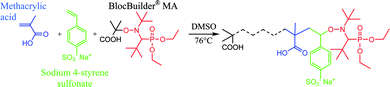DOI:
10.1039/B9PY00371A
(Paper)
Polym. Chem., 2010,
1, 720-729
Received
10th December 2009
, Accepted 5th February 2010
First published on
4th March 2010
Abstract
The SG1-mediated copolymerization of methacrylic acid (MAA) and 4-styrene sulfonate (SS) was studied in dimethylsulfoxide solution at 76 °C, first, to determine the reactivity ratios in such conditions and second, to check the living character of the reaction at low molar fraction of SS. The reactivity ratios in the terminal model were rMAA = 0.44 and rSS = 1.34 indicating a favored incorporation of SS at the beginning of the copolymerization. All characteristics of a controlled/living system were observed, in good agreement with an efficient deactivation of the propagating radicals by the nitroxide SG1, via probable formation of an SS terminal subunit-based alkoxyamine. The method was shown to be particularly well-suited for the design of living polymers intended to be used as hydrophilic macroinitiators for the synthesis of amphiphilic block copolymers. This was demonstrated in both solution polymerization and ab initio, batch emulsion polymerization. The latter process allowed well-defined block copolymer nanoparticles to be formed at low temperature, in a single step, by simultaneous chain growth and self-assembling.
Introduction
In the past decade, the advent of controlled/living free-radical polymerization (CRP)1,2 based on the reversible deactivation of the propagating radicals has revolutionized the domain of polymer chemistry and opened the door to the possibility of designing new polymer architectures and creating new materials with targeted properties. Among the various CRP techniques, nitroxide-mediated polymerization (NMP)3 is acknowledged to be simple and efficient as it requires a limited number of reactants, i.e. a simple alkoxyamine, which is activated by temperature, without the need for additional catalyst or radical initiator. This is important for the design of block copolymers, in particular when those are synthesized in heterogeneous conditions like emulsion polymerization.4–9 Indeed, one of the challenges in such multiphasic systems is to locate the propagation and activation-deactivation steps within the formed particles and avoid the formation of non-controlled polymer chains in the other phases. This sometimes becomes difficult when multiple reactants are needed and exhibit different solubilities and partition coefficients.
In our group, we developed the elaboration of amphiphilic block copolymer nanoparticles in water using soluble alkoxyamine macroinitiators based on SG1 as a nitroxide for in situ chain extension with a hydrophobic monomer.10–15 Due to the above mentioned advantages of NMP, chain growth and self-assembly take place simultaneously and the system is quite robust, in terms of both colloidal and macromolecular characteristic control. With SG1 as a nitroxide,16 the number of monomers exhibiting homopolymerization with controlled/living character is quite high now, among the families of styrenics, acrylates and dialkylacrylamides.3 This allowed for the development of amphiphilic block copolymer particles composed of poly(acrylic acid) as the stabilizing shell and of polystyrene, poly(n-butyl acrylate), or cross-linked poly(N,N-diethylacrylamide) as the core.10,12–15 With methacrylic esters, a controlled/living polymerization was achieved in the presence of a small percentage of styrene.17–22 The same principle applied for methacrylic acid (MAA) and hydrophilic methacrylic esters.23–25 This discovery allowed us to design new water-soluble macroalkoxyamines based on poly(methacrylic acid) with less than 9 mol% of styrene,23 and to perform the heterophase copolymerization of methyl methacrylate with a low percentage of styrene at low temperature (below 90 °C).25,26 Those macroalkoxyamines were particularly efficient and allowed very small particles to be designed, composed of well-defined amphiphilic diblock copolymers. However, the presence of styrene as a comonomer with methacrylic acid in the hydrophilic block may lead to hydrophobic interactions, resulting in colloidal stability issues at high solids content. To overcome this drawback, we need to replace styrene with a more hydrophilic comonomer. It could be acrylonitrile as shown very recently27 and this will be the topic of a forthcoming article. Here we were interested in testing sodium 4-styrene sulfonate (SS), the homopolymerization of which can be well controlled via NMP.28–31 First, it was important to study the SG1-mediated copolymerization of methacrylic acid and sodium 4-styrene sulfonate (Scheme 1) via determination of the reactivity ratios under NMP conditions and to find the best conditions to synthesize living copolymers. Then the poly(methacrylic acid-co-sodium 4-styrene sulfonate) (P(MAA-co-SS)) macroalkoxyamines were used as macroinitiators in solution and aqueous emulsion polymerizations (Scheme 2) to induce the formation of amphiphilic block copolymers, able to self-assemble in water. In the latter application the advantage of replacing styrene by 4-styrene sulfonate on the polymer colloid stability was evaluated.
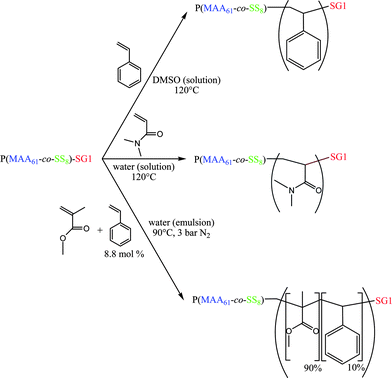 |
| | Scheme 2 A schematic representation of the polymerizations initiated by the P(MAA61-co-SS8)-SG1 macroalcoxyamine in solution and emulsion. | |
Experimental
1 Materials
Methacrylic acid (MAA, purest grade, Acros, stabilized with 250 ppm of methylethylhydroquinone) and sodium 4-styrene sulfonate (SS, purity ≥ 90%, Fluka) were used without further purification. Styrene (S, 99%, Aldrich), N,N-dimethylacrylamide (DMA, 99%, Aldrich) and methyl methacrylate (MMA, 99%, Aldrich) were distilled under reduced pressure before use. The N-(2-methylpropyl)-N-(1-diethylphosphono-2,2-dimethylpropyl)-O-(2-carboxyl prop-2-yl) hydroxylamine initiator (the so-called BlocBuilder-MA®, 99%) and the N-tert-butyl-N-(1-diethyl phosphono-2,2-dimethylpropyl) nitroxide (SG1, 85%) were kindly supplied by Arkema. Dimethylsulfoxide d6 (DMSO d6, Euriso-top®), dimethylsulfoxide (DMSO, Prolabo, technical), ethanol (Prolabo, pure), trimethylsilyldiazomethane (2.0 M in diethyl ether, Aldrich), sodium carbonate (Na2CO3, Prolabo, pure) and sodium hydroxide (NaOH, SDS, 1 M solution in water) were used as received. For the emulsion polymerizations, deionized water was used (USF Regeneration).
The copolymerization reactions were carried out directly in Young tubes of 5 mm in diameter in the 500 MHz NMR spectrometer heated at 76 °C. For a typical experiment (Table 1, experiment 4): in a separate flask, the monomers SS (0.138 g, 5.55 × 10−1 mol L−1, initial molar fraction of SS in the comonomer mixture: fSS,0 = 0.198) and MAA (0.209 g, 2.01 mol L−1) were mixed in DMSO d6 (1.19 mL) and deoxygenated with an argon stream for 30 min at room temperature. During this time, the BlocBuilder® initiator (0.0135 g, 2.93 × 10−2 mol L−1) introduced in the NMR tube was deoxygenated under an argon stream during 30 min. With a syringe, the monomer solution was also introduced in the NMR tube; the latter was sealed under argon and maintained in an ice bath before starting the experiment. The tube was then introduced into the spectrometer already heated at 76 °C and the first spectrum was recorded, marking the time 0 of the polymerization. The spectra (64 scans during 64 s) were recorded every 10 min during an overall polymerization time of a few hours (typically 3.8 h for the experiment 4). In order to calculate the monomer conversions, the solvent peak (DMSO, δ = 2.54 ppm) was used as an internal reference (peak integral: IDMSO). The conversion of SS (xSS) was calculated on the basis of the 3 vinylic H peaks (δ = 6.75 ppm, δ = 5.80 ppm and δ = 5.25 ppm), and the integral Iss for 1 H was the average of the 3 peak integrals. For MAA (conversion = xMAA), the integral IMAA for 1 H was the average of the 2 = CH2 proton peak integrals at δ = 6.00 ppm and δ = 5.56 ppm. For kinetic analysis, the overall conversion considered was the molar conversion directly accessible via the NMR analysis. It can also be calculated from the individual monomer conversions according to the relationship: xmol = xSS × fSS,0 + xMAA × fMAA,0 (fSS,0 and fMAA,0 are the initial molar fractions of SS and MAA, respectively, in the reaction mixture). For the plots representing Mn as a function of the overall conversion, one used the weight conversion, that can be calculated according to: xw = xSS × wSS,0 + xMAA × wMAA,0 (wSS,0 and wMAA,0 are the initial weight fractions of SS and MAA, respectively, in the reaction mixture).
| Expt |
Symbol |
[MAA]0/mol L−1 |
[SS]0/mol L−1 |
f
SS,0
|
[BlocBuilder®]0/mol L−1 |
Target Mnb at 100% conversion/g mol−1 |
Overall time/h |
|
Initial molar fraction of sodium 4-styrene sulfonate.
Theoretical number-average molar mass, calculated according to Mn = MM(BlocBuilder®) + conversion × initial weight of monomers/initial mol number of BlocBuilder®, with conversion = 1 and molar mass of the initiator, MM(BlocBuilder®) = 381 g mol−1.
higher dilution to ensure a good solubility for SS.
|
|
1
|
■ |
2.03 |
0.062 |
0.021 |
1.84 × 10−2 |
10 500 |
7.1 |
|
2
|
● |
2.07 |
0.142 |
0.055 |
1.94 × 10−2 |
11 000 |
9.0 |
|
3
|
Δ |
2.01 |
0.215 |
0.078 |
2.28 × 10−2 |
9 800 |
10.8 |
|
4
|
◆ |
2.01 |
0.555 |
0.198 |
2.93 × 10−2 |
9 900 |
3.8 |
|
5
|
× |
1.21 |
0.897 |
0.394 |
4.05 × 10−2 |
6 700 |
7.5 |
The copolymerization reactions were carried out in dimethylsulfoxide at 76 °C (Table 2). In a typical experiment (experiment 6 in Table 2), a mixture of MAA (24.0 g, 2.0 mol L−1) and SS (6.167 g, 1.9 × 10−1 mol L−1, initial molar fraction of SS in the comonomer mixture: fSS,0 = 0.088) in DMSO (127.3 g) was deoxygenated with a nitrogen stream for 20 min at room temperature. The BlocBuilder® initiator (1.215 g, 2.2 × 10−2 mol L−1) was then added and nitrogen bubbling was carried out for 10 more minutes. The solution was introduced with a syringe into 6 Schlenk tubes under nitrogen; the latter were immersed in a thermostated oil bath, which marked the time zero of the reaction. The tubes were collected at regular time intervals over a period of 2 h and quenched by immersion in an ice-water bath. For each sample, the monomer overall molar conversion (xmol) was determined by 1H NMR analysis of the raw solution. For the copolymers characterized by aqueous phase size exclusion chromatography (Table 2, experiments 6–8), all samples were precipitated in cooled acetone. The copolymers were collected by filtration and dried under vacuum at room temperature for 3 days to eliminate residual MAA and solvent. For the copolymer from experiment 9 (Table 2) analyzed by size exclusion chromatography (SEC) in DMF (with 1 g L−1 LiBr) no precipitation was performed. The copolymer further used as a macroinitiator (Table 2, experiment 10) was precipitated in cooled diethyl ether, collected by filtration and dried under vacuum at room temperature for 3 days. Before injecting in SEC DMF those copolymers 9 and 10 containing methacrylic acid units, a reaction of methylation was performed to turn the acid groups into methyl esters, using trimethylsilyldiazomethane32,33 in tetrahydrofuran and water solution (90/10, v/v) under stirring during 3 h at room temperature. The dried methylated copolymers were then solubilized in the SEC solvent.
| Expt |
Symbol |
[SS]0/mol L−1 |
f
SS,0
|
[BlocBuilder®]0/mol L−1 |
Target Mnb at 100% conversion/g mol−1 |
Overall time/h |
|
Initial molar fraction of sodium 4-styrene sulfonate.
Theoretical number-average molar mass, calculated according to Mn = MM(BlocBuilder®) + conversion × initial weight of monomers/initial mol number of BlocBuilder®, with conversion = 1 and molar mass of the initiator, MM(BlocBuilder®) = 381 g mol−1.
SEC analysis in water solution + 5M LiNO3 for the precipitated copolymers.
SEC analysis in DMF solution + 1 g L−1 LiBr for the raw copolymer.
SEC analysis in DMF solution + 1 g L−1 LiBr for the precipitated copolymer.
|
|
6
|
● |
1.93 × 10−1 |
0.088 |
2.23 × 10−2 |
9 900 |
2.0 |
|
7
|
× |
1.27 × 10−1 |
0.059 |
2.09 × 10−2 |
9 950 |
1.5 |
|
8
|
▲ |
9.93 × 10−2 |
0.045 |
1.97 × 10−2 |
10 050 |
0.8 |
|
9
|
■ |
1.89 × 10−1 |
0.088 |
2.15 × 10−2 |
10 070 |
2.1 |
|
10
|
|
1.88 × 10−1 |
0.086 |
2.22 × 10−2 |
9 900 |
0.5 |
4 Chain extension experiments in solution
For a typical experiment (chain extension with styrene, Table 3, experiment 11), the P(MAA61-co-SS8)-SG1 macroinitiator (Table 2, experiment 10; Mn = 6900 g mol−1, purity = 80%) (0.132 g, 4.4 × 10−3 mol L−1) was dissolved at room temperature in dimethylsulfoxide (DMSO, 2.482 g) in the presence of styrene (1.133 g, 3.1 mol L−1). The solution was introduced into a 10 mL round-bottom flask. After degassing for 20 min under nitrogen, the polymerization was carried out at 120 °C for 7.5 h; the final polymerization medium was very viscous. The flask was then sunk into an ice-water bath to quench the reaction. The copolymer was dried in a ventilated oven at 70 °C until constant weight, to remove the solvent and residual monomer. Monomer conversion was determined by gravimetry and the methylated copolymer was analyzed by SEC in DMF.
| Expt |
Monomer (concentration in mol L−1) |
[P(MAA61-co-SS8)-SG1]0a/mol L−1 |
Solvent |
Target Mnb at 100% conversion/g mol−1 |
Overall time (conversion)/h |
|
MAA = methacrylic acid and SS = sodium 4-styrene sulfonate.
Theoretical number-average molar mass, calculated according to Mn = Mn(macroinitiator) + conversion × initial weight of monomers/initial mol number of initiator, with conversion = 1 and Mn(macroinitiator) = 6 900 g mol−1.
|
|
11
|
S (3.1) |
4.4 × 10−3 |
DMSO |
80 900 |
7.5 (50%) |
|
12
|
DMA (3.0) |
3.8 × 10−3 |
water |
85 000 |
7.0 (100%) |
The emulsion polymerization of MMA with a small percentage of S was carried out at 90 °C under 3 bar pressure of nitrogen, in a 300 mL thermostated glass Parr® reactor, stirred at 300 rpm for 30 min and then at 150 rpm (Table 4, experiment 13). A mixture of water (74.0 g), the P(MAA61-co-SS8)-SG1 macroinitiator (Mn = 6900 g mol−1; 5.68 g, 6.0 × 10−3 mol L−1aq), NaOH 1M (36.5 g, 3.3 × 10−1 mol L−1aq, i.e. 1 equiv. NaOH based on the carboxylic acid groups) and Na2CO3 (0.41 g, 3.5 × 10−2 mol L−1) was introduced into a 250 mL round-bottom flask and stirred at room temperature until the P(MAA61-co-SS8)-SG1 macroinitiator was well dissolved. Methyl methacrylate (25.2 g, 2.3 mol L−1aq) and styrene (2.62 g, 2.3 × 10−1 mol L−1aq; initial molar fraction of S in the comonomer mixture: fS,0 = 0.091) (monomer content of 19.3 wt.% based on the overall mass of the reaction medium), were then added to this homogeneous mixture and the obtained unstable biphasic system was introduced into the preheated and stirred reactor, after nitrogen bubbling for 20 min at room temperature. Time zero of the polymerization was taken when temperature of the mixture reached 60 °C. The polymerization was allowed to proceed for 3.5 h. Samples were periodically withdrawn to follow monomer conversion by gravimetry and to analyze the copolymers and the particle size.
| Expt |
Monomer content (wt%) |
Target Mn at 100% conversion b/g mol−1 |
[Macroinitiator]0/mmol L−1water |
|
[Na2CO3]0 = 35 mmol L−1water; 1 equivalent NaOH based on the COOH groups; pH = 8; P = 3 bar.
Theoretical number-average molar mass, calculated according to Mn = Mn(macroinitiator) + conversion × initial weight of monomers/initial mol number of initiator, with conversion = 1 and Mn(macroinitiator) = 6 900 g mol−1.
|
|
13
|
19.3 |
49 130 |
6.0 |
|
14
|
18.9 |
35 800 |
8.7 |
6 Analytical techniques
1H NMR spectroscopy for in situ kinetic analysis was performed in 5 mm diameter Young tube in DMSO d6 at 76 °C in a Bruker Avance 500 (500 MHz) spectrometer. The chemical shift scale was calibrated on the basis of the solvent peak (δ = 2.54 ppm). 1H NMR spectroscopy for kinetic analysis (monomer conversion) of samples from experiments 6 to 10 (Table 2) was performed in 5 mm diameter tubes in DMSO d6 at 25 °C in a Bruker Avance 200 (200 MHz) spectrometer. The chemical shift scale was calibrated on the basis of the solvent peak (δ = 2.54 ppm).
13C NMR spectroscopy analysis of the copolymers was performed in 10 mm diameter tubes in DMSO d6 at 25 °C on a Bruker Avance III 500 spectrometer, operating at a frequency of 125.76 MHz. Spectra were recorded applying the following conditions, allowing quantitative analysis: spectral width 248 ppm with 64 K data points, flip angle of 30°, relaxation delay of 15 s, digital resolution of 0.47 Hz.pt−1 and suppression of the NOE. The chemical shift scale was calibrated on the solvent peak (δ = 40.45 ppm).
Size exclusion chromatography (SEC) analyses in water (+ LiNO3, 0.5 M) were performed at 35 °C at a flow rate of 0.7 mL min−1. All polymers (turned into sodium salt by the addition of concentrated NaOH aqueous solution) were analyzed at a concentration of 5.0 mg mL−1 after filtration through a 0.45 μm pore size membrane. The steric exclusion was carried out on three Shodex OH Pack columns (PAK SB-806M HQ, 1000 Å and exclusion limit = 106 g mol−1). The system was equipped with a triple detection system from Viscotek (TDA 302), i.e. differential refractive index (RI), viscosimetry and light scattering (LS). The number-average molar mass (Mn), the weight-average molar mass (Mw), and the molar mass distribution (polydispersity index: PDI = Mw/Mn) were determined using the triple detection system at a fixed copolymer concentration (5 mg mL−1). The system was calibrated with a poly(ethylene oxide) universal standard from Viscotek (refractive index increment dn/dc = 0.132 mL g−1, molar mass at the peak maximum Mp = 21 917 g mol−1 and intrinsic viscosity [η] = 0.384 dl g−1; refractive index of solvent = 1.33).
The SEC analyses in DMF (+ LiBr, 1 g L−1 and toluene as a flow rate marker) were performed at 60 °C at a flow rate of 0.8 mL min−1. Before injecting the polymer samples, a reaction of methylation was performed, to turn the acid groups into methyl esters, using trimethylsilyldiazomethane.32,33 All polymers were analyzed at a concentration of 5 mg mL−1 after filtration through a 0.45 μm pore size membrane. The separation was carried out on a Polymer Laboratories Gel precolumn (50 × 7.5 mm) and three Polymer Standards Service columns (GRAM columns, 30–1000 Å). The set-up was equipped with a refractive index (RI) detector from Viscotek (Dual 250). The average molar masses (Mn and Mw) and the polydispersity index of all samples were derived from the RI signal by a calibration curve based on poly(methyl methacrylate) (PMMA) standards (from Polymer Standards Service).
The intensity-average diameter (Di) of the latex particles and the dispersity factor (σ) were measured by dynamic light scattering (DLS) at a temperature of 25 °C and an angle of 90° using a Zetasizer Nano Series (S90) from Malvern Instrument. Before measurements, the latex samples were diluted in deionized water or in water at controlled pH. The particles were also visualized by transmission electron microscopy (TEM, JEOL 100 Cx II at 100 keV equipped with a high resolution CCD camera Keen View from SIS).
Results and discussion
1 Determination of the reactivity ratios
Although the literature reports one example of values for the reactivity ratios of methacrylic acid and sodium 4-styrene sulfonate in classical radical copolymerization34 those were measured in conditions that were too far from our own system to be used here. In this work, the monomer reactivity ratios were determined via a thorough kinetic study of the copolymerization of MAA with sodium SS in NMP conditions, initiated by the BlocBuilder® alkoxyamine at 76 °C. In a controlled copolymerization system, it is indeed important to determine the reactivity ratios over a broad conversion range (and not only at low conversion like classically performed studies) to avoid an effect of the low molar mass initiator reactivity and to fit with the long-chain hypothesis. To monitor the kinetics, we decided to recourse to direct NMR analysis of the individual monomer conversions in the reaction medium (see the Experimental for details). To meet the criteria that were previously defined for the NMP of MMA or MAA with a low percentage of styrene, the amount of SS in the comonomer mixture was kept rather low and maintained in the 2.1 to 39.4% range (see Table 1 for the experimental conditions). In contrast to the previous systems, no free SG1 was added in the copolymerization mixture, to avoid the influence of this paramagnetic species on the NMR spectrum quality. The initiator concentration was close to 0.02 mol L−1, and adapted to target a similar final Mn of approximately 10 000 g mol−1.
It appears in Fig. 1 that, although the temperature was very low for an NMP (i.e. 76 °C), the overall conversion rate was rather large and the final conversion reached values as high as 80% in a few hours only. Due to the slight difference in alkoxyamine initiator concentration, a comparison of the copolymerization kinetics cannot be very accurate, but it anyhow appears that the higher the fraction of SS, the lower the copolymerization rate was. This result is in good agreement with the previous conclusions showing a decrease of the propagating radical concentration with the increase in the molar fraction of the styrenic comonomer.17,20 Moreover, there was no final plateau at intermediate conversion, typical of the extensive formation of dead chains as observed in the radical homopolymerization of methacrylate monomers in the presence of SG1.19,35 A downward curvature with a trend toward a plateau at c.a. 60% conversion after 1 h of reaction was only observed for the lowest SS molar fraction of 2.1% (experiment 1). These results indicate that styrene sulfonate played a crucial role in the copolymerization and exhibited a favorable influence on the activation-deactivation equilibrium and on the formation of alkoxyamines, while decreasing the impact of the irreversible side-reactions.
 |
| | Fig. 1 Effect of the molar fraction of sodium 4-styrene sulfonate (SS) in the monomer feed for the copolymerization with methacrylic acid (MAA) in DMSO d6 at 76 °C monitored by direct NMR analysis of the reaction mixture: overall molar conversion vs. time for the experiments 1(■, fss,0 = 0.021), 2 (●,fss,0 = 0.055), 3 (△, fss,0 = 0.078), 4 (◆,fss,0 = 0.198), and 5 (×,fss,0 = 0.394). | |
It is possible to determine the reactivity ratios on the basis of the knowledge of the individual comonomer conversions as a function of time.36 We applied here the terminal model and to reach the best accuracy, we decided to use two different methods, both based on the NMR analysis of the individual and overall molar conversions. In the first step, the reactivity ratio of MAA, rMAA, was determined using the Jaacks method,37 which is valid when the molar fraction of SS is kept low. In the second step, the Skeist equation38,36 was employed via a plot of the molar fraction of SS in the comonomer mixture, fSS, as a function of the monomer overall conversion and determination of the best theoretical equation in which the reactivity ratio of styrene sulfonate, rSS, was used as an adjustable parameter for fitting the data.
Reactivity ratio of methacrylic acid (rMAA).
The reactivity ratio of MAA was calculated from the logarithmic plot of the individual comonomer conversions represented in Fig. 2. According to the Jaacks method,37 a linear relationship as given in eqn (1) should be obtained when the molar fraction of SS is kept low enough to favor the MAA-ended propagating macroradicals. This was actually done for the experiments with the initial molar fraction of SS ranging from fSS,0 = 2.1% to fSS,0 = 19.8%. Fig. 2 indeed shows such a good linearity in the −Ln(1 − xMAA) versus −Ln(1 − xSS) plot and the slope calculated from the overall experimental data, which superimpose quite well, was rMAA = 0.44.| | | −Ln(1 − xMAA) = rMAA × (−Ln(1 − xSS)) | (1) |
Reactivity ratio of sodium 4-styrene sulfonate (rSS).
Following the principle of the Skeist method,38 the variation of fSS, as a function of the monomer overall molar conversion was plotted in Fig. 3 for the five experiments depicted in Table 1, with the initial value fSS,0 ranging from 2.1% to 39.4%. In all cases, a depletion of the proportion of SS in the comonomer mixture was observed indicating a consumption of this monomer faster than that of methacrylic acid. In batch conditions, this trend might be detrimental to the quality of the NMP control, as a decreasing proportion of SS will eventually lead to an increase of the concentration of propagating radicals and hence of the irreversible termination reactions (it might be corrected via a continuous addition of SS throughout the reaction). This point will be discussed later, but one should keep in mind that such a polymerization should not be conducted until complete conversion if one intends to produce macroinitiators for the synthesis of block copolymers.
 |
| | Fig. 3 Experimental points of the molar fraction of sodium 4-styrene sulfonate (fSS) as a function of the overall molar conversion for the controlled radical copolymerization of methacrylic acid with sodium 4-styrene sulfonate. fSS,0 = 2.1% (experiment 1, ■), fSS,0 = 5.5% (experiment 2, ●), fSS,0 = 7.8% (experiment 3, Δ), fSS,0 = 19.8% (experiment 4, ◆) and fSS,0 = 39.4% (experiment 5, ×). Full lines = theoretical curves calculated using the Skeist equation (eqn (2)), with rMAA = 0.44 and rSS = 1.34. | |
For the five series of data points, the theoretical curves from the Skeist equation (see eqn (2)) were superimposed using rMAA = 0.44 for methacrylic acid, and taking rSS as an adjustable parameter. The best fit for all curves simultaneously was found using a simple non-linear least-square method, and gave rSS = 1.34. When fitting the data using both rMAA and rSS as adjustable parameters, the results were rMAA = 0.43 and rSS = 1.30.
| |  | (2) |
Influence of the reactivity ratios on the chain microstructure.
With the reactivity ratios rMAA = 0.44 and rSS = 1.34 in the terminal model, it is possible to estimate the chain compositional microstructure, assuming the system behaves similarly to a conventional radical copolymerization.36 In particular, the majority of the SS subunits should be isolated units (the calculated initial proportion of SS isolated units is 81%, for an initial composition of fSS,0 = 0.078 and it is 85% for an initial composition of fSS,0 = 0.058) and the other units should be essentially in the dyad form. The proportion of isolated units would even increase with the polymerization progress due to the continuous decrease of SS molar fraction in the medium. This is an important outcome for the controlled character of the NMP, as discussed earlier.18,20 Indeed, a chain-end structure of the type MAA-SS-SG1 would be quite favorable to an enhanced alkoxyamine dissociation rate due the penultimate unit effect from the MAA unit, hence allowing the polymerization to proceed at low temperature. Inversely, the SS-SS-SG1 dyad-ended alkoxyamine would hardly redissociate at 76 °C and consequently, the chains would appear as dead species.
Several copolymerizations were performed with the aim of testing the controlled character of the system for low molar mass target copolymers (i.e., Mn = 10 000 g mol−1). The experimental conditions are given in Table 2. For a first series of experiments (experiments 6–8), the initial molar fraction of sodium SS was varied from fSS,0 = 0.088 to fSS,0 = 0.045 and the copolymers were analyzed by SEC in water solution after purification by precipitation. For the experiment 9 similar to experiment 6, the SEC analysis was performed in DMF solution without any purification, but after methylation of the acid units. Finally, the copolymer 10 was prepared according to the same experimental conditions with short polymerization time to design a well-defined macroinitiator.
The plot of the number-average molar mass, Mn, and the polydispersity index, Mw/Mn, as a function of the overall weight conversion is represented in Fig. 4. Note that in a living copolymerization system, the representation of Mnvs. conversion is only expected to be linear when the latter is calculated as a weight conversion. This is particularly needed when both comonomers have very different molar masses. It appears here that a linear evolution was observed, but the experimental Mn values remained rather far above the theoretical ones. This denotes the difficulty we had in finding appropriate conditions for determining Mn and Mw/Mn of our copolymers, i.e. appropriate solvent and appropriate detection and/or calibration. The conditions may not be optimal for absolute values. For instance, a difference was observed between the aqueous SEC results of copolymer 6 and the DMF SEC results of copolymer 9 (besides the potential difference induced by the precipitation of copolymer 6). The discrepancy between the experimental and the expected data may also come from the formation of alkoxyamines based on SS dyads or triads as mentioned above. They would not grow or grow extremely slowly, hence leading to low molar mass species that may not be taken into account within the integration limits of the SEC peaks. The controlled character of the copolymerization was anyhow revealed by the continuous decrease of the polydispersity indices with monomer conversion, toward values as low as 1.2 (Fig. 4) and by the continuous shift of the SEC peaks toward higher molar masses with the progress of the polymerization as shown in Fig. 5. It is important to note that an amount of SS of 5.9 mol% (experiment 7) did not alter the quality of the copolymerization control in comparison to experiment 6 with 8.8 mol%, whereas with 4.5 mol% (experiment 8), the polydispersity indexes were higher. In conclusion, the good control of the polymerization has to be related to the appropriate reactivity of 4-styrene sulfonate, leading to a favorable incorporation of this comonomer from the very beginning of the reaction.
 |
| | Fig. 4 Number-average molar mass, Mn, and polydispersity index, Mw/Mn, versus the overall weight conversion for the polymers resulting from the copolymerization of methacrylic acid with sodium 4-styrene sulfonate (SS) in DMSO at 76 °C with fSS,0 = 0.088 (experiment 6, ● and ○), fSS,0 = 0.059 (experiment 7, × and +), fSS,0 = 0.045 (experiment 8, ▲ and △), and fSS,0 = 0.088 (experiment 9, ■ and □) (straight line: theoretical Mnvs. conversion). The experimental Mn values determined either with methyl methacrylate units in the copolymer (SEC in DMF) or with sodium methacrylate units (SEC in water) were all recalculated on the basis of methacrylic acid units. | |
The copolymer from the experiment 10 was purified by precipitation in diethyl ether after 0.5 h reaction and 35% overall molar conversion and further characterized via SEC in DMF solution. The experimental Mn was 6 900 g mol−1 and the polydispersity index was 1.25. Analysis of the copolymer composition via13C NMR (12 mol% in SS) allowed the average number of each monomer unit per chain to be determined as P(MAA61-co-SS8)-SG1. The living character of this low molar mass copolymer was tested via chain extension in solution and it was then used as a water-soluble macroinitiator in surfactant-free emulsion polymerization.
3 Chain extension in solution
To test the living character of a polymer, chain extension is usually an appropriate method, when the experimental conditions are chosen in such a way that the shift of the SEC peak of the macroinitiator should be complete. Here, we performed two different experiments, one with styrene in DMSO solution and the other one with N,N-dimethylacrylamide in water. Both were performed at 120 °C and the target molar mass at full conversion was higher than 80 000 g mol−1 (see Table 3).
As shown in Fig. 6, the chain extension experiments led to a complete shift of the SEC peak of the macroinitiator, without any visible trace at the initial elution volume, indicating a very efficient reinitiation, toward block copolymers, i.e. P(MAA61-co-SS8)-b-PS and P(MAA61-co-SS8)-b-PDMA. Those results support a high chain-end functionalization of the copolymer with an SG1-based alkoxyamine. Like in the previous case when styrene was used as a comonomer in the synthesis of the macroinitiator instead of sodium 4-styrene sulfonate,23 and in agreement with the calculated microstructure based on the reactivity ratios, one supposes that the chain-end structure is of the type: MAA-SS-SG1. This explains both the low polymerization temperature for the synthesis of the macroinitiator and the living character of the so-formed chains.
 |
| | Fig. 6 Chain extension of the P(MAA61-co-SS8)-SG1 alkoxyamine macroinitiator via SG1-mediated polymerization of styrene (S) in DMSO and of N,N-dimethylacrylamide (DMA) in water at 120 °C: SEC chromatograms of the P(MAA61-co-SS8)-SG1 macroinitiator (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ) and of the copolymers (⋯), (a) P(MAA61-co-SS8)-b-PS from experiment 11 and (b) P(MAA61-co-SS8)-b-PDMA from experiment 12. The SEC analyses were performed in DMF solution. ) and of the copolymers (⋯), (a) P(MAA61-co-SS8)-b-PS from experiment 11 and (b) P(MAA61-co-SS8)-b-PDMA from experiment 12. The SEC analyses were performed in DMF solution. | |
Fig. 7 shows that both polymerizations proceeded in a fast manner and reached high although incomplete conversions (ca. 80%). This was already seen with the macroinitiators based on styrene as a comonomer. This result might be explained by an accumulation of SG1 due to irreversible termination reactions at such high conversions, for which styrene proportion in the polymerization medium is very low, and might be overcome by a continuous feeding in styrene. The polymerizations were however very well controlled as shown by the molar mass and polydispersity index results, as well as the shift of the size exclusion chromatograms throughout the polymerization reaction (Table 5 and Fig. 8).
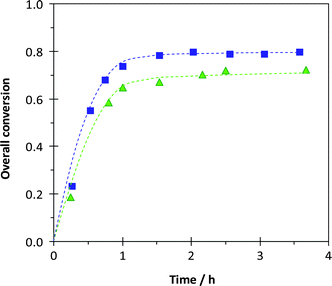 |
| | Fig. 7 Surfactant-free, ab initio, batch emulsion polymerizations of methyl methacrylate with 9.0 mol% of styrene initiated by the P(MAA61-co-SS8)-SG1 macroinitiator at 90 °C. Conversion versus time plots for experiment 13 (■, 6.0 mmol L−1 in macroinitiator) and for experiment 14 (▲, 8.7 mmol L−1 in macroinitiator). | |
| Expt |
Time/h |
Conversion (%) |
M
n,th
/g mol−1 |
M
n,SEC
/g mol−1 |
M
w/Mn |
Dic/nm |
σ |
|
Theoretical Mn calculated at the experimental conversion.
Raw polymer analyzed after methylation by size exclusion chromatography in DMF with LiBr (without recalculation to turn the methyl methacrylate units into methacrylic acid units).
Intensity-average diameter of the final latex from DLS (large aggregates appear in the intensity distribution but are negligible in the number distribution, which explains the large values of σ).
|
|
13
|
3.5 |
80 |
41 600 |
44 200 |
1.37 |
43 |
0.29 |
|
14
|
3.6 |
72 |
28 650 |
30 100 |
1.34 |
29 |
0.43 |
 |
| | Fig. 8 Surfactant-free, ab initio, batch emulsion polymerization of methyl methacrylate with 9.0 mol% of styrene initiated by the P(MAA61-co-SS8)-SG1 macroinitiator at 90 °C. (a) Mn and Mw/Mn (SEC in DMF, methylated copolymers, PMMA calibration) versus conversion plot for the experiment 13 (■ and □, 6.0 mmol L−1 in macroinitiator) and for the experiment 14 (▲ and Δ, 8.7 mmol L−1 in macroinitiator) (straight lines: theoretical Mnvs. conversion); (b) size exclusion chromatograms as a function of conversion: P(MAA61-co-SS8)-SG1 () and P(MAA61-co-SS8)-b-P(MAA-co-S) block copolymers (⋯) for the experiment 13. | |
The system thus led to amphiphilic diblock copolymers with a short hydrophilic segment and a long hydrophobic one. Due to the insolubility of the P(MMA-co-S) copolymer block in water, a self-assembly process took place simultaneously to the growth step and led to block copolymer micelles with very low hydrodynamic diameter and narrow size distribution as shown by the TEM picture (Fig. 9). The presence of a very small proportion of aggregates was revealed by the dispersity factor of the DLS analysis (σ in Table 5), which was above 0.1. The DLS number distribution presented one single narrow peak for both experiments, whereas the intensity distribution exhibited shoulders at large diameters.
In order to evaluate the colloidal stability of the particles as a function of pH, a variation of pH was applied to the aqueous solution used for diluting the samples in DLS. A broad range of values, i.e. from 2 to 10, was studied and the results are shown in Fig. 10. As observed previously with the poly(methacrylic acid) macroinitiators containing a small percentage of styrene,26 the pH had no significant effect on the average particle size, i.e. the hydrophilic corona had no propensity to fully extend. However the replacement of styrene with 4-styrene sulfonate had a strong influence on the colloidal stability at low pH. Indeed, the particles remained stable at a pH of 2, whereas they started to lose stability at pH below 5 when styrene was present in the hydrophilic chain and completely coagulated at pH = 4 and below. This is easily explained by the strong acid character of 4-styrene sulfonic acid, which brings a sufficiently high charge density to the particle surface at low pH, and hence maintains their electrostatic repulsion even after the collapse of the poly(methacrylic acid) segments. It is also important to mention that the particles of experiment 13 and 14 kept the same diameter upon storage for more than 3 months in concentrated conditions at room temperature, indicating a very good stability in time.
 |
| | Fig. 10 Surfactant-free, ab initio, batch emulsion polymerization of methyl methacrylate with 9.0 mol% of styrene initiated by the P(MAA61-co-SS8)-SG1 macroinitiator at 90 °C. Effect of the pH of the aqueous solution used for diluting the sample on the intensity-average diameter from DLS for the experiment 13 (■) and the experiment 14 (▲). | |
Conclusion
The SG1-mediated copolymerization of methacrylic acid with a small percentage of sodium 4-styrene sulfonate performed in DMSO solution at 76 °C was shown to exhibit all the characteristics of a controlled/living system. The reactivity ratios in the terminal model, i.e.rMAA = 0.44 and rSS = 1.34, indicate a favored incorporation of SS at the beginning of the copolymerization, enabling an efficient deactivation of the propagating radicals by the nitroxide SG1. However, a rapid depletion in this comonomer may induce a loss of control at very high conversion. The method is nevertheless particularly well-suited for the design of living polymers intended to be used as hydrophilic macroinitiators for the synthesis of amphiphilic block copolymers. This was demonstrated in both solution polymerization and ab initio, batch emulsion polymerization. The latter process allowed well-defined, self-stabilized block copolymer nanoparticles to be formed at low temperature, in a single step, by simultaneous chain growth and self-assembling. The advantage of 4-styrene sulfonate over styrene was an enhanced colloidal stability over a broad pH range, including very low pH, at which the collapse of the PMAA shell would normally lead to extensive flocculation.
Acknowledgements
The authors wish to thank Arkema for the financial support of Ségolène Brusseau's PhD thesis and for kindly providing the nitroxide SG1 and the BlocBuilder® MA alkoxyamine. They are grateful to Patricia Beaunier for transmission electron microscopy analysis, to Agnès Pallier for aqueous SEC analysis at PPMD/ESPCI and to Jutta Rieger for SEC analysis in DMF at the Laboratoire de Chimie des Polymères.
References
-
Handbook of Radical Polymerization, ed. K. Matyjaszewski and T. Davis, 2002, John Wiley & Sons, Hoboken, New Jersey Search PubMed.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661 CrossRef CAS.
-
M. Monteiro, B. Charleux, in Chemistry and Technology of Emulsion Polymerization, ed. A. van Herk, Blackwell Publishing Ltd, Oxford, 2005, ch. 5, pp. 111 Search PubMed.
- M. Save, Y. Guillaneuf and R. G. Gilbert, Aust. J. Chem., 2006, 59, 693 CrossRef CAS.
-
B. Charleux, F. Ganachaud, in Macromolecular Engineering: From Precise Macromolecular Synthesis to Macroscopic Materials Properties and Application, ed. K. Matyjaszewski, Y. Gnanou, L. Leibler, Wiley-VCH, 2007 Search PubMed.
- M. F. Cunningham, Prog. Polym. Sci., 2008, 33, 365 CrossRef CAS.
- P. B. Zetterlund, Y. Kagawa and M. Okubo, Chem. Rev., 2008, 108, 3747 CrossRef CAS.
- J. K. Oh, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6983 CrossRef CAS.
- B. Charleux and J. Nicolas, Polymer, 2007, 48, 5813 CrossRef CAS.
- G. Delaittre, J. Nicolas, C. Lefay, M. Save and B. Charleux, Chem. Commun., 2005, 614 RSC.
- G. Delaittre, J. Nicolas, C. Lefay, M. Save and B. Charleux, Soft Matter, 2006, 2, 223 RSC.
- G. Delaittre, M. Save and B. Charleux, Macromol. Rapid Commun., 2007, 28, 1528 CrossRef CAS.
- G. Delaittre and B. Charleux, Macromolecules, 2008, 41, 2361 CrossRef CAS.
- G. Delaittre, C. Dire, J. Rieger, J. L. Puteaux and B. Charleux, Chem. Commun., 2009, 2887 RSC.
-
D. Benoit, S. Grimaldi, J.-P. Finet, P. Tordo, M. Fontanille, Y. Gnanou, in ACS Symp. Series: Controlled Radical Polymerization, ed. K. Matyjaszewski, 1998, 685, pp. 225 Search PubMed.
- B. Charleux, J. Nicolas and O. Guerret, Macromolecules, 2005, 38, 5485 CrossRef CAS.
- J. Nicolas, C. Dire, L. Mueller, J. Belleney, B. Charleux, S. R. A. Marque, D. Bertin, S. Magnet and L. Couvreur, Macromolecules, 2006, 39, 8274 CrossRef CAS.
- C. Dire, J. Belleney, J. Nicolas, D. Bertin, S. Magnet and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6333 CrossRef CAS.
- J. Nicolas, L. Mueller, C. Dire, K. Matyjaszewski and B. Charleux, Macromolecules, 2009, 42, 4470 CrossRef CAS.
- B. Lessard and M. Maric, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2574 CrossRef CAS.
- B. Lessard, C. Tervo, S. De Wahl, F. Clerveaux Jr., K. K. Tang, S. Yasmine, S. Andjeli, A. D'Alessandro and M. Maric, Macromolecules, 2010, 43, 868 CrossRef CAS.
- C. Dire, B. Charleux, S. Magnet and L. Couvreur, Macromolecules, 2007, 40, 1897 CrossRef CAS.
- J. Nicolas, P. Couvreur and B. Charleux, Macromolecules, 2008, 41, 3758 CrossRef CAS.
- C. Dire, J. Nicolas, S. Brusseau, B. Charleux, S. Magnet and L. Couvreur, ACS Symp. Series, 2009, 1024, 303 CAS.
- C. Dire, S. Magnet, L. Couvreur and B. Charleux, Macromolecules, 2009, 42, 95 CrossRef CAS.
- J. Nicolas, S. Brusseau and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 34 CrossRef CAS.
- M. Bouix, J. Gouzi, B. Charleux, J.-P. Vairon and P. Guinot, Macromol. Rapid Commun, 1998, 19, 209 CrossRef CAS.
- W. Huang, B. Charleux, R. Chiarelli, L. Marx, A. Rassat and J.-P. Vairon, Macromol. Chem. Phys., 2002, 203, 1715 CrossRef CAS.
- T. N. T. Phan and D. Bertin, Macromolecules, 2008, 41, 1886 CrossRef CAS.
- R. Nicolaÿ, L. Marx, P. Hémery and K. Matyjaszewski, Macromolecules, 2007, 40, 6067 CrossRef CAS.
- L. Couvreur, C. Lefay, J. Belleney, B. Charleux, O. Guerret and S. Magnet, Macromolecules, 2003, 36, 8260 CrossRef CAS.
- C. Lefay, J. Belleney, B. Charleux, O. Guerret and S. Magnet, Macromol. Rapid Commun., 2004, 25, 1215 CrossRef CAS.
- J. Belleney, G. Hélary and V. Migonney, Eur. Polym. J., 2002, 38, 439 CrossRef CAS.
- R. McHale, F. Aldabbagh and P. B. Zetterlund, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2194 CrossRef CAS.
-
G. Odian, Principles of Polymerization, John Wiley & Sons, New York, 1994 Search PubMed.
- V. Jaacks, Makromol. Chem., 1972, 161, 161 CrossRef CAS.
- I. Skeist, J. Am. Chem. Soc., 1946, 68, 1781 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.