 2004, 126, 7041 CrossRef CAS; (b) X. Chen, J. L. Liao, Y. Liang, M. O. Ahmed, H. Tseng and S. A. Chen, J. Am. Chem. Soc., 2003, 125, 636 CrossRef CAS; (c) H. Y. Zhen, C. Luo, W. Yang, W. Y. Song, B. Du, J. X. Jiang, C. Y. Jiang, Y. Zhang and Y. Cao, Macromolecules, 2006, 39, 1693 CrossRef CAS; (d) J. Jiang, C. Jiang, W. Yang, H. Zhen, F. Huang and Y. Cao, Macromolecules, 2005, 38, 4072 CrossRef CAS; (e) M. Suzuki, S. Tokito, F. Sato, T. Igarashi, K. Kondo, T. Koyama and T. Yamaguchi, Appl. Phys. Lett., 2005, 86, 103507 CrossRef; (f) L. Deng, P. T. Furuta, S. Garon, J. Li, D. Kavulak, M. E. Thompson and J. M. J. Fréchet, Chem. Mater., 2006, 18, 386 CrossRef CAS; (g) P. T. Furuta, L. Deng, S. Garon, M. E. Thompson and J. M. J. Fréchet, J. Am. Chem. Soc., 2004, 126, 15388 CrossRef CAS; (h) E. Tekin, E. Holder, V. Marin, B. J. de Gans and U. S. Schubert, Macromol. Rapid Commun., 2005, 26, 293 CrossRef CAS; (i) G. L. Schulz, X. W. Chen, S. A. Chen and S. Holdcroft, Macromolecules, 2006, 39, 9157 CrossRef CAS.
- (a) S.-C. Lo, N. A. H. Male, J. P. J. Markham, S. W. Magennis, P. L. Burn, O. V. Salata and I. D. W. Samuel, Adv. Mater., 2002, 14, 975 CrossRef CAS; (b) T. D. Anthopoulos, M. J. Frampton, E. B. Namdas, P. L. Burn and I. D. W. Samuel, Adv. Mater., 2004, 16, 557 CrossRef CAS; (c) S.-C. Lo, T. D. Anthopoulos, E. B. Namdas, P. L. Burn and I. D. W. Samuel, Adv. Mater., 2005, 17, 1945 CrossRef CAS; (d) S.-C. Lo, R. N. Bera, R. E. Harding, P. L. Burn and I. D. W. Samuel, Adv. Funct. Mater., 2008, 18, 3080 CrossRef CAS; (e) J. Li and D. Liu, J. Mater. Chem., 2009, 19, 7584 RSC.
- W.-Y. Lai, J. W. Levell, P. L. Burn, S.-C. Lo and I. D. W. Samuel, J. Mater. Chem., 2009, 19, 4952 RSC.
- J. M. Pollino, L. P. Stubbs and M. Weck, Macromolecules, 2003, 36, 2230 CrossRef CAS.
- J. D. Rule and J. S. Moore, Macromolecules, 2002, 35, 7878 CrossRef CAS.
- M. J. Frampton, E. B. Namdas, S.-C. Lo, P. L. Burn and I. D. W. Samuel, J. Mater. Chem., 2004, 14, 2881 RSC.
- Z. Xiong, N. Wang, M. Dai, A. Li, J. Chen and Z. Yang, Org. Lett., 2004, 6, 3337 CrossRef CAS.
- A. B. Tamayo, B. D. Alleyne, P. I. Djurovich, S. Lamansky, I. Tsyba, N. N. Ho, R. Bau and M. E. Thompson, J. Am. Chem. Soc., 2003, 125, 7377 CrossRef CAS.
- S.-C. Lo, E. B. Namdas, P. L. Burn and I. D. W. Samuel, Macromolecules, 2003, 36, 9721 CrossRef CAS.
- D. D. Manning, L. E. Strong, X. Hu, P. J. Beck and L. L. Kiessling, Tetrahedron, 1997, 53, 11937 CrossRef CAS.
- J. A. Love, J. P. Morgan, T. M. Trnka and R. H. Grubbs, Angew. Chem., Int. Ed., 2002, 41, 4035 CrossRef CAS.
- A. Kimyonok, B. Domercq, A. Haldi, J.-Y. Cho, J. R. Carlise, X.-Y. Wang, L. E. Hayden, S. C. Jones, S. Barlow, S. R. Marder, B. Kippelen and M. Weck, Chem. Mater., 2007, 19, 5602 CrossRef CAS.
- E. B. Namdas, A. Ruseckas, I. D. W. Samuel, S.-C. Lo and P. L. Burn, J. Phys. Chem. B, 2004, 108, 1570 CrossRef CAS.
- J. R. Carlise, X.-Y. Wang and M. Weck, Macromolecules, 2005, 38, 9000 CrossRef CAS.
- J.-C. Ribierre, S. G. Stevenson, I. D. W. Samuel, S. V. Staton and P. L. Burn, J. Disp. Technol., 2007, 3, 233 Search PubMed.
- J. N. Demas and G. A. J. Crosby, J. Phys. Chem., 1971, 75, 991 CrossRef.
- N. C. Greenham, I. D. W. Samuel, G. R. Hayes, R. T. Phillips, Y. Kessener, S. C. Moratti, A. B. Holmes and R. H. Friend, Chem. Phys. Lett., 1995, 241, 89 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |